

Abstract
We previously reported that the sympathetic neurotransmitter neuropeptide Y (NPY) is potently angiogenic, primarily through its Y2 receptor, and that endogenous NPY is crucial for capillary angiogenesis in rodent hindlimb ischemia. Here we sought to identify the source of NPY responsible for revascularization and its mechanisms of action. At d 3, NPY−/− mice demonstrated delayed recovery of blood flow and limb function, consistent with impaired collateral conductance, while ischemic capillary angiogenesis was reduced (∼70%) at d 14. This biphasic temporal response was confirmed by 2 peaks of NPY activation in rats: a transient early increase in neuronally derived plasma NPY and increase in platelet NPY during late-phase recovery. Compared to NPY-null platelets, collagen-activated NPY-rich platelets were more mitogenic (∼2-fold vs. ∼1.6-fold increase) for human microvascular endothelial cells, and Y2/Y5 receptor antagonists ablated this difference in proliferation. In NPY+/+ mice, ischemic angiogenesis was prevented by platelet depletion and then restored by transfusion of platelets from NPY+/+ mice, but not NPY−/− mice. In thrombocytopenic NPY−/− mice, transfusion of wild-type platelets fully restored ischemia-induced angiogenesis. These findings suggest that neuronally derived NPY accelerates the early response to femoral artery ligation by promoting collateral conductance, while platelet-derived NPY is critical for sustained capillary angiogenesis.—Tilan, J. U., Everhart, L. M., Abe, K., Kuo-Bonde, L., Chalothorn, D., Kitlinska, J., Burnett, M. S., Epstein, S. E., Faber, J. E., Zukowska, Z. Platelet neuropeptide Y is critical for ischemic revascularization in mice.
Keywords: angiogenesis, NPY
Revascularization in occlusive disease is comprised of two processes, arteriogenesis and angiogenesis. Arteriogenesis is an ischemia-independent process driven by shear stress and hemodynamic forces and serves to bypass the existing occlusion (1). The early response to an occlusion, remodeling (enlargement) of preexisting collateral vessels, can occur as early as 12 h after femoral artery ligation (FAL; ref. 2). Several days after FAL, vascular cell proliferation increases in association with sprouting angiogenesis (1). In contrast to arteriogenesis, this process is primarily driven by tissue ischemia and highly dependent on hypoxia inducible factors and vascular endothelial growth factor (VEGF; ref. 3). Angiogenesis begins with the degradation of extracellular matrix, followed by the migration, proliferation, and differentiation of endothelial cells, resulting in capillary formation and ultimately a new capillary network (3). Recently, basic fibroblast growth factor (bFGF) has been shown to regulate VEGF-induced angiogenesis in mice (4), highlighting the complexity of these multifaceted processes. Similarly, our laboratory has also shown that neuropeptide Y (NPY) is upstream of VEGF signaling (5). While endothelial cells play a significant role in both processes of revascularization, vascular smooth muscle cell (VSMC) proliferation is critical to the outward remodeling of native collaterals.
Although VEGF and bFGF are among the best-described factors involved in collateral vessel development, there is growing evidence that neuronal factors may be crucial for this process (6, 7). Catecholamines have been shown to play a potential role in augmenting collateral vessel growth, as well as angiogenesis in tissue ischemia (8), while other researchers have shown a role for them in hypertrophic remodeling (9) and stimulating VSMC proliferation (10). Moreover, NPY, a sympathetic neurotransmitter coreleased with norepinephrine, has recently emerged as a vascular growth factor, acting in a receptor- and cell-specific manner. It is potently angiogenic by activating its Gi-coupled receptors, the Y2 and Y5 receptors (Y2R and Y5R; refs. 11, 12), and mitogenic for VSMCs via Y1R and Y5R (13). Although NPY is primarily released from sympathetic nerves, there are ≥2 extraneuronal sources of NPY. The endothelium possesses an autocrine system [the peptide, a serine protease, dipeptidyl peptidase IV (DPPIV), which forms a Y2/Y5R agonist, NPY3–36, as well as NPY receptors] that is involved in endothelial cell proliferation, migration, and differentiation (11). Also, the peptide has been identified in megakaryocytes and platelets (14–16) and released from platelets at sites of vascular remodeling in rats (14, 17). Furthermore, our laboratory has recently shown that increased activation of platelet NPY is associated with enhanced atherosclerotic lesion burden and vulnerability in patients with peripheral arterial disease (PAD; ref. 18).
As a rich source of angiogenic growth factors, a role for circulating platelets in tumor angiogenesis was proposed, in part, as a potential explanation for the effectiveness of anticoagulants in cancer therapy (19). Well documented and studied in tumor angiogenesis since then, circulating platelets have also been shown to provide delivery of arterio-/angiogenic factors under ischemic conditions (20, 21). In addition, several groups have demonstrated platelet-derived factors, such as bFGF, VEGF, and matrix metalloproteinases, to mediate arteriogenic and angiogenic processes in humans and in animal models of PAD (22, 23). These studies suggest that platelet activation may release stored factors that contribute to both the shear-stress-driven arteriogenic process and the ischemic-driven angiogenic process.
We and others have shown that exogenous NPY and NPY3–36 have potent angiogenic activity in both in vitro (5) and in vivo (24, 25) models of ischemia and tumor growth (26, 27), and that inhibition or loss of Y2Rs and DPPIV severely impairs capillary angiogenesis in these situations. In neural crest-derived tumors, NPY stimulated the formation of vessels possessing endothelial and VSMCs, resembling normal vessels (28). While these studies have established NPY as an angiogenic factor, Y2R activation also increases collateral-dependent blood flow (29), suggesting a role in collateral growth or dilation. These findings were recently supported in a chronic model of myocardial ischemia, demonstrating exogenous NPY3–36-mediated improved myocardial function, along with increased proangiogenic growth factor expression and decreased antiangiogenic factor expression (30). While these studies have primarily focused on neurogenic ischemic revascularization, the temporal relationship of angiogenic NPY expression and the source of endogenous NPY involved in this process have not been determined. Thus, the purpose of our study was to elucidate the role of endogenous NPY and its receptors in ischemic capillary revascularization and identify its source and mechanism of actions.
MATERIALS AND METHODS
Animals
The following male animals were used: 4- to 12-wk-old NPY−/− mice and their wild-type control 129S1/SvImJ mice (Jackson Laboratory, Bar Harbor, ME, USA), and 6- to 8-wk-old 129X1/SvJ (Jackson Laboratory) and Wistar rats (150–175 g; Charles River, Boston, MA, USA). All protocols were approved by the Animal Care and Use Committees for Ethics in Animal Experiments of Georgetown University and the MedStar Research Institute.
Unilateral hindlimb ischemia
Animals were anesthetized with Avertin (250 mg/kg) intraperitoneally. Hair was removed from the hindquarters with depilating cream, with care taken to avoid erythema. The femoral artery was exposed aseptically through a 2-mm incision and isolated from the femoral vein and nerve, with care taken to avoid damage to vessels or nerve. The femoral artery was ligated as described previously (5). Briefly, 6-0 sutures were used to ligate proximal to the bifurcation of the profundus and femoral arteries. A similar procedure was carried out for sham-treated controls without ligation of the femoral artery.
As described previously for hindlimb ischemia (31), noninvasive measurements of superficial hindlimb perfusion were obtained before ligation, immediately after ligation, and 3, 7, and 14 d after ligation using a scanning laser-Doppler perfusion imager (model LDI2-IR; Moor Instruments, Wilmington, DE, USA). Calculated perfusion was expressed as the laser Doppler perfusion imaging (LDPI) ratio of the ischemic to contralateral nonischemic limb.
In vivo assessment of limb function and ischemic damage
Modification of a clinical standard score for ischemic lower extremities (32) was used serially for semiquantitative assessment of limb function (0: normal, 1: plantar flexion and grasping, 2: plantar flexion, and 3: dragging of foot). Semiquantitative measurement of the ischemic damage was also assessed (0: normal, 1: mild discoloration, 2: severe discoloration, 3: necrosis, and 4: amputation; ref. 33).
Tissue processing, immunohistochemistry, and image analysis
Skeletal muscle samples were frozen with liquid nitrogen (Roberts Oxygen, Rockville, MD, USA). Slides with 5-μm sections were prepared using a cryostat and stored at −80°C. Morphology was studied using sections stained with hematoxylin-eosin.
Immunohistochemistry
Immunostaining was carried out according to the manufacturer's protocols using the following antibodies (Abs): CD31/PECAM-1 anti-rat and anti-mouse (1:100; BD Pharmingen, San Diego, CA, USA) with 2° Abs biotinylated goat anti-mouse (1:25; Vector Laboratories, Burlingame, CA, USA) and biotinylated goat anti-rat (1:100; (BD Pharmingen), respectively. Staining was performed on frozen slides with biotin-avidin peroxidase (ABC Kit; Vector Laboratories) for visualization. Sections were collected and evaluated using a Nikon Eclipse E600 photomicroscope (Nikon, Melville, NY, USA), and images were analyzed using U.S. National Institutes of Health (NIH) Scion Image software (Scion Corp., Frederick, MD, USA).
Assessment of revascularization
Capillary vascularization was determined by the density of PECAM-1/CD31 positively stained vessels, quantified as the number of capillaries per fiber in 4 to 6 fields/muscle.
qRT-PCR of NPY, NPY receptors, and DPPIV mRNAs
RNA was isolated using Tri Reagent (Sigma-Aldrich, St. Louis, MO, USA), and cDNA was synthesized by Superscript II Reverse Transcriptase (Invitrogen Life Technologies, Carlsbad, CA, USA) using random hexamers (Perkin Elmer, Foster City, CA, USA) or iScript (Bio-Rad, Hercules, CA, USA). Quanitative RT-PCR was performed using the iCycler iQ real-time PCR detection system (Bio-Rad). Samples were run in duplicate, each consisting of 2 μl of cDNA amplified in 20 μl of 1× TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA) containing primers (300 nM each) and FAM-labeled TaqMan MGB probe (200 nM) predesigned by Applied Biosystems. cDNA samples were amplified by PCR for 40 cycles (95°C for 45° s, 58°C for 45 s, 60°C for 1 min). Results were analyzed using iCycler iQ 3.0 software, provided by Bio-Rad, and expression levels were calculated by the comparative CT method using β-actin as an endogenous reference gene.
Blood
Immediately before euthanasia, blood was collected from the vena cava (1-ml syringe with 23-gauge needle) and processed for ELISA: platelet-rich plasma (PRP), 150 g for 10 min at 4°C; platelet-poor plasma (PPP), 10,000 g for 2 min at 4°C. For platelet treatment of endothelial cells with collagen-activated PRP, following isolation of PRP, this fraction was transferred to a fresh tube and incubated with collagen (Chrono-Log Corp., Havertown, PA, USA) at 5 μg/μl for 6 min at room temperature, inverting throughout incubation. The mixture was then centrifuged at 2000 g for 10 min at 4°C. The supernatant was then collected and snap-frozen.
NPY enzyme immunoassay (EIA)
The NPY EIA kit (S-1145) was purchased from Bachem (Torrance, CA, USA), and the measurements were performed according to the manufacturer's procedure. Prior to assay, plasma extraction with C-18 Sep-Columns (Bachem) was performed according to the manufacturer's procedure.
Platelet count
Harvested blood, as described above, from the inferior vena cava was centrifuged at 150 g for 10 min at 4°C in order to isolate the PRP, which was then centrifuged at 2000 g for 10 min at 4°C and the supernatant discarded. The remaining pellet (platelets) was washed and resuspended with 1× PBS. Resuspended platelets were diluted and counted on a hemacytometer using a phase-contrast microscope.
Cell culture
Adult dermal human microvascular endothelial cells (HMVEC-dAd) were purchased from Lonza at passage 6 and cultured up to passage 8 in EBM-2MV medium in a 5% CO2 humidified incubator. HMEVCs were split at a 1:5 ratio after each passage and allowed to grow to confluence.
DNA synthesis assay
HMVECs were plated onto 96-well dishes (1500 cells/well), cultured for 2 d, growth-arrested in serum-free medium for 24 h and then treated with medium supplemented with 1.5% of PPP, PRP, or collagen-activated PRP plasma derived from NPY+/+ and NPY−/− mice (for preparation of plasma; see below). Serum-free medium and standard EBM-2MV medium served as negative and positive controls, respectively. Four hours after treatment, 0.5 μCi [3H]-thymidine/well was added. Twenty hours later, the cells were washed twice in PBS, trypsinized, and lysed by freezing and thawing under hypotonic conditions. Then cell lysates were transferred to filtermats (Wallac, Turku, Finland) in a 96-well harvester (Harvester 96 Mach II; Tomtec, Orange, CT, USA), and radioactivity was counted in a Betaplate Liquid Scintillation Counter (model 1205; Wallac). Alternatively, the proliferation levels were measured using MTS-based CellTiter 96AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA). Levels of proliferation were normalized to low serum control.
Platelet depletion and transfer
An adaptation of the Kuter et al. (34) nonimmune method of sustained thrombocytopenia followed by platelet transfusion to study neointimal hyperplasia (35), established in our laboratory (unpublished results), was used here to isolate the effect of platelet-derived NPY on ischemic capillary angiogenesis. Chemical-induced thrombocytopenia was established and maintained with busulfan (25 mg/kg; Sigma) injections at d −4, −2, +1, +8, relative to FAL d 0. Based on previous cell counts, platelets were transfused at the time of surgery and on d +7 at 4 × 105 platelets/ml via tail vein injection.
Statistical analysis
All data are presented as means ± se. Based on comparison, nonparametric data were analyzed by the Mann-Whitney U test, while parametric data were analyzed by 1-way or 2-way (repeated measures) ANOVA with post hoc multiple comparison t tests using Dunnet's, Bonferroni's, or Tukey's test using GraphPad Prism 5.00 for Windows (GraphPad Software, San Diego, CA, USA); P < 0.05 was considered statistically significant for the indicated n per group. For mRNA analysis with real time RT-PCR, genes from individual samples not detected were given a maximum value of 40 cycles as the limit of detection.
RESULTS
Ischemic revascularization is impaired in NPY−/− mice with hindlimb ischemia
To test the hypothesis that endogenous NPY stimulates revascularization of distal tissues in response to ischemia, systemic knockouts and wild-type controls were observed over 14 d following FAL, a known model of hindlimb ischemia. Serial blood flow measurements were acquired by LDPI of the plantar foot in both hindpaws (Fig. 1A, B). Immediately following FAL, blood flow in the ligated hindpaw of both groups dropped to ∼25% of the nonligated contralateral hindpaw (Fig. 1B). At 3 d, perfusion in NPY+/+ mice improved by almost 3-fold from levels immediately after FAL, while it was delayed in the NPY−/− group and doubled from following ligation. By 7 d, the NPY−/− group's recovery matched that of the NPY+/+ group. At termination of the experiment, both groups had attained similar levels of recovery.
Figure 1.
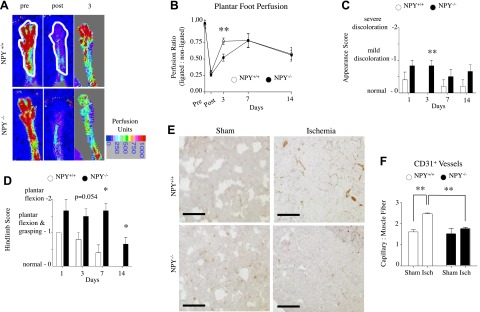
Revascularization in NPY−/− mice. A) Laser Doppler perfusion images of representative plantar hindpaw in NPY+/+ and NPY−/− mice. B) Quantification of perfusion determined for anatomically defined region of interest (white outline) in hindpaws pre- and post-FAL and 3, 7, and 14 d after. Values are expressed as a ratio of perfusion units in ligated limb to nonligated limb. **P < 0.01; Student's t test. C) Foot appearance score (index of ischemia): 0, normal; 1, mild discoloration; 2, severe discoloration; 3, necrosis; 4, amputation. All NPY+/+ mice were normal on d 3. Data are presented as means ± se. **P < 0.01; Mann-Whitney U test. D) Hindlimb usage score (index of muscle function): 0, normal; 1, plantar flexion and grasping; 2, plantar flexion; 3, dragging. A higher score indicates impaired use or appearance. All NPY+/+ mice were normal on d 14. Data are presented as means ± se. *P < 0.05; Mann-Whitney U test. E) Cross section of gastrocnemius muscle from sham-operated or ischemic NPY+/+ and NPY−/− mice at 14 d immunostained with CD31 for microvessel density. Scale bars = 100 μm. F) Quantification of microvessel density. Data are presented as means ± se; n = 5–6/group. **P < 0.01; 1-way ANOVA with post hoc Bonferroni's test.
To evaluate the functional consequences of FAL in NPY−/− and NPY+/+ mice, clinical-like use and appearance scores of the hindpaw were obtained. For appearance (Fig. 1C), mice were given a score from 0 to 4 at each time point. Consistent with the delay in flow recovery, the hindpaw appearance of NPY−/− mice of the ligated limb was worse than NPY+/+ mice, with differences evident at d 3 and all NPY+/+ mice appearing normal (score=0). These differences were confirmed by hindlimb use scores (Fig. 1D). For use (Fig. 1D), mice were given a clinical score from 0 to 3 at each time point. All NPY+/+ mice regained normal use of the hindpaw, achieving a score of 0 14 d after FAL. In contrast, NPY−/− failed to display full use of the paw by d 14, with significant differences beginning at d 7. These data suggest that a lack of endogenous NPY delays flow recovery within the first week after ligation.
At d 14 following ligation, CD31 staining demonstrated an ischemia-induced increase in capillary-to-muscle fiber ratio in the gastrocnemius muscle of the ligated limb in NPY+/+ mice (Fig. 1E, F). In contrast, no increase was observed in NPY−/− mice. This decreased ischemic response in NPY−/− animals is consistent with the essential role of endogenous NPY in capillary angiogenesis (5).
Circulating levels of platelet NPY increase in hindlimb ischemia
The above data suggest that endogenous NPY is involved in recovery of blood flow in hindlimb ischemia. NPY is secreted from the sympathetic nerves in proportion to nerve activity (36), which is known to increase during ischemia (5) and stress (37). As a result, circulating plasma levels of the peptide accurately represent neuronal release. However, platelets are also a source for NPY. To determine whether circulating NPY levels were elevated during hindlimb ischemia, and if so, in which blood fraction, peptide levels in plasma and platelets as well as mRNA expression in megakaryocytes were measured in rodents.
As previously reported, in murine (38) and human (18) models, basal NPY immunoreactivity (NPY-ir) levels in PPP and PRP are indistinguishable. However, conditions known to activate the NPY system, such as stress and atherosclerosis, stimulate accumulation of NPY specifically in platelets, resulting in increased levels of the peptide in PRP, as compared to PPP fraction (18, 38). To determine whether a similar phenomenon is associated with ischemia, NPY levels in PPP and PRP were compared in mice 21 d following FAL (Fig. 2A). Given platelet turnover of ∼5 d, this extended period after ligation allowed for complete renewal of the platelet population to reflect ischemia-induced changes in megakaryocytes. Indeed, increased NPY-ir in PRP as compared to PPP indicated that platelets might serve as an additional source of NPY, supplementing soluble sympathetic-neuron-derived peptide. In a separate group of mice, we observed that circulating platelet counts increase in response to FAL (Fig. 2B). To determine the source of NPY-ir from platelets, we looked at NPY mRNA levels within whole bone marrow. Since changes in megakaryocytes would precede those in platelets, we measured NPY mRNA at an earlier time point of 14 d. Although the increase in NPY mRNA was not statistically significant at d 14 (Fig. 2C), the trend is consistent with significant increases in NPY-ir of PRP (Fig. 2D) and circulating platelet counts (Fig. 2B) at later time points following FAL. This time course is in line with platelet turnover time of approximately 1 wk. Together, these data suggest that hindlimb ischemia up-regulates circulating platelet NPY levels via an increase in both platelet count and peptide synthesis in megakaryocytes.
Figure 2.
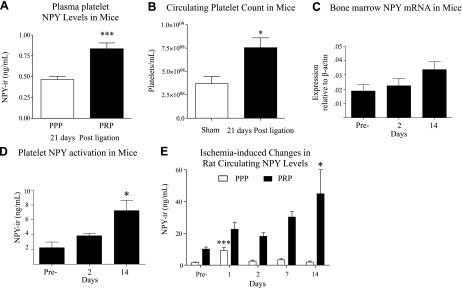
Ischemia activates platelet-derived NPY. A) NPY-ir levels of PPP (white) and PRP (black) 21 d after FAL in 129S/SvImJ NPY+/+ mice. Data are presented as means ± se n = 7–8/group. ***P < 0.001; Student's t test. B) Circulating platelet counts in sham-operated and 21 d post-FAL 129S/SvImJ mice. Data are presented as means ± se; n = 3–4/time point. *P < 0.05; Student's t test. C) NPY mRNA expression in isolated whole bone marrow (n=3–4/time point) from 129×1/SvJ mice preligation and 2 and 14 d after ligation. D) NPY-ir levels of PRP (n=5–12/time point) from 129X1/SvJ mice preligation and 2 and 14 d after ligation. E) NPY-ir levels of PPP and PRP following FAL in Wistar rats; n = 3–8/time point. *P < 0.05, ***P < 0.001 vs. d 0; 1-way ANOVA with post hoc Bonferroni's test.
As shown above, mice lacking NPY exhibit impaired perfusion and hindlimb function in the early phase and decreased capillary density in the late phase following FAL. To determine whether these time-dependent NPY effects are due to parallel changes in its levels in plasma (PPP), reflecting release from the sympathetic nerves (39), or in platelets (reflected in PRP) we used a rat model of hindlimb ischemia. Rats express high levels of NPY in platelets (17), thereby allowing us a greater range to evaluate changes over time in response to FAL. Furthermore, we have shown that this model has robust NPY-dependent angiogenesis (5). Following FAL, endogenous plasma levels of NPY-ir increased on d 1 before returning to basal levels on d 2 (Fig. 2E). In contrast, and in line with normal platelet turnover of ∼5 d, platelet NPY-ir levels were activated later, and steadily increased to a 4-fold elevation at 14 d (Fig. 2E). As observed in mice, these data suggest that hindlimb ischemia increases circulating NPY levels in platelets. Furthermore, they suggest that sympathetic-derived NPY is activated early on in the recovery phase.
Hindlimb ischemia induces time-dependent changes in activation of NPY, its receptors, and DPPIV in rats
To further study the involvement of NPY in response to FAL, we measured mRNA levels of NPY, Y1R, Y2R, and DPPIV in the adductor and gastrocnemius muscles of Wistar rats by real-time RT-PCR. Interestingly, a coordinated peak in expression of NPY, Y2R, and DPPIV was observed 2 d after FAL in both tissues that, however, experience distinct primary stimuli: fluid shear stress in the adductor muscle and hypoxia in the gastrocnemius muscle (Fig. 3A, B). However, peaks in the adductor muscle did not reach significance. Y1R expression was unchanged early but trended toward a late increase after ligation in the adductor (Fig. 3A). In the gastrocnemius, ischemia increased Y2 (16-fold) and DPPIV mRNA (4-fold; P=0.0581) at 2 d (Fig. 3B).
Figure 3.
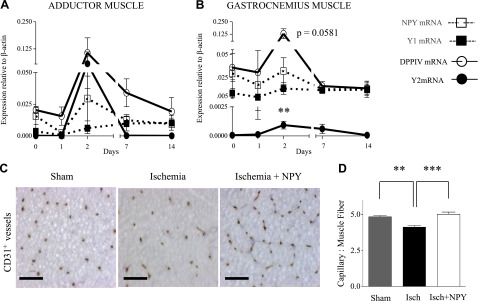
Time-dependent expression of the NPY system in rat ischemia. A, B) mRNA expression levels at similar time points of adductor (A) and gastrocnemius (B) muscles following FAL in Wistar rats, measured by real time RT-PCR. Data are presented as mean ± se expression relative to β-actin (n=3–8/time point). **P < 0.01 vs. d 0; 1-way ANOVA with post hoc Bonferroni's test. C) Representative cross sections of gastrocnemius muscle from Wistar rats 5 d following sham operation, FAL or FAL and NPY (slow-release pellet, 1 μg/pellet/14 d) immunostained with CD31 for microvessel density. Scale bars = 50 μm. D) Quantification of CD31+ vessels. Data are presented as mean ± se capillary-to-muscle fiber ratio (n=6/group). **P < 0.01, ***P < 0.001; 1-way ANOVA with post hoc Bonferroni's test.
Immunohistological analyses revealed decreased capillarity in ischemic rat gastrocnemius muscle at d 5 following ligation, which has been previously reported (5), while exogenous NPY restored the number of capillaries per muscle fiber to that of sham-operated hindlimbs (Fig. 3C, D). In contrast, no differences in vascularity between these tissues were observed at d 14 following FAL (data not shown). These observations, along with the peak of Y2R and DPPIV mRNA activation at 2 d following occlusion, suggest a role for NPY in early stages of revascularization. In contrast, during the later stages when elevated levels of platelet NPY may saturate the system, addition of the exogenous peptide has no effect.
Platelet NPY stimulates endothelial cell proliferation
Based on different temporal peaks observed in NPY-ir in PPP and PRP in the rat model, we hypothesized that platelet NPY is important for blood flow recovery during hindlimb ischemia. Platelet-derived angiogenic growth factors reside in platelet α-granules (40) that have been preferentially released by collagen activation (41). To determine a potential mechanism by which platelets could serve as an angiogenic source of NPY, we isolated platelet and plasma fractions from mice 14 d following FAL and tested the effects of these fractions on endothelial cell proliferation in vitro (Fig. 4). In growth-arrested HMVECs exposed for 24 h to either PPP or collagen-activated PRP from NPY+/+ mice, all fractions were proliferative (Fig. 4A). Interestingly, ischemia increased the proliferative potential of collagen-activated PRP but not PPP (Fig. 4A).
Figure 4.
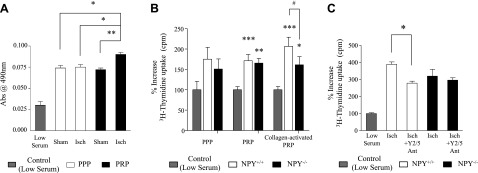
Increased angiogenic potential of ischemic platelets. A) HMVECs were treated with PPP or collagen-activated PRP from mice sham-operated or ischemic (Isch) at 14 d after ligation. Number of viable cells (MTS assay) was measured following each treatment. Data are presented as means ± se. All treatments were significantly increased relative to low-serum control. *P < 0.05, **P < 0.01; 1-way repeated measures ANOVA with post hoc Bonferroni's test. B) HMVECs were treated with PPP, PRP, or collagen-activated (5 μg/ml) PRP from ischemic mice 14 d after ligation, and 3H-thymidine uptake was measured. Data are presented as means ± se. *P < 0.05, **P < 0.01, ***P < 0.001 vs. low serum control; #P < 0.05; 1-way repeated measures ANOVA with post hoc Bonferroni's test. C) HMVECs were treated with collagen-activated PRP from either ischemic NPY+/+ or NPY−/− mice at 14 d after ligation with and without Y2/Y5 antagonists, and 3H-thymidine uptake was measured. Data are presented as mean ± se. *P < 0.05; 1-way repeated measures ANOVA with post hoc Bonferroni's test.
To determine whether specific release of angiogenic factors from platelet α-granules is required, and whether platelet-derived NPY has a direct effect on endothelial cell proliferation, platelet fractions were further separated into one in which platelets were stimulated to degranulate by collagen and another that was not activated by collagen; supernatants of both platelet preparations were assayed (Fig. 4B). Under similar conditions, HMVECs were treated with PPP, PRP, or collagen-activated PRP isolated from NPY+/+ and NPY−/− mice 14 d after ligation. Interestingly, among plasma fractions isolated from NPY+/+ mice, the mitogenic activity of nonactivated PRP was comparable to that of PPP, while collagen-activated PRP exhibited an augmented proliferative effect, suggesting that NPY needs specific activation for its release from platelets. In line with this observation, the increased angiogenic effect was not observed with collagen activation of NPY-null PRP (Fig. 4B).
To confirm that an increase in mitogenic activity of collagen-activated PRP from NPY+/+ mice is indeed NPY dependent, a mitogenic assay was performed in the presence of antagonists blocking NPY, Y2R, and Y5R. Y2/Y5R blockade reduced the proliferative effect of collagen-activated PRP from NPY+/+ ischemic mice to levels observed with collagen-activated NPY-null PRP (Fig. 4C).
Platelets are a critical source for angiogenic activity of NPY in vivo
To determine the role of platelet NPY in revascularization of ischemic tissues, we conducted a platelet depletion and transfer study using NPY−/− mice and their according wild-type controls. Chemical depletion of platelets was achieved by busulfan, which inhibits megakaryoblast/cyte formation (34) and thus, production of platelets. At 5 d after busulfan treatment, nonischemic mice exhibited severe thrombocytopenia (40% of basal platelet levels; Fig. 5A), which was maintained through at least 14 d. At this same time point, we detected decreased NPY-ir in PRP by 81% in NPY+/+ nonischemic mice (Fig. 5B), demonstrating a diminished NPY potential in these platelet-depleted mice.
Figure 5.
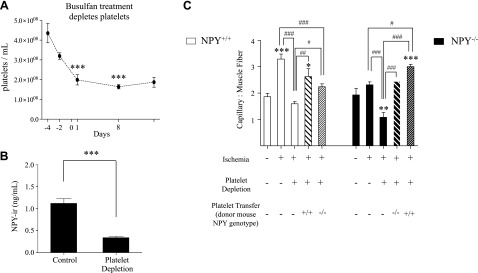
Platelet-derived NPY regulates angiogenesis. A) Busulfan-induced thrombocytopenia. NPY+/+ mice were injected with busulfan (25 mg/kg) at d −4, −2, +1, and +8 relative to experimental FAL on d 0. Platelets were isolated and counted with a hemacytometer. Time course of the experiment ran 14 d after d 0 in order to match the experimental design of the platelet transfer study. Data are presented as means ± se. ***P < 0.001 vs. d −4; 1-way ANOVA with post hoc Bonferroni's test. B) NPY-ir levels of PRP in control and platelet-depleted mice at d 14, corresponding with termination of experiment as described above. Data are presented as means ± se. ***P < 0.001 vs. ischemia alone; Student's t test. C) Quantification of microvessel density (CD31+ vessels) in gastrocnemius 14 d after ligation in NPY+/+ and NPY−/− mice in a platelet transfer study. Platelets were transfused at 4 × 105 cells/ml. *P < 0.05, **P < 0.01, ***P < 0.001 vs. sham treatment; #P < 0.05, ##P < 0.01, ###P < 0.001; 1-way ANOVA with post hoc Bonferroni's test.
At 14 d after ligation of thrombocytopenic and non-busulfan-treated mice, capillary angiogenesis was assessed as the capillary-to-muscle fiber ratio (Fig. 5C). NPY+/+ mice increased the capillary-to-muscle fiber ratio in gastrocnemius muscle relative to sham-treated mice. This response was ablated by platelet depletion. Autologous transfer of NPY-containing platelets into thrombocytopenic NPY+/+ mice restored the increase in capillary density observed in mice without busulfan treatment. Transfusion of NPY-null platelets into thrombocytopenic NPY+/+ mice slightly increased muscle vascularization, but only to levels observed in ischemic NPY−/− and sham-treated NPY+/+ mice. Thus, in contrast to autologous transfer of NPY+/+ platelets, transfer of NPY−/− platelets did not increase capillary density as compared to the sham-treated controls.
In NPY−/− mice, basal capillarity was similar to that in NPY+/+ mice; however, no increase was observed in response to FAL. Muscle vascularization in NPY−/− mice was further compromised by platelet depletion. Autologous transfer of NPY-null platelets into thrombocytopenic NPY−/− mice restored capillary density to levels observed without platelet depletion, but did not increase capillarity relative to sham-treated mice. However, transfer of NPY-containing platelets into NPY−/− mice depleted of their own platelets restored the ischemic response to levels observed in NPY+/+ mice and increased vessel density, as compared to the sham-treated controls (Fig. 5C). These data demonstrate that platelet-derived NPY is required for ischemic angiogenesis and provide further support of a role for NPY in late-phase recovery following FAL.
DISCUSSION
Our findings demonstrate an important role for endogenous NPY in ischemic angiogenesis, contributing to both the early and late response to FAL. We further show that different sources of NPY contribute to this response, initially from sympathetic nerves, followed by platelets. Furthermore, delayed blood flow recovery via plantar LPDI in mice lacking NPY is representative of deficient collateral-dependent blood flow (8). This impairment is consistent with deficiencies in hindpaw appearance and use. In addition, we show that NPY-deficient mice demonstrate impaired ischemic capillary angiogenesis in the gastrocnemius muscle. Essential to our observations of the ischemic response is the increase in megakaryocyte production and subsequent platelet release of NPY. We observed for the first time that ischemic activation of megakaryocytes provides late angiogenic phase delivery of NPY from platelets, associated with near complete recovery of flow in ischemic tissue. Therefore, we hypothesize that NPY released from sympathetic nerves contributes to the initial response to FAL by promoting collateral dilation, while at later time points, platelet NPY promotes continued collateral growth and capillary angiogenesis. Thus, the early peak of neuronal NPY release accompanied by increases in DPPIV and Y2R expression favors a Y2R-mediated increase in collateral flow and diminishes Y1R-mediated inhibition of NO production (42), facilitating vasodilation of adductor collaterals. During the later peak of the peptide action, however, NPY-mediated angiogenesis in the distal tissues is platelet dependent.
Delayed early improvement of hindpaw perfusion observed in NPY−/− mice 3 d after FAL suggests reduced collateral enlargement. Recovery of hindlimb flow after FAL is mediated primarily by remodeling of the collateral network in the adductor, the primary site of flow-limiting resistance after FAL, since the capillary bed only accounts for ∼15% of resistance (43). Although not exact, laser-Doppler plantar perfusion correlates well with overall leg blood flow, as measured by microspheres (8). Also, collateral remodeling becomes evident as early as 12 h after FAL (2), whereas ischemic capillary angiogenesis requires 5–7 d to become evident (43). Consistent with this timeline, the impaired recovery of perfusion is accompanied by worse hindlimb ischemic appearance and use scores at d 3 that persist thereafter, since the initial ischemic injury is largely lasting, as has been routinely reported, e.g., ref. 43. Accordingly, although we did not measure collateral remodeling by histology, our findings of decreased perfusion and worse appearance in NPY−/− mice at d 3 after FAL consistently support delayed recovery of collateral flow. This is in line with the previously reported stimulatory effect of NPY on collateral-dependent recovery of perfusion mediated by Y2R (29). In contrast, differences in capillary density in gastrocnemius muscle and hindlimb usage 14 d after FAL are due to the impaired angiogenesis in NPY−/− mice.
Although perfusion was only significantly lower 3 d after FAL in NPY−/− mice, this is the time when tissue injury is greatest due to low flow. Consistent with this, NPY−/− mice had significantly greater and sustained ischemic injury and use impairment. Furthermore, a similar pattern of early differences in perfusion and limb appearance, followed in time by a diminution in differences in these parameters in association with changes in capillary density has been reported by others using the mouse FAL model (44, 45). Collectively, our perfusion and use and appearance data support the notion that NPY contributes to early collateral dilation and later angiogenic processes.
In NPY+/+ mice, the early response to ischemia is associated with a transient increase in plasma NPY, suggesting that neuronal release of the peptide increased collateral conductance. Consistently, NPY has been shown to increase collateral-dependent flow following FAL via activation of its Y2R (29). Moreover, monocytes/macrophages, which are recruited to the site of the vascular injury, have been shown to mediate collateral growth, specifically in an NO-dependent manner (46). NPY was recently shown to inhibit IL-1β-induced NO production and iNOS expression from microglial cells in a Y1R-dependent manner (42). Moreover, NPY is also vasoconstrictive via Y1R (47). Therefore, these findings are in line with our early-phase observations of trends for increased Y2R and DPPIV and decreased Y1R mRNAs in adductor muscle of rats following FAL. Since DPPIV converts NPY to a Y2/Y5R-selective agonist that is not active at Y1Rs, these changes in the NPY system will prevent Y1R-mediated actions of the peptide, such as vasoconstriction, while promoting its Y2R-driven effects, including collateral enlargement and angiogenesis. Therefore, we propose a shift in NPY receptor-specific actions during the early-phase response to FAL away from Y1R-mediated vasoconstriction of conductance vessels in the upper hind limb, and toward Y2R-activated opening of preexisting collaterals and proangiogenic processes associated with the activation of endothelial cells in the distal hind limb (Fig. 6). The increasing trend of Y1R during the late-phase response suggests collateralization via VSMC migration, proliferation, and differentiation (13). Altogether, these mechanisms will lead to blood flow recovery in ischemic limb, a process that was impaired in NPY−/− mice.
Figure 6.

Differential role of NPY in different phases of ischemic revascularization. Under basal conditions, neuronally derived NPY decreases blood flow by stimulating vasoconstriction and prevents proarteriogenic activity of monocytes by inhibiting NO synthesis. Both of these processes are Y1R mediated. In early-phase ischemic recovery (∼d 0–3), up-regulated DPPIV converts NPY to the Y2/Y5R agonist, NPY3–36. Consequently, the Y1R-driven actions of NPY are inhibited, thereby decreasing vasoconstriction and enabling proarteriogenic processes triggered by immune cells. Moreover, the truncated form of NPY activates Y2R, which facilitates opening of preexisting collaterals. During early-phase recovery (∼d 5), the same product of DPPIV cleavage triggers the Y2R-mediated angiogenic effects of NPY. Megakaryocyte-platelet activation is also occurring then, increasing circulating levels of NPY. This increased bioavailability of NPY from platelets in combination with DPPIV provides a critical role for sustained ischemic capillary angiogenesis. Last, NPY contributes to vessel maturation as a VSMC mitogen during late-phase recovery (∼d 14). +, activation of downstream effects of receptor activation; −, inhibition of downstream effects of receptor activation.
The second phase of the NPY-dependent response to ischemia involves angiogenesis, which is evident ∼5 d after FAL (ref. 43 and Fig. 6). The decreased density of vessels in gastrocnemius muscle 5 d after FAL, which could be overcome by the addition of exogenous NPY, suggested that the immediate release of neuronally derived NPY during the early phase of rat hindlimb ischemia was insufficient to restore the vasculature despite increased expression of Y2R and the processing enzyme DPPIV. In contrast, 14 d after FAL in rats, complete spontaneous revascularization of ischemic tissues was achieved, while exogenous NPY had no effect, suggesting that the system was already saturated. This spontaneous recovery was associated with a marked increase in NPY-ir of PRP by d 14, which is supported by an increasing trend of NPY mRNA in the bone marrow. These synchronized data demonstrate the ischemic activation of megakaryocyte/platelet-derived NPY. Altogether, these observations suggest that platelets serve as a critical source of angiogenic NPY, necessary for complete revascularization of the ischemic tissues. This notion was confirmed by impaired ischemic angiogenesis in mice depleted of platelets, as well as by the reduced revascularization of ischemic tissues in NPY−/− mice that was rescued by the transfer of platelets from ischemic NPY+/+ mice.
In line with the platelet transfer results, collagen-activated PRP from ischemic animals exerted an enhanced proliferative effect on HMVECs, which could be inhibited by Y2/Y5R antagonists or prevented by NPY depletion (NPY−/−). Notably, endothelial cell proliferation was used here solely as a measure of the late-phase angiogenic potential at d 14, while in fact other processes induced by platelet NPY, such as endothelial cell migration and tube formation, are likely also contributing to its proangiogenic actions (5, 11, 48). The effect of platelet-derived NPY can be mimicked at early stages of the ischemic response by local exogenous NPY treatment, as shown by NPY-induced acceleration of angiogenesis observed 5 d after FAL in rats. Furthermore, the complete inhibition of ischemic angiogenesis in mice depleted of NPY-positive platelets clearly indicates them as the major source of NPY involved in this process. Thus, we propose that this later delivery of NPY by circulating platelets drives NPY-mediated ischemic revascularization by its effects on endothelial cells.
Last, a trending increase in adductor muscle expression of Y1 mRNA during late-stage recovery may contribute to the maturation of new vessels, as well as development of a select number of large-caliber conductance vessels and simultaneous “pruning” (49) mediated by Y1R-dependent VSMC proliferation.
As alluded to above, inflammatory cells are also present at this platelet-endothelium interface. In particular, T-lymphocyte (33) and monocyte (50) activation and subsequent release of growth factors and chemokines contribute to arteriogenesis (51) and angiogenesis (52). Associated activation of inflammatory and thrombotic pathways has provided a hemostatic challenge in response to shear-stress forces and vascular injury. Hindlimb ischemia as a model for PAD must take into consideration the thrombotic role of platelets, in addition to their role in angiogenesis. Balancing thrombosis and hemorrhage underscores a common obstacle with therapeutic targets, balancing the localized and systemic effects of a particular agent.
It has been recently demonstrated, however, that inhibition of arterial thrombosis without compromising hemostasis in response to arterial vascular injury can be achieved (53). On the other hand, this duality applies to other competing mechanisms and has been described as the Janus phenomenon (54). Specifically, it identifies the challenges of decoupling detrimental atherogenic pathways with beneficial proangiogenic or proarteriogenic enhancements that are mediated by a single agent. The appeal of NPY as a therapeutic agent is, in part, due to its receptor-specific effects. While Y1R activation increases neointimal formation (55), ischemic capillary angiogenesis is mediated by Y2R (5). Furthermore, differential organization and targeted release of pro- and antiangiogenic factors from α-granules within platelets (40, 56) adds another potential layer of specificity to NPY-mediated revascularization. However, simultaneous intraplatelet regulation of the release of thrombotic and angiogenic factors has yet to be explored with respect to NPY. Organization and progression of platelet content involves several processes that include differential biosynthesis and packaging of granular components, particularly in the α-granule (57). Furthermore, it has been recently shown that platelets can selectively sequester angiogenic regulators in vivo (tumor model) and in vitro (58), along with further α-granule selective stimulation and release of proangiogenic proteins. With regard to platelet activation and the release of its contents, the relationship of platelets to the endothelium not only provides a foundation for the coagulation cascade, but due to α-granule contents such as positive mediators VEGF, bFGF, and platelet-derived growth factor (PDGF) (59–62) and negative regulation with thrombospondin and platelet factor 4 (63, 64), platelets are also critical to de novo angiogenesis.
The clinical relevance of our findings is supported by data showing NPY mRNA and protein in human megakaryocytes and platelets (15), while activation of platelet NPY has been associated with increased atherosclerotic lesion burden and vulnerability in male PAD patients (18). A similar activation of the platelet NPY system has been shown in animals subjected to chronic stress (38). Thus, further studies could examine the NPY system and bone marrow-derived cell activation in remote ischemia, specifically, whether megakaryocyte NPY content increases following SDF-1 stimulation, known to mobilize endothelial progenitor cells and increase ischemic revascularization (52, 65). While our studies established mechanisms of NPY action in a healthy animal state, these findings can be translated into relevant disease models such as diabetes or aging. Moreover, despite the crucial role of platelet NPY demonstrated in our study, other sources of NPY may also contribute to our observations. In particular, inflammatory/immune cells such as monocytes (66, 67) and the endothelium itself (11) synthesize NPY. The specific contributions of these sources of the peptide to tissue revascularization are important aspects to address in future studies.
In summary, ischemic activation of the NPY system is a complex, coordinated response of different tissues under different temporal regulation (Fig. 6). Sympathetic nerves are the primary source of NPY in the early response to hindlimb ischemia. This release of NPY is associated with increased DPPIV expression, which converts the peptide to NPY3–36 inactive at Y1Rs. In this way, the Y1R-mediated inhibition of NO release and its vasoconstrictive effects are reduced, while activation of Y2R stimulates endothelial cell proliferation and possibly contributes to its role in releasing proarteriogenic factors. During the late stages of ischemia, megakaryocyte activation increases circulating levels of NPY in platelets. Results of our study center on platelet-derived NPY as the focal point of revascularization of ischemic tissue, and suggest platelet NPY as the primary driver of Y2/Y5-mediated angiogenic and Y1/Y5-mediated arteriogenic processes in the later response to ligation. This receptor-regulated capability to stimulate vessel formation and maturation make NPY an exciting therapeutic target. Furthermore, accumulation of NPY in platelets has been shown to be an integral part of the stress response (38). Stress, on the other hand, is known to exacerbate disorders associated with increased angiogenesis, such as obesity and atherosclerosis (38, 55). Therefore, our findings provide a potential mechanism underlying these deleterious effects of stress and may translate into new therapeutic strategies designed to alleviate them.
Acknowledgments
This work was supported by U.S. National Institutes of Health (NIH) grants HL067357 and HL055310 to Z.Z. and, in part, CA123211 to J.K.
The authors thank Anton Wellstein, Thomas G. Sherman, and Allan V. Kalueff for their insightful comments and editing. The authors declare no conflicts of interest.
A brilliant scientist, dedicated colleague, caring mentor, and friend has been dearly missed with the passing of Dr. Zofia Zukowska on April 15, 2012. However, her contribution to science will continue through those she has influenced with her wisdom, passion, and scholarship.
Footnotes
- bFGF
- basic fibroblast growth factor
- DPPIV
- dipeptidyl peptidase
- FAL
- femoral artery ligation
- HMVEC
- human microvascular endothelial cell
- LDPI
- laser Doppler perfusion imaging
- NPY
- neuropeptide Y
- NPY-ir
- neuropeptide Y immunoreactivity
- PAD
- peripheral arterial disease
- PPP
- platelet-poor plasma
- PRP
- platelet-rich plasma
- VEGF
- vascular endothelial growth factor
- VSMC
- vascular smooth muscle cell
- Y1R
- Y1 receptor
- Y2R
- Y2 receptor
- Y5R
- Y5 receptor
REFERENCES
- 1. Schaper W. (2009) Collateral circulation: past and present. Basic Res. Cardiol. 104, 5–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scholz D., Ito W., Fleming I., Deindl E., Sauer A., Wiesnet M., Busse R., Schaper J., Schaper W. (2000) Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis). Virchows Arch. 436, 257–270 [DOI] [PubMed] [Google Scholar]
- 3. Potente M., Gerhardt H., Carmeliet P. (2011) Basic and therapeutic aspects of angiogenesis. Cell 146, 873–887 [DOI] [PubMed] [Google Scholar]
- 4. Murakami M., Nguyen L. T., Hatanaka K., Schachterle W., Chen P. Y., Zhuang Z. W., Black B. L., Simons M. (2011) FGF-dependent regulation of VEGF receptor 2 expression in mice. J. Clin. Invest. 121, 2668–2678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee E. W., Michalkiewicz M., Kitlinska J., Kalezic I., Switalska H., Yoo P., Sangkharat A., Ji H., Li L., Michalkiewicz T., Ljubisavljevic M., Johansson H., Grant D. S., Zukowska Z. (2003) Neuropeptide Y induces ischemic angiogenesis and restores function of ischemic skeletal muscles. J. Clin. Invest. 111, 1853–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Torres-Vazquez J., Gitler A. D., Fraser S. D., Berk J. D., Van N. P., Fishman M. C., Childs S., Epstein J. A., Weinstein B. M. (2004) Semaphorin-plexin signaling guides patterning of the developing vasculature. Dev. Cell. 7, 117–123 [DOI] [PubMed] [Google Scholar]
- 7. Carmeliet P., Tessier-Lavigne M. (2005) Common mechanisms of nerve and blood vessel wiring. Nature 436, 193–200 [DOI] [PubMed] [Google Scholar]
- 8. Chalothorn D., Zhang H., Clayton J. A., Thomas S. A., Faber J. E. (2005) Catecholamines augment collateral vessel growth and angiogenesis in hindlimb ischemia. Am. J. Physiol. Heart Circ. Physiol. 289, H947–H959 [DOI] [PubMed] [Google Scholar]
- 9. Vecchione C., Fratta L., Rizzoni D., Notte A., Poulet R., Porteri E., Frati G., Guelfi D., Trimarco V., Mulvany M. J., Agabiti-Rosei E., Trimarco B., Cotecchia S., Lembo G. (2002) Cardiovascular influences of alpha1b-adrenergic receptor defect in mice. Circulation 105, 1700–1707 [DOI] [PubMed] [Google Scholar]
- 10. Hu Z. W., Shi X. Y., Lin R. Z., Chen J., Hoffman B. B. (1999) Alpha1-adrenergic receptor stimulation of mitogenesis in human vascular smooth muscle cells: role of tyrosine protein kinases and calcium in activation of mitogen-activated protein kinase. J. Pharmacol. Exp. Ther. 290, 28–37 [PubMed] [Google Scholar]
- 11. Zukowska-Grojec Z., Karwatowska-Prokopczuk E., Rose W., Rone J., Movafagh S., Ji H., Yeh Y., Chen W. T., Kleinman H. K., Grouzmann E., Grant D. S. (1998) Neuropeptide Y: a novel angiogenic factor from the sympathetic nerves and endothelium. Circ. Res. 83, 187–195 [DOI] [PubMed] [Google Scholar]
- 12. Herzog H. (2003) Neuropeptide Y and energy homeostasis: insights from Y receptor knockout models. Eur. J. Pharmacol. 480, 21–29 [DOI] [PubMed] [Google Scholar]
- 13. Pons J., Kitlinska J., Ji H., Lee E. W., Zukowska Z. (2003) Mitogenic actions of neuropeptide Y in vascular smooth muscle cells: synergetic interactions with the beta-adrenergic system. Can. J. Physiol. Pharmacol. 81, 177–185 [DOI] [PubMed] [Google Scholar]
- 14. Ericsson A., Schalling M., McIntyre K. R., Lundberg J. M., Larhammar D., Seroogy K., Hokfelt T., Persson H. (1987) Detection of neuropeptide Y and its mRNA in megakaryocytes: enhanced levels in certain autoimmune mice. Proc. Natl. Acad. Sci. U. S. A. 84, 5585–5589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hancock V., Martin J. F., Lelchuk R. (1993) The relationship between human megakaryocyte nuclear DNA content and gene expression. Br. J. Haematol. 85, 694–697 [DOI] [PubMed] [Google Scholar]
- 16. Myers A. K., Torres Duarte A. P., Zukowska-Grojec Z. (1993) Immunoreactive neuropeptide Y (NPY) in plasma and platelets of rat and mouse strains and human volunteers. Regul. Pept. 47, 239–245 [DOI] [PubMed] [Google Scholar]
- 17. Myers A. K., Farhat M. Y., Vaz C. A., Keiser H. R., Zukowska-Grojec Z. (1988) Release of immunoreactive-neuropeptide by rat platelets. Biochem. Biophys. Res. Commun. 155, 118–122 [DOI] [PubMed] [Google Scholar]
- 18. Li L., Najafi A. H., Kitlinska J. B., Neville R., Laredo J., Epstein S. E., Burnett M. S., Zukowska Z. (2011) Of mice and men: neuropeptide Y and its receptors are associated with atherosclerotic lesion burden and vulnerability. J. Cardiovasc. Trans. Res. 4, 351–362 [DOI] [PubMed] [Google Scholar]
- 19. Pinedo H. M., Verheul H. M., D'Amato R. J., Folkman J. (1998) Involvement of platelets in tumour angiogenesis? Lancet 352, 1775–1777 [DOI] [PubMed] [Google Scholar]
- 20. Amano H., Hackett N. R., Rafii S., Crystal R. G. (2005) Thrombopoietin gene transfer-mediated enhancement of angiogenic responses to acute ischemia. Circ. Res. 97, 337–345 [DOI] [PubMed] [Google Scholar]
- 21. Iba O., Matsubara H., Nozawa Y., Fujiyama S., Amano K., Mori Y., Kojima H., Iwasaka T. (2002) Angiogenesis by implantation of peripheral blood mononuclear cells and platelets into ischemic limbs. Circulation 106, 2019–2025 [DOI] [PubMed] [Google Scholar]
- 22. Kondoh K., Koyama H., Miyata T., Takato T., Hamada H., Shigematsu H. (2004) Conduction performance of collateral vessels induced by vascular endothelial growth factor or basic fibroblast growth factor. Cardiovasc. Res. 61, 132–142 [DOI] [PubMed] [Google Scholar]
- 23. Hershey J. C., Baskin E. P., Glass J. D., Hartman H. A., Gilberto D. B., Rogers I. T., Cook J. J. (2001) Revascularization in the rabbit hindlimb: dissociation between capillary sprouting and arteriogenesis. Cardiovasc. Res. 49, 618–625 [DOI] [PubMed] [Google Scholar]
- 24. Ekstrand A. J., Cao R., Bjorndahl M., Nystrom S., Jonsson-Rylander A. C., Hassani H., Hallberg B., Nordlander M., Cao Y. (2003) Deletion of neuropeptide Y (NPY) 2 receptor in mice results in blockage of NPY-induced angiogenesis and delayed wound healing. Proc. Natl. Acad. Sci. U. S. A. 100, 6033–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kitlinska J., Lee E. W., Movafagh S., Pons J., Zukowska Z. (2002) Neuropeptide Y-induced angiogenesis in aging. Peptides 23, 71–77 [DOI] [PubMed] [Google Scholar]
- 26. Lu C., Tilan J. U., Everhart L., Czarnecka M., Soldin S. J., Mendu D. R., Jeha D., Hanafy J., Lee C. K., Sun J., Izycka-Swieszewska E., Toretsky J. A., Kitlinska J. (2011) Dipeptidyl peptidases as survival factors in ewing sarcoma family of tumors: implications for tumor biology and therapy. J. Biol. Chem. 286, 27494–27505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu C., Everhart L., Tilan J., Kuo L., Sun C. C., Munivenkatappa R. B., Jonsson-Rylander A. C., Sun J., Kuan-Celarier A., Li L., Abe K., Zukowska Z., Toretsky J. A., Kitlinska J. (2010) Neuropeptide Y and its Y2 receptor: potential targets in neuroblastoma therapy. Oncogene 29, 5630–5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kitlinska J., Abe K., Kuo L., Pons J., Yu M., Li L., Tilan J., Everhart L., Lee E. W., Zukowska Z., Toretsky J. A. (2005) Differential effects of neuropeptide Y on the growth and vascularization of neural crest-derived tumors. Cancer Res. 65, 1719–1728 [DOI] [PubMed] [Google Scholar]
- 29. Cruze C. A., Su F., Limberg B. J., Deutsch A. J., Stoffolano P. J., Dai H. J., Buchanan D. D., Yang H. T., Terjung R. L., Spruell R. D., Mittelstadt S. W., Rosenbaum J. S. (2007) The Y2 receptor mediates increases in collateral-dependent blood flow in a model of peripheral arterial insufficiency. Peptides 28, 269–280 [DOI] [PubMed] [Google Scholar]
- 30. Robich M. P., Matyal R., Chu L. M., Feng J., Xu S. H., Laham R. J., Hess P. E., Bianchi C., Sellke F. W. (2010) Effects of neuropeptide Y on collateral development in a swine model of chronic myocardial ischemia. J. Mol. Cell. Cardiol. 49, 1022–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kinnaird T., Stabile E., Burnett M. S., Shou M., Lee C. W., Barr S., Fuchs S., Epstein S. E. (2004) Local delivery of marrow-derived stromal cells augments collateral perfusion through paracrine mechanisms. Circulation 109, 1543–1549 [DOI] [PubMed] [Google Scholar]
- 32. Rutherford R. B., Baker J. D., Ernst C., Johnston K. W., Porter J. M., Ahn S., Jones D. N. (1997) Recommended standards for reports dealing with lower extremity ischemia: revised version. J. Vasc. Surg. 26, 517–538 [DOI] [PubMed] [Google Scholar]
- 33. Stabile E., Burnett M. S., Watkins C., Kinnaird T., Bachis A., la Sala A., Miller J. M., Shou M., Epstein S. E., Fuchs S. (2003) Impaired arteriogenic response to acute hindlimb ischemia in CD4-knockout mice. Circulation 108, 205–210 [DOI] [PubMed] [Google Scholar]
- 34. Kuter D. J., Rosenberg R. D. (1995) The reciprocal relationship of thrombopoietin (c-mpl ligand) to changes in the platelet mass during busulfan-induced thrombocytopenia in the rabbit. Blood 85, 2720–2730 [PubMed] [Google Scholar]
- 35. Sirois M. G., Simons M., Kuter D. J., Rosenberg R. D., Edelman E. R. (1997) Rat arterial wall retains myointimal hyperplastic potential long after arterial injury. Circulation 96, 1291–1298 [DOI] [PubMed] [Google Scholar]
- 36. Westfall T. C. (2004) Prejunctional effects of neuropeptide Y and its role as a cotransmitter. In Neuropeptide Y and Related Peptides (Michel M., ed) pp. 137–183, Springer, Berlin [Google Scholar]
- 37. Zukowska-Grojec Z., Vaz A. C. (1988) Role of neuropeptide Y (NPY) in cardiovascular responses to stress. Synapse 2, 293–298 [DOI] [PubMed] [Google Scholar]
- 38. Kuo L. E., Kitlinska J. B., Tilan J. U., Li L., Baker S. B., Johnson M. D., Lee E. W., Burnett M. S., Fricke S. T., Kvetnansky R., Herzog H., Zukowska Z. (2007) Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat. Med. 13, 803–811 [DOI] [PubMed] [Google Scholar]
- 39. Lundberg J. M., Rudehill A., Sollevi A., Theodorsson-Norheim E., Hamberger B. (1986) Frequency- and reserpine-dependent chemical coding of sympathetic transmission: differential release of noradrenaline and neuropeptide Y from pig spleen. Neurosci. Lett. 63, 96–100 [DOI] [PubMed] [Google Scholar]
- 40. Italiano J. E., Jr., Richardson J. L., Patel-Hett S., Battinelli E., Zaslavsky A., Short S., Ryeom S., Folkman J., Klement G. L. (2008) Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood 111, 1227–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harrison P., Cramer E. M. (1993) Platelet alpha-granules. Blood Rev. 7, 52–62 [DOI] [PubMed] [Google Scholar]
- 42. Ferreira R., Xapelli S., Santos T., Silva A. P., Cristovao A., Cortes L., Malva J. O. (2010) Neuropeptide Y modulation of interleukin-1β (IL-1β)-induced nitric oxide production in microglia. J. Biol. Chem. 285, 41921–41934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chalothorn D., Clayton J. A., Zhang H., Pomp D., Faber J. E. (2007) Collateral density, remodeling, and VEGF-A expression differ widely between mouse strains. Physiol. Genomics 30, 179–191 [DOI] [PubMed] [Google Scholar]
- 44. Brenes R. A., Jadlowiec C. C., Bear M., Hashim P., Protack C. D., Li X., Lv W., Collins M. J., Dardik A. (2012) Toward a mouse model of hind limb ischemia to test therapeutic angiogenesis. J. Vasc. Surg. 56, 1669–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tirziu D., Moodie K. L., Zhuang Z. W., Singer K., Helisch A., Dunn J. F., Li W., Singh J., Simons M. (2005) Delayed arteriogenesis in hypercholesterolemic mice. Circulation 112, 2501–2509 [DOI] [PubMed] [Google Scholar]
- 46. Troidl K., Tribulova S., Cai W. J., Ruding I., Apfelbeck H., Schierling W., Troidl C., Schmitz-Rixen T., Schaper W. (2010) Effects of endogenous nitric oxide and of DETA NONOate in arteriogenesis. J. Cardiovasc. Pharmacol. 55, 153–160 [DOI] [PubMed] [Google Scholar]
- 47. Wahlestedt C., Hakanson R., Vaz C. A., Zukowska-Grojec Z. (1990) Norepinephrine and neuropeptide Y: vasoconstrictor cooperation in vivo and in vitro. Am. J. Physiol. 258, R736–742 [DOI] [PubMed] [Google Scholar]
- 48. Movafagh S., Hobson J. P., Spiegel S., Kleinman H. K., Zukowska Z. (2006) Neuropeptide Y induces migration, proliferation, and tube formation of endothelial cells bimodally via Y1, Y2, and Y5 receptors. FASEB J. 20, 1924–1926 [DOI] [PubMed] [Google Scholar]
- 49. Schaper W., Schaper J. (2004) Arteriogenesis, Kluwer Academics, London [Google Scholar]
- 50. Hoefer I. E., Grundmann S., van Royen N., Voskuil M., Schirmer S. H., Ulusans S., Bode C., Buschmann I. R., Piek J. J. (2005) Leukocyte subpopulations and arteriogenesis: specific role of monocytes, lymphocytes and granulocytes. Atherosclerosis 181, 285–293 [DOI] [PubMed] [Google Scholar]
- 51. Tritsaris K., Myren M., Ditlev S. B., Hubschmann M. V., van der Blom I., Hansen A. J., Olsen U. B., Cao R., Zhang J., Jia T., Wahlberg E., Dissing S., Cao Y. (2007) IL-20 is an arteriogenic cytokine that remodels collateral networks and improves functions of ischemic hind limbs. Proc. Natl. Acad. Sci. U. S. A. 104, 15364–15369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tan Y., Shao H., Eton D., Yang Z., Alonso-Diaz L., Zhang H., Schulick A., Livingstone A. S., Yu H. (2007) Stromal cell-derived factor-1 enhances pro-angiogenic effect of granulocyte-colony stimulating factor. Cardiovasc. Res. 73, 823–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Andre P., Morooka T., Sim D., Abe K., Lowell C., Nanda N., Delaney S., Siu G., Yan Y., Hollenbach S., Pandey A., Gao H., Wang Y., Nakajima K., Parikh S. A., Shi C., Phillips D., Owen W., Sinha U., Simon D. I. (2011) Critical role for syk in responses to vascular injury. Blood 118, 5000–5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Epstein S. E., Stabile E., Kinnaird T., Lee C. W., Clavijo L., Burnett M. S. (2004) Janus phenomenon: the interrelated tradeoffs inherent in therapies designed to enhance collateral formation and those designed to inhibit atherogenesis. Circulation 109, 2826–2831 [DOI] [PubMed] [Google Scholar]
- 55. Li L., Lee E. W., Ji H., Zukowska Z. (2003) Neuropeptide Y-induced acceleration of postangioplasty occlusion of rat carotid artery. Arterioscler. Thromb. Vasc. Biol. 23, 1204–1210 [DOI] [PubMed] [Google Scholar]
- 56. Ma L., Perini R., McKnight W., Dicay M., Klein A., Hollenberg M. D., Wallace J. L. (2005) Proteinase-activated receptors 1 and 4 counter-regulate endostatin and VEGF release from human platelets. Proc. Natl. Acad. Sci. U. S. A. 102, 216–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sehgal S., Storrie B. (2007) Evidence that differential packaging of the major platelet granule proteins von willebrand factor and fibrinogen can support their differential release. J. Thromb. Haemost. 5, 2009–2016 [DOI] [PubMed] [Google Scholar]
- 58. Klement G. L., Yip T. T., Cassiola F., Kikuchi L., Cervi D., Podust V., Italiano J. E., Wheatley E., Abou-Slaybi A., Bender E., Almog N., Kieran M. W., Folkman J. (2009) Platelets actively sequester angiogenesis regulators. Blood 113, 2835–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Brunner G., Nguyen H., Gabrilove J., Rifkin D. B., Wilson E. L. (1993) Basic fibroblast growth factor expression in human bone marrow and peripheral blood cells. Blood 81, 631–638 [PubMed] [Google Scholar]
- 60. Mohle R., Green D., Moore M. A., Nachman R. L., Rafii S. (1997) Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc. Natl. Acad. Sci. U. S. A. 94, 663–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pintucci G., Froum S., Pinnell J., Mignatti P., Rafii S., Green D. (2002) Trophic effects of platelets on cultured endothelial cells are mediated by platelet-associated fibroblast growth factor-2 (FGF-2) and vascular endothelial growth factor (VEGF). Thromb. Haemost. 88, 834–842 [PubMed] [Google Scholar]
- 62. Wartiovaara U., Salven P., Mikkola H., Lassila R., Kaukonen J., Joukov V., Orpana A., Ristimaki A., Heikinheimo M., Joensuu H., Alitalo K., Palotie A. (1998) Peripheral blood platelets express VEGF-C and VEGF which are released during platelet activation. Thromb. Haemost. 80, 171–175 [PubMed] [Google Scholar]
- 63. Iruela-Arispe M. L., Bornstein P., Sage H. (1991) Thrombospondin exerts an antiangiogenic effect on cord formation by endothelial cells in vitro. Proc. Natl. Acad. Sci. U. S. A. 88, 5026–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Maione T. E., Gray G. S., Petro J., Hunt A. J., Donner A. L., Bauer S. I., Carson H. F., Sharpe R. J. (1990) Inhibition of angiogenesis by recombinant human platelet factor-4 and related peptides. Science 247, 77–79 [DOI] [PubMed] [Google Scholar]
- 65. Yamaguchi J., Kusano K. F., Masuo O., Kawamoto A., Silver M., Murasawa S., Bosch-Marce M., Masuda H., Losordo D. W., Isner J. M., Asahara T. (2003) Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation 107, 1322–1328 [DOI] [PubMed] [Google Scholar]
- 66. Holler J., Zakrzewicz A., Kaufmann A., Wilhelm J., Fuchs-Moll G., Dietrich H., Padberg W., Kuncova J., Kummer W., Grau V. (2008) Neuropeptide Y is expressed by rat mononuclear blood leukocytes and strongly down-regulated during inflammation. J. Immunol. 181, 6906–6912 [DOI] [PubMed] [Google Scholar]
- 67. Schwarz H., Villiger P. M., von Kempis J., Lotz M. (1994) Neuropeptide Y is an inducible gene in the human immune system. J. Neuroimmunol. 51, 53–61 [DOI] [PubMed] [Google Scholar]