

Abstract
The potent inflammatory mediator prostaglandin E2 (PGE2) is implicated in the pathogenesis of several chronic inflammatory conditions, including periodontitis. The inducible enzyme microsomal prostaglandin E synthase-1 (mPGES-1), catalyzing the terminal step of PGE2 biosynthesis, is an attractive target for selective PGE2 inhibition. To identify mPGES-1 inhibitors, we investigated the effect of aminothiazoles on inflammation-induced PGE2 synthesis in vitro, using human gingival fibroblasts stimulated with the cytokine IL-1β and a cell-free mPGES-1 activity assay, as well as on inflammation-induced bone resorption in vivo, using ligature-induced experimental periodontitis in Sprague-Dawley rats. Aminothiazoles 4-([4-(2-naphthyl)-1,3-thiazol-2-yl]amino)phenol (TH-848) and 4-(3-fluoro-4-methoxyphenyl)-N-(4-phenoxyphenyl)-1,3-thiazol-2-amine (TH-644) reduced IL-1β-induced PGE2 production in fibroblasts (IC50 1.1 and 1.5 μM, respectively) as well as recombinant mPGES-1 activity, without affecting activity or expression of the upstream enzyme cyclooxygenase-2. In ligature-induced experimental periodontitis, alveolar bone loss, assessed by X-ray imaging, was reduced by 46% by local treatment with TH-848, compared to vehicle, without any systemic effects on PGE2, 6-keto PGF1α, LTB4 or cytokine levels. In summary, these results demonstrate that the aminothiazoles represent novel mPGES-1 inhibitors for inhibition of PGE2 production and reduction of bone resorption in experimental periodontitis, and may be used as potential anti-inflammatory drugs for treatment of chronic inflammatory diseases, including periodontitis.—Kats, A., Båge, T., Georgsson, P., Jönsson, J., Quezada, H. C., Gustafsson, A., Jansson, L., Lindberg, C., Näsström, K., Yucel-Lindberg, T. Inhibition of microsomal prostaglandin E synthase-1 by aminothiazoles decreases prostaglandin E2 synthesis in vitro and ameliorates experimental periodontitis in vivo.
Keywords: cyclooxygenase-2, gingival fibroblasts, interleukin-1β, PGE2, mPGES-1 inhibitor, anti-inflammatory
Periodontitis is a common disease that affects the supporting structures of the tooth and is characterized by a destruction of periodontal ligaments, gingival connective tissue, and alveolar bone, which can eventually lead to tooth loss (1, 2). The disease is initiated by microbial biofilm in the periodontal pocket, which instigates a host immune response resulting in the activation of inflammatory cells. These cells, together with resident cells, such as gingival fibroblasts, produce inflammatory mediators, including prostaglandins, cytokines, and proteolytic enzymes, that collectively contribute to connective tissue destruction and alveolar bone resorption (3–5).
Prostaglandins, especially prostaglandin E2 (PGE2), play a significant role in the inflammatory response, contributing to the pathogenesis of several chronic inflammatory conditions, including periodontitis, rheumatoid arthritis, and cardiovascular inflammatory diseases (6–8). The inflammatory mediator PGE2 exerts a range of biological activities that include stimulation of inflammation-associated bone resorption (5). Biosynthesis of PGE2 is regulated via 3 groups of enzymes: phospholipase A2, cyclooxygenase (COX), and prostaglandin E synthase (PGES). The phospholipase A2 enzymes catalyze the conversion of membrane lipids to arachidonic acid, which is further converted to prostaglandin H2 (PGH2) by the two COX isoforms (COX-1 and COX-2). The terminal step from PGH2 to PGE2 is catalyzed by PGES enzymes (9). Three different isoforms of PGES have been identified: the glutathione-dependent membrane-associated microsomal PGES (mPGES)-1, the glutathione-independent mPGES-2, and the cytosolic PGES (cPGES) (10–12). The first discovered PGES, mPGES-1, is reported to be induced by inflammatory stimuli and considered to be the PGES mainly responsible for inflammation-induced PGE2 production (10, 13).
The expression of PGES has been reported previously in various tissues, including gastric ulcer tissue, rheumatoid arthritis-affected synovial tissue, and recently also in periodontitis-affected gingival tissue (14–16). In our report showing that all 3 PGES are expressed in the gingival tissue of patients with periodontitis, we also demonstrated that the mPGES-1 isoform was induced by the inflammatory mediators interleukin-1β (IL-1β) and tumor necrosis factor α (TNF-α) in gingival fibroblasts and smooth muscle cells (14). We further reported that the anti-inflammatory steroid dexamethasone, as well as the anti-inflammatory and antiplaque agent triclosan, inhibited cytokine-induced mPGES-1 mRNA and protein expression accompanied by abolished PGE2 production in gingival fibroblasts (17, 18), highlighting the significance of mPGES-1 in the regulation of PGE2 synthesis in gingival tissue.
The enzyme COX-2 has been considered an attractive target for PGE2 inhibition and thereby for therapeutic intervention in the management of chronic inflammation-associated diseases (19). The selective COX-2 inhibitors, as well as traditional nonsteroid anti-inflammatory drugs (NSAIDs), have been reported to provide symptomatic relief to patients with rheumatoid arthritis and osteoarthritis (20). It has also been demonstrated that inhibition of COX-2 reduces bone loss and cartilage destruction associated with joint inflammation in rodent models (21). However, in addition to their beneficial effects, NSAIDs targeting COX-1 and COX-2 have been reported to cause side effects, such as gastrointestinal toxicity (22, 23). Although the COX-2-specific inhibitors have reduced gastrointestinal toxicity compared to NSAIDs, these drugs are reported to cause increased cardiovascular risk due to inhibition of prostacyclin synthesis (23–25). In light of this, the mPGES-1 enzyme, acting downstream of COX-2, represents an attractive target for new classes of drugs selectively inhibiting inflammation-induced PGE2 production with potentially safer profiles than COX-2 inhibitors.
Several studies have implicated the involvement of PGE2 in the pathogenesis of periodontal disease due to increased concentrations of PGE2 in periodontal tissue and in gingival crevicular fluid of patients exhibiting periodontal disease, compared to periodontally healthy controls (8, 26, 27). In addition, the enhanced levels of PGE2 in periodontitis correlate well with disease severity as measured by attachment loss (28). Moreover, in experimental periodontitis, PGE2 has been shown to exacerbate the disease, in contrast to the structurally related lipid mediator resolvin E1, which has been shown to protect from osteoclast-mediated bone destruction and restore tissue homeostasis in a rabbit model (29, 30). The involvement of PGE2 in periodontitis is further supported by reports that NSAIDs, as well as selective COX-2 inhibitors, known to inhibit PGE2 synthesis, decreased periodontal disease in terms of alveolar bone resorption (31, 32). Furthermore, the antiplaque agent triclosan, shown to reduce PGE2 production and mPGES-1 expression in gingival fibroblasts (18), has been demonstrated to have a beneficial effect on periodontal disease by reducing gingival inflammation (33). It has also been reported that toothpaste containing triclosan reduced alveolar bone loss in experimental periodontitis in rats (34). One of the earliest identified inhibitors of mPGES-1 was the 5-lipoxygenase-activating protein inhibitor 1-[(4-chlorophenyl)methyl]-3-[(1,1-dimethylethyl)thio]-α,α-dimethyl-5-(1-methylethyl)-1H-indole-2-propanoic acid (MK-886), demonstrated to inhibit mPGES-1 enzyme activity and protein expression (35, 36). Furthermore, the substance curcumin from the turmeric plant, with anti-inflammatory and anticarcinogenic activities, has been reported to reduce IL-1β-stimulated mPGES-1 expression in in vitro studies (37, 38). Recently, curcumin was shown to inhibit TNF-α, IL-6, and COX-2 expression in gingival tissues of rats with induced experimental periodontitis (39).
Extensive research is underway to identify and develop specific mPGES-1 inhibitors, but to our knowledge, there is still a lack of clinically useful mPGES-1 inhibitors for treatment and prevention of chronic inflammatory diseases. For periodontal treatment, therapy today is mainly focused on the management of the microbial biofilm, not taking into account the central role of inflammation in causing tissue damage, which makes this therapy only partly effective (40). New and more efficient treatment options, based on modulation of the inflammatory response together with direct control of the microbial biofilm, are needed for management of periodontal disease (40).
Thiazole compounds have previously been reported to exhibit some anti-inflammatory properties and also to decrease disease progression of collagen-induced arthritis in mice (41–44), although their effects on PGE2 synthesis and bone resorption in periodontitis have not been sufficiently clarified. In this study, we aimed to investigate the effect of aminothiazole derivatives as potential mPGES-1 inhibitors on the regulation of PGE2 in gingival fibroblasts, as well as the effect of an aminothiazole derivative on experimental periodontitis in rats.
MATERIALS AND METHODS
Materials
Dulbecco's modified Eagle medium (DMEM), penicillin, streptomycin, fetal calf serum (FCS), trypsin, HEPES, phosphate-buffered saline (PBS), and DMSO, as well as Superscript II and AmpliTaq Gold DNA polymerase, were purchased from Invitrogen Life Technologies (Paisley, UK). Aminothiazole derivatives 4-([4-(2-naphthyl)-1,3-thiazol-2-yl]amino)phenol (TH-848) and 4-(3-fluoro-4-methoxyphenyl)-N-(4-phenoxyphenyl)-1,3-thiazol-2-amine hydrobromide (TH-644) were purchased from ChemBridge Corp. (San Diego, CA, USA). The cytokine IL-1β, the fluorescently labeled secondary goat anti-mouse phycoerythrin (PE) antibody for flow cytometry, monoclonal mouse anti-rat IL-1β antibodies, and the cell and tissue staining kit were obtained from R&D Systems (Minneapolis, MN, USA). PGE2 enzyme immunoassay (EIA) kit, PGH2, mPEGS-1 (human recombinant), MK-886, 5-bromo-2-(4-fluorophenyl)-3-[4-(methylsulfonyl)phenyl]-thiophene (DuP-697), COX inhibitor screening assay kit, PGE2 monoclonal EIA kit, leukotriene B4 (LTB4) express EIA kit, 6-keto prostaglandin F1α (PGF1α) EIA kit, and the antibodies for mPGES-1 (monoclonal mouse IgG), mPGES-2 (polyclonal rabbit IgG), cPGES (polyclonal rabbit IgG) and COX-2 (monoclonal mouse IgG) used for flow cytometry were purchased from Cayman Chemical (Ann Arbor, MI, USA). CytoTox 96 nonradioactive cytotoxicity assay was from Promega (Madison, WI, USA). The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assay kit was obtained from Abnova (Taipei, Taiwan). Dexamethasone, triclosan, celecoxib, glutathione (GSH), FeCl2, saponin, and Triton X-100 were purchased from Sigma-Aldrich (St. Louis, MO, USA). The fluorescently labeled secondary goat anti-mouse fluorescein isothiocyanate (FITC) antibody was obtained from DakoCytomation (Glostrup, Denmark) and the fluorescently labeled secondary sheep anti-rabbit PE antibody from Serotec (Oxford, UK). Formaldehyde, xylene, and hematoxylin and eosin (H&E) were purchased from HistoLab (Gothenburg, Sweden) and the RNeasy Mini Kit from Qiagen Inc. (Valencia, CA, USA). The primers for PCR amplification for mPGES-1, 5′-CCAAGTGAGGCTGCGGAAGAA-3′ and 3′-GCTTCCCAGAGGATCTGCAGA-5′ (GenBank accession number NM_004878); COX-2, 5′-TTCAAATGAGATTGTGGGAAAATTGCT-3′ and 3′-TTCTATGAGTCCGTCTCTACTAGA-5′ (GenBank NM_000963); and GAPDH, 5′-AAAGGGTCATCATCTCTGCC-3′ and 5′-GACAAAGTGGTCGTTGAGG-3′, were supplied by CyberGene AB (Stockholm, Sweden); and GelRed for electrophoresis was obtained from Bio-Nuclear Scandinavia (Stockholm, Sweden). For susceptibility tests, the microorganisms Streptococcus mutans IB, S. mutans OMZ, Streptococcus sobrinus, Streptococcus sanguis, Streptococcus salivarius, Lactobacillus brevis, Lactobacillus casei, Actinomyces naeslundii, and Actinomyces viscosus were obtained from the Department of Cariology, Institute of Odontology (University of Gothenburg, Gothenburg, Sweden); the microorganisms Porphyromonas gingivalis and Fusobacterium nucleatum were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA; ATCC 25586 and ATCC 33277, respectively); and Actinobacillus (Aggregatibacter) actinomycetemcomitans strain Y4 was originally obtained from Dr. Sigmund S. Socransky (Forsyth Dental Center, Boston, MA, USA). Brain heart infusion (BHI) agar was obtained from Difco Laboratories (Detroit, MI, USA) and horse blood agar (HBA) from Sahlgrenska University Hospital (Gothenburg, Sweden). Antibiotic sensitivity discs were obtained from Biodisc AB (Solna, Sweden) and the anaerobic pouch system (AnaeroGen compact) from Thermo Fisher Scientific (Oxoid, Basingstoke, UK). Sprague-Dawley rats were purchased from Harlan Laboratories (Indianapolis, IN, USA) and silk ligatures (4-0 perma-hand, nonabsorbable, 12×45 cm) from Johnson & Johnson International (New Brunswick, NJ, USA). Gantrez was obtained from International Specialty Products (Köln, Germany) and the Bio-Plex Rat Cytokine Assay from Bio-Rad Laboratories (Hercules, CA, USA).
Docking studies
Docking studies were performed using the software Autodock Vina (45) and the 3-dimensional crystal structure of mPGES-1 [Protein Data Bank (PDB) ID code 3DWW; ref. 46]. Aminothiazole derivatives were identified as potential mPGES-1 inhibitors using the structures of the well-known mPGES-1 inhibitor MK-886 (35) and triclosan, previously reported by our group as an mPGES-1 inhibitor (18), as starting points. The aminothiazoles TH-848 and TH-644 were further evaluated in experimental studies. Calculated affinities for the dockings were −5.8 kcal/mol for PGH2, −6.7 kcal/mol for MK-886, −6.8 kcal/mol for triclosan, −8.7 kcal/mol for TH-848, and −9.1 kcal/mol for TH-664.
Gingival fibroblasts
Primary human gingival fibroblasts were established from gingival biopsies obtained from 8 healthy patients (ages 6–12) with no clinical signs of periodontal disease. The study was approved by the Regional Ethical Review Board in Stockholm. Gingival fibroblasts were established and cultured as described previously (47). For the experiments, fibroblasts (6×105) were seeded in Petri dishes (6 cm) in DMEM supplemented with penicillin (50 U/ml), streptomycin (50 μg/ml), and FCS (5%) and cultured at 37°C for 24 h. The cell layers were rinsed twice with serum-free DMEM, followed by the addition of serum-free DMEM containing the cytokine IL-1β in the absence or presence of the aminothiazoles TH-848 or TH-644 or the PGE2 inhibitors dexamethasone, celecoxib, triclosan, or MK-886. The cells were incubated for 6 h for mRNA expression analysis or 24 h for cytotoxicity analyses, protein expression analysis, or subsequent PGE2 determination. The cell monolayers were collected for protein analysis using flow cytometry or for total RNA isolation. Control cells were treated with only culture medium or IL-1β, as indicated in the figures.
Rat gingival fibroblasts were established from healthy gingiva of 4 rats. The fibroblasts were obtained and cultured in the same manner as the human gingival fibroblasts described above. For the experiments, cells (4×104) were seeded in 24-well plates and cultured for 24 h at 37°C. The cell layers were rinsed twice with serum-free DMEM, followed by addition of DMEM containing the cytokine IL-1β (0.3 ng/ml) alone or in combination with TH-848 (2 μM). Control cells were treated with DMEM or IL-1β only. After an incubation period of 24 h, the cells were further used for MTT assay, or medium was removed and stored at −20°C for later PGE2 determination.
Cytotoxicity analysis
CytoTox 96 nonradioactive cytotoxicity assay was used to measure the release of lactate dehydrogenase (LDH), according to the manufacturer's instructions. Human gingival fibroblasts (1×104) were seeded in 96-well plates in 4 replicates and treated for 24 h with TH-848 in concentrations ranging from 0.5 to 10 μM, or the cytokine IL-1β (0.5 ng/ml) alone or in combination with TH-848 (2.5 μM). Treatment with serum-free culture medium served as a control for spontaneous release of LDH. The LDH release in the cell culture supernatants was expressed as percentage of maximum LDH release. The MTT cell proliferation assay kit was used to assess the viability of human and rat gingival fibroblasts, according to the manufacturer's instructions. Human gingival fibroblasts were seeded in 24-well plates in 4 replicates and treated for 24 h with TH-848 in concentrations ranging from 0.5 to 10 μM, or the cytokine IL-1β (0.5 ng/ml) alone or in combination with TH-848 (2.5 μM). Rat gingival fibroblasts were seeded in 24-well plates and treated for 24 h with TH-848 (2 μM) alone or in combination with IL-1β (0.5 ng/ml) in culture medium with 1% FCS. Cells killed with saponin were used as a negative control. Fibroblasts were incubated with MTT reagent for 3 h; the resulting formazan crystals were solubilized and spectrophotometrically quantified at 570 nm. Absorbance values were expressed as relative to unstimulated control cells.
mPGES-1 activity assay
The in vitro mPGES-1 activity assay was performed using a modified previously described method (48). Briefly, human recombinant mPGES-1 enzyme (0.1 mg) was mixed with TH-848 (200 μM), TH-644 (200 μM), or MK-886 (100 μM) in PBS containing 5 mM Triton X-100, 1 mM EDTA, and 2.5 mM GSH in a final volume of 120 μl, and incubated for 15 min at room temperature. PGH2 (40 μM) was added to initiate the reaction, and after 20 s of incubation (on ice), the reaction was stopped by removing 10 μl of the mixture to 490 μl of 4 mg/ml FeCl2 solution containing 0.1 mM HCl in PGE2 EIA buffer. The PGE2 concentrations in the samples were determined using EIA, and the activity of mPGES-1 was expressed as recombinant mPGES-1 activity (relative to control).
COX-2 activity assay
TH-848 (200 μM), TH-644 (200 μM), MK-886 (100 μM), and the COX-2 inhibitor DuP-697 (0.1 μM) were tested for COX-2 enzyme activity. The assay was performed using the COX inhibitor screening assay kit according to the manufacturer's instructions, and the results were expressed as recombinant COX-2 activity (relative to control).
Flow cytometry
Human gingival fibroblasts were seeded and grown as described above. After treatment with IL-1β alone or in combination with the substances TH-848, TH-644,or MK-886, the cells were collected by trypsinization and washed with PBS. The cells were then fixed in 2% paraformaldehyde and permeabilized with PBS containing 0.1% saponin and 0.01 M HEPES. Flow cytometric analysis was performed as described previously (36). Briefly, the cells were incubated with primary antibodies against PGE synthases and COX-2 and fluorescently labeled secondary antibodies. For each sample, 10,000–20,000 events were acquired by a FACSVerse flow cytometer using the FACSuite flow cytometry software (Becton Dickinson, San Jose, CA, USA). The cells were analyzed with regard to PGE synthases and COX-2; results are presented as histograms of cell counts vs. fluorescence intensity.
RNA preparation and RT-PCR
Fibroblasts were seeded and cultured as described above. After 6 h of treatment with IL-1β, in agreement with our previous studies (47, 49), in the absence or presence of TH-848 or TH-644, total RNA was isolated from the cells by the commercially available RNeasy Mini Kit. The amount of total RNA was quantified using a NanoVue Plus spectrophotometer (GE Healthcare, Uppsala, Sweden), and first-strand cDNA was obtained by reverse transcription of 1.0 μg of total RNA using Superscript II in a total volume of 20 μl. The PCR amplification for mPGES-1, COX-2, and GAPDH was performed as described previously (36, 47). The PCR reaction products were separated on a 2% agarose gel containing GelRed. For each experiment, PCR amplifications were also carried out in samples without cDNA as negative controls. The RT-PCR products were detected using the molecular imager ChemiDoc XRS+ system (Bio-Rad Laboratories), and the intensities of the bands were quantified with the software Image Lab (Bio-Rad Laboratories).
Measurements of PGE2, LTB4, and 6-keto PGF1α
The amount of PGE2 in the cell culture medium, the supernatants of mPGES-1 activity assays, and the rat blood plasma samples were determined using the PGE2 monoclonal EIA kit according to the manufacturer's instructions. Detection limit for the PGE2 EIA kit was 15 pg/ml. Levels of LTB4 in rat blood plasma samples were determined using the LTB4 Express EIA kit. Levels of 6-keto PGF1α, the stable breakdown product of prostacyclin, were determined using the 6-keto PGF1α EIA kit. The detection limits for LTB4 and 6-keto PGF1α were 45 and 6 pg/ml, respectively.
Antimicrobial experiments
The antimicrobial susceptibility of 12 bacterial strains to the aminothiazoles was performed using the disc diffusion assay, as described previously (50, 51). The first group of strains, the facultative anaerobic gram-positive microorganisms (S. mutans IB, S. mutans OMZ, S. sobrinus, S. sanguis, S. salivarius, L. brevis, L. casei, A. naeslundii, and A. viscosus) was cultured on BHI agar, and the second group of bacteria, the anaerobic gram-negative microorganisms (P. gingivalis, A. actinomycetemcomitans and F. nucleatum) were cultured on HBA. All strains were cultured overnight, washed once in sterile phosphate buffer, and diluted to an optical density of 0.5 at 500 nm, as measured in a spectrophotometer. The suspension was then diluted 10 times, after which Petri dishes with BHI agar or HBA were flooded with 3 ml of the bacterial suspension, and excess fluid was removed by a sterile pipette. The surfaces were allowed to dry at 37°C for 30 min, and the circular antibiotic sensitivity discs, containing 10 μl of TH-848 or TH-644 (50 μM–1.0 mM), or the positive controls triclosan (0.03%) and chlorhexidine (2 mg/ml), were placed on the surface of the agar plates. The negative control consisted of DMSO diluted in 70% ethanol, which was also used for preparation and administration of the aminothiazoles. The plates were thereafter kept for 30 min at room temperature and directly thereafter incubated, at 37°C, anaerobically for 24 h (gram-positive strains) in candle jars or 72 h (gram-negative strains) in candle jars using the anaerobic pouch system to obtain a strict anaerobic condition.
Animals and the induction of experimental periodontitis
In total, 80 Sprague-Dawley rats weighing 270–350 g were used in this study. The rats were kept at the animal facility at the Karolinska University Hospital, housed in temperature-controlled rooms and receiving water and food ad libitum. Prior to starting, all studies including animals were approved by the Stockholm North Animal Ethics Committee. For induction of experimental periodontitis, the rats were anesthetized using isoflurane, and silk ligatures were placed around the cervical part of the second upper molar on both sides of 60 rats. The rats were divided into 4 groups with 20 rats in each group: nonligated control group, experimental periodontitis group treated with the aminothiazole TH-848, experimental periodontitis group treated with vehicle, and experimental periodontitis group receiving only ligatures. Blood samples were collected from the tail of all rats both at the start and the end of the experimental period, and from the heart of the rats at the end of the experimental period.
Preparation and administration of TH-848
The aminothiazole derivative TH-848 (100 μM), resuspended in DMSO and diluted in 70% ethanol, was initially injected in the gingiva (20 μl/side), buccaly next to the ligature-equipped teeth, immediately after ligature placement in the TH-848 group. In the following days of the experiment, topical treatment with a gel containing TH-848 (150 μM, 50 μl/side) was administrated to the TH-848 group 1×/d. For the vehicle group, the injection was prepared and administered in the same way, containing the corresponding amount of DMSO diluted in ethanol. For the preparation of the test gel, TH-848 resuspended in DMSO was dissolved in ethanol containing SDS and then mixed into a gel base (4% gantrez gel containing SDS) to a final concentration of 150 μM. The vehicle gel was prepared in the same way, using DMSO solution without TH-848. The experimental periodontitis group receiving only ligatures (20 rats) and the nonligated control group (20 rats) did not receive any topical treatment. After 8 d, all rats were euthanized using CO2, blood samples were collected, and the jaws were isolated for measurement of alveolar bone levels and collection of gingival biopsies for establishment of gingival fibroblasts.
Measurement of alveolar bone levels
The rat jaws were stored in formaldehyde and photographed under a light microscope for a clinical overview. Thereafter, the upper jaws were evenly separated at the intermaxillary suture and dissected. For the assessment of radiological alveolar bone levels, a standard intraoral X-ray unit (Planmeca, Helsinki, Finland) was used. The pieces of jaw were placed on a Planmeca ProSensor with the occlusal surface perpendicular to the sensor and exposed to 50 kV and 8 mA for 0.16 s, and the radiographs were analyzed using the supported software Planmeca Romexis. Alveolar bone levels were measured from a straight line marking and extrapolating the level of the occlusal surface of the second upper molar. From this line, the distance to the marginal bone was measured at the interproximal sites, mesial and distal, of the tooth. The measurements were performed by 2 dentists in a blinded procedure. To assess measurement reliability, calculations were performed of interexaminer correlation (0.80; P<0.0001), intraindividual deviation (0.017±0.096 mm, mean±sd), from repeated blinded measurements of 20 teeth) and interindividual deviation (0.018±0.110 mm). Approximated mean bone degradation was calculated by subtracting the mean bone level of the nonligated control group from the mean bone levels of the ligature-treated groups. Teeth that lost their ligatures during the experimental period were excluded from the study. After the radiographic analysis, some of the jaws were also defleshed, stained with aqueous methylene blue (1%), and photographed under a light microscope for visualization of the marginal bone.
Determination of cytokines in blood plasma
A total of 15 cytokines were analyzed in blood plasma collected on the first and last days of the experiments from the tail of selected rats. Selection was based on the sufficient amount of blood plasma for analysis at both time points, and whether the ligatures were intact at the end of the experiment. The cytokine measurements were performed using Luminex technology on a Bioplex Suspension Array System (Bio-Rad Laboratories) with a commercial Bio-Plex rat cytokine assay. Data on the detection levels of the assay can be obtained from the manufacturer.
Statistical analyses
Statistical analyses were performed using the statistical package IBM SPSS Statistics 19.0 (IBM, Armonk, NY, USA). All cell culture experiments were analyzed in a minimum of triplicates, and reproducible data representing one of ≥3 independent experiments was demonstrated. Results are expressed as means ± sd. Student's t test (2-tailed) was used in the statistical analysis. In the animal experiments, Pearson's correlation coefficient was used in order to calculate the interexaminer agreement regarding alveolar bone level measurements. Within rats, the mean values of alveolar bone level at mesial and distal sites were calculated using the measurements of both observers. One-way variance analysis was adopted for comparisons between groups regarding alveolar bone level, with rats as the unit of analysis. Pairwise comparisons within groups were performed using Scheffé's method. For levels of cytokines, PGE2, 6-keto PGF1α, and LTB4 in blood plasma, comparisons of levels in blood plasma before and after treatment within treatment groups were performed using the Wilcoxon paired signed-rank test, while the Kruskal-Wallis test was applied for comparisons between treatment groups regarding change after treatment. Bonferroni correction was used to compensate for multiple comparisons. Results were considered statistically significant at P < 0.05.
RESULTS
Aminothiazoles decrease PGE2 production in human gingival fibroblasts
Aminothiazole derivatives, identified by computer docking studies as described in Materials and Methods, were screened for their ability to inhibit PGE2 production in cell cultures of gingival fibroblasts. The compounds TH-848 and TH-644, with the capacity to inhibit PGE2 production, were further explored with regard to inflammation-induced PGE2 production, mPGES-1, and COX-2 activity, as well as protein and mRNA expression. To investigate the effect of TH-848 and TH-644 on inflammation-induced PGE2 production, human gingival fibroblasts were stimulated with the cytokine IL-1β (0.5 ng/ml) in combination with different doses of the aminothiazoles for 24 h. In agreement with our previous findings, the inflammatory mediator IL-1β increased the production of PGE2 compared to unstimulated control cells (Fig. 1). Treatment of the cells with TH-848 or TH-644 at different doses (0.5, 1, 2, and 5 μM) significantly (P<0.05) reduced the IL-1β-stimulated PGE2 production in a dose-dependent manner (Fig. 1A, B), with IC50 1.1 μM for TH-848 and 1.5 μM for TH-644. Neither TH-848 nor TH-644 by itself, at the indicated concentrations, significantly affected the levels of PGE2 production as compared to untreated control cells (Fig. 1A, B); moreover, TH-848 was nontoxic to gingival fibroblasts at the tested doses (Table 1).
Figure 1.
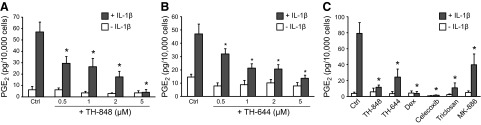
Effect of aminothiazoles on PGE2 production stimulated with IL-1β. A, B) Gingival fibroblasts were treated with TH-848 (0.5, 1, 2, and 5 μM; A) or TH-644 (0.5, 1, 2 and 5 μM; B), alone or in combination with IL-1β (0.5 ng/ml) for 24 h, and PGE2 was analyzed in culture medium. C) Effect of dexamethasone (dex; 1 μM), celecoxib (10 μM), triclosan (3.5 μM), or MK-886 (4 μM), as well as the aminothiazoles TH-848 (2 μM) and TH-644 (2 μM) alone or in combination with IL-1β (0.5 ng/ml) on PGE2 production. Control (Ctrl) cells were not treated with any inhibitors. Results are presented as means ± sd (n=3). All results represent ≥3 independent experiments. *P < 0.05 vs. IL-1β-stimulated cells.
Table 1.
Cytotoxicity of TH-848 in gingival fibroblasts
| Treatment | TH-848 (μM) | Extracellular LDH (% of LDH maximum) | MTT assay (% of control) |
|---|---|---|---|
| LDH maximum | − | 100.0 ± 10.8 | − |
| Saponin | − | − | 12.4 ± 2.4* |
| Control | − | 19.7 ± 12.5 | 100.0 ± 2.6 |
| TH-848 | 0.5 | 14.3 ± 5.4 | 104.2 ± 6.0 |
| TH-848 | 1.0 | 18.8 ± 11.5 | 112.0 ± 6.1* |
| TH-848 | 2.0 | 21.9 ± 8.5 | 123.2 ± 9.6* |
| TH-848 | 5.0 | 18.4 ± 7.8 | 124.5 ± 25.9 |
| TH-848 | 10.0 | 15.9 ± 10.8 | 114.9 ± 10.7 |
| IL-1β | − | 15.4 ± 6.8 | 117.2 ± 15.6 |
| IL-1β + TH-848 | 2.5 | 12.6 ± 4.1 | 129.9 ± 13.5 |
Human gingival fibroblasts were challenged with different concentrations of TH-848, or the cytokine IL-1β (0.5 ng/ml) alone or in combination with TH-848, for 24 h. Cytotoxicity was analyzed by assessment of LDH release and by MTT assay. Extracellular released LDH is presented as percentage of LDH maximum and MTT assay as viability compared to untreated control cells, values are means values ± sd. All analyses were performed in ≥4 replicates. Results shown represent ≥3 independent experiments.
P <0.05 vs. untreated control cells.
To further evaluate the inhibitory effect of TH-848 and TH-644 on PGE2 synthesis, we compared the effects of the aminothiazoles to those of dexamethasone, celecoxib, triclosan, and MK-886, which have previously been reported to inhibit PGE2 production (17, 18, 50). The anti-inflammatory substances dexamethasone (1.0 μM), celecoxib (10 μM), triclosan (3.5 μM), and the mPGES-1 inhibitor MK-886 (4 μM) reduced the stimulatory effect of IL-1β on PGE2 production, similar to the aminothiazoles TH-848 and TH-644 (Fig. 1C).
TH-848 and TH-644 decreased mPGES-1 but not COX-2 enzyme activity
The effect of TH-848 and TH-644 on the enzyme activity of recombinant mPGES-1 was investigated using an in vitro activity assay measuring the conversion of PGH2 to the end product PGE2, as described in Materials and Methods. The results showed that both TH-848 (200 μM) and TH-644 (200 μM) decreased the mPGES-1 enzyme activity (Fig. 2A). In addition, the well-known mPGES-1 activity inhibitor MK-886 (100 μM; refs. 35, 36), used as a positive control, also decreased the activity of mPGES-1 in the assay (Fig. 2A). To investigate the specificity of TH-848 and TH-644 as mPGES-1 inhibitors, we also tested the effect of the aminothiazole derivatives on COX-2 activity. The enzyme activity of COX-2, measuring the conversion of arachidonic acid to prostaglandins, was not affected in the presence of TH-848 (200 μM) or TH-644 (200 μM), in contrast to the COX-2 inhibitor DuP-697 (0.1 μM), used as a positive control (51), which strongly inhibited the COX-2 enzyme activity (Fig. 2B). The mPGES-1 inhibitor MK-886 (100 μM), similar to TH-848 and TH-644, did not affect the COX-2 activity (1.0±0.1 relative to control).
Figure 2.

Effect of TH-848 and TH-644 on mPGES-1 and COX-2 protein activity. A) An in vitro cell free mPGES-1 activity assay, measuring the conversion of PGH2 to PGE2, was performed using human recombinant mPGES-1 enzyme alone [control (Ctrl)] or in combination with TH-848 (200 μM), TH-644 (200 μM) or MK-886 (100 μM). The amount of PGE2 produced in the assay is expressed as mPGES-1 activity relative to control. B) An in vitro COX-2 activity assay, measuring the conversion of arachidonic acid to prostaglandins, was performed using human recombinant COX-2 enzyme alone (Ctrl) or in combination with TH-848 (200 μM), TH-644 (200 μM), or DuP-697 (0.1 μM). Results are expressed as COX-2 activity relative to Ctrl. Values are presented as means ± sd (n=3). All results represent ≥3 independent experiments. *P < 0.05 vs. Ctrl.
Effect of TH-848 and TH-644 on protein expression of PGES enzymes and COX-2
The effect of TH-848 and TH-644 on protein expression was also investigated regarding all 3 PGES isoforms, mPGES-1 mPGES-2 and cPGES, as well as the upstream enzyme COX-2, using flow cytometry. In agreement with previous findings (14, 36), IL-1β (0.5 ng/ml) increased the protein expression of mPGES-1 and COX-2 in human gingival fibroblasts (Fig. 3A, B). TH-848 (2 μM) slightly decreased the IL-1β-increased mPGES-1 protein expression in the cells (Fig. 3A), in contrast to TH-644 (2 μM), which did not affect the expression of mPGES-1 (Fig. 3A). Similar to TH-848, MK-886 (4 μM), decreased the IL-1β-increased protein expression of mPGES-1 (Fig. 3A). Neither TH-848, TH-644, nor MK-886 affected the IL-1β-increased COX-2 protein expression as compared to cells stimulated with IL-1β only (Fig. 3B). In addition, TH-848, TH-644, and MK-886 did not affect the protein expression of the constitutively expressed isoforms of PGES, mPGES-2, and cPGES, as compared to untreated control cells (Fig. 3C, D).
Figure 3.

Effects of TH-848, TH-644, and MK-886 on protein expression of the PGES isoenzymes mPGES-1, mPGES-2, and cPGES, as well as the upstream enzyme COX-2. Gingival fibroblasts were treated with IL-1β (0.5 ng/ml) in the absence or presence of TH-848 (2 μM), TH-644 (2 μM) or MK-886 (4 μM) for 24 h. Control (Ctrl) cells were treated with culture medium only. Protein expression of mPGES-1 (A), COX-2 (B), mPGES-2 (C), and cPGES (D) was analyzed using flow cytometry and presented as histograms of cell counts vs. logarithmic fluorescence intensity. Results shown represent ≥3 independent experiments.
Aminothiazoles TH-848 and TH-644 did not affect mRNA expression of mPGES-1 and COX-2
The effect of the aminothiazoles on mPGES-1 and COX-2 mRNA expression was also evaluated. Gingival fibroblasts were treated with IL-1β alone or in combination with TH-848 or TH-644 for 6 h, and the mRNA expression was analyzed using RT-PCR. The results showed that TH-848 (2 μM) and TH-644 (2 μM) did not affect the IL-1β (0.5 ng/ml)-induced mPGES-1 mRNA expression in 6 h cultures (Fig. 4A). Similarly, COX-2 mRNA expression induced by IL-1β was unaffected by TH-848 or TH-644 (Fig. 4B).
Figure 4.

Expression of mPGES-1 (A) and COX-2 (B) mRNA in gingival fibroblasts treated with aminothiazoles. Cells were treated with IL-1β (0.5 ng/ml) alone or in combination with TH-848 (2 μM) or TH-644 (2 μM) for 6 h. Control (Ctrl) cells were treated with culture medium only. mRNA expression was analyzed by RT-PCR using GAPDH as an internal control. Graphs show semiquantification of mRNA expression in relation to GAPDH, expressed as relative to control cells. Results are presented as means ± sd from ≥3 independent experiments.
Aminothiazoles TH-848 and TH-644 did not display antimicrobial activity
As the aminothiazoles were partially based on the structure of triclosan, which is a known antibacterial agent, the antibacterial effects of TH-848 and TH-644 were also investigated. Neither TH-848 nor TH-644 revealed any visible antibacterial action against the 9 gram-positive strains, as demonstrated for A. naeslundii (Fig. 5A) or the 3 gram-negative periodontitis-associated bacterial strains, as demonstrated for A. actinomycetemcomitans (Fig. 5B). In contrast, both triclosan and chlorhexidine, used as positive controls, inhibited the growth of all strains tested, including A. naeslundii and A. actinomycetemcomitans (Fig. 5).
Figure 5.
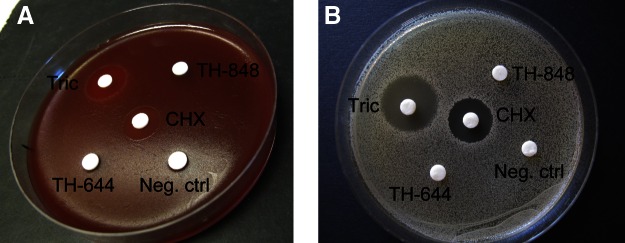
Antimicrobial assessment of TH-848 and TH-644. Antibacterial effects of TH-848 and TH-644 were investigated on several different oral bacterial strains (as indicated in Materials and Methods) using the disc diffusion assay. Photographs show representative disc diffusion test displaying the effect of TH-848 (1 mM), TH-644 (1 mM), triclosan (Tric; 0.03%), chlorhexidine (CHX; 2 mg/ml) and negative control on the gram-negative periodontal pathogen A. actinomycetemcomitans (A) or the gram-positive oral bacterial strain A. naeslundii (B).
Alveolar bone resorption was decreased by TH-848 in experimental periodontitis
In the next series of experiments, we proceeded to evaluate the effect of the aminothiazole TH-848 on bone resorption in experimental periodontitis as a clinically relevant in vivo model to explore the potential of the aminothiazoles as possible adjuncts for treatment of periodontitis. The ligature model has been previously shown to cause an acute inflammation with concomitant alveolar bone destruction similar to that observed in the chronic inflammation of periodontitis (52). Sprague-Dawley rats with ligature-induced experimental periodontitis (Fig. 6A) were treated with TH-848 or a corresponding vehicle. Clinically, gingival tissue destruction could be observed around the ligated teeth in rats with induced experimental periodontitis compared to control rats with no ligatures (Fig. 6B, C). When the soft tissue was removed from the jaws, the alveolar bone levels for nonligated control rats, rats with experimental periodontitis receiving only ligatures, vehicle-treated rats with experimental periodontitis, and TH-848-treated rats with experimental periodontitis could be observed visually (Fig. 6D–G).
Figure 6.
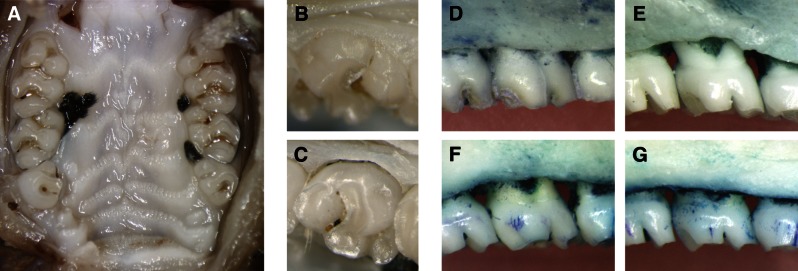
Photographs of ligature-induced periodontitis. A) Overview of ligature placement around the upper second molars of the rat. B, C) Clinical photographs from a control tooth without ligature (B) and a tooth with ligature-induced experimental periodontitis receiving only ligature (C). D–G) Photographs of defleshed jaws illustrating the marginal alveolar bone of a nonligated control (D), ligature-induced experimental periodontitis receiving only ligature (E), ligature-induced periodontitis treated with vehicle gel (F),and ligature-induced periodontitis treated with gel containing TH-848 (G).
Dental radiographs were taken of all jaws for measurements of alveolar bone levels, and representative radiographs from the 4 treatment groups are presented in Fig. 7A–D. The distributions of alveolar bone level according to treatment are illustrated in Fig. 7E. When comparing the mean values of bone measurements at group level, all 3 ligature-induced experimental periodontitis groups exhibited significantly (P<0.0001) higher alveolar bone loss compared to the nonligated controls (Table 2). Treatment of ligature-induced experimental periodontitis with the aminothiazole TH-848 significantly (P<0.0001) decreased alveolar bone loss compared with the group treated with vehicle or the group receiving ligatures only (Table 2). The approximated mean bone degradation in the group treated with TH-848 was 44% less than that of the group receiving ligature only, and 46% less than that of the vehicle-treated group. No significant differences in bone levels were observed between the experimental periodontitis group with only ligatures and the vehicle-treated group (Table 2).
Figure 7.
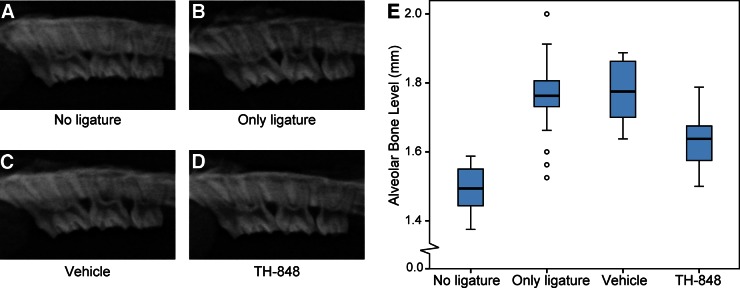
TH-848 ameliorates experimental periodontitis in rats. A–D) Representative radiographs of jaw from a tooth with no ligature (A), experimental periodontitis receiving only ligature (B), experimental periodontitis treated with gel containing vehicle (C), and experimental periodontitis treated with gel containing TH-848 (D). E) Distributions of mean alveolar bone levels according to treatment groups: no ligature (n=20), only ligature (n=20), vehicle-treated experimental periodontitis (n=18) and TH-848-treated experimental periodontitis (n=20). Small circles indicate outliers.
Table 2.
Alveolar bone levels in rats
| Treatment | n | Alveolar bone level (mm) |
|---|---|---|
| No ligature | 20 | 1.49 ± 0.016* |
| Only ligature | 20 | 1.76 ± 0.025# |
| Vehicle | 18 | 1.77 ± 0.020# |
| TH-848 | 20 | 1.64 ± 0.018* |
Alveolar bone levels are means ± se
P < 0.0001 vs. all other groups;
P < 0.0001 vs. no ligature and TH-848 groups.
TH-848 did not affect the levels of PGE2, 6-keto PGF1α, LTB4, or cytokines in blood
The effect of experimental periodontitis, as well as the effect of TH-848 treatment on systemic levels of PGE2, LTB4, and 6-keto PGF1α, which is the stable breakdown product of prostacyclin, was analyzed in blood plasma samples collected from the rats. The results showed no significant differences between treatment groups for PGE2, 6-keto PGF1α, or LTB4 in heart blood plasma (Table 3) or for PGE2 or 6-keto PGF1α in tail blood plasma measuring the differences between the groups on the first and last day of the experiment (data not shown). In addition, the systemic effect of local treatment with TH-848 on 15 cytokines was also investigated in blood plasma samples. The levels of the cytokines did not significantly differ between the first and last day within the different groups, or between groups (Table 4).
Table 3.
Levels of PGE2, 6-keto PGF1α, and LTB4 in blood plasma
| Treatment | PGE2 [pg/ml (n)] | 6-keto PGF1α [pg/ml (n)] | LTB4 [pg/ml (n)] |
|---|---|---|---|
| No ligature | 1970 ± 885 (11) | 738 ± 527 (9) | 10714 ± 6074 (12) |
| Only ligature | 2486 ± 1713 (11) | 647 ± 409 (8) | 10352 ± 4430 (11) |
| Vehicle | 2432 ± 1374 (11) | 586 ± 466 (8) | 10358 ± 5542 (11) |
| TH-848 | 2307 ± 1341 (10) | 669 ± 611 (8) | 10491 ± 6288 (10) |
Levels were measured in heart blood collected at the end of the experimental period. Analyte concentrations are means ± sd. No significant differences were found between the treatment groups for any of the analytes.
Table 4.
Levels of cytokines in rat blood plasma
| Cytokine | NL, n = 7 |
OL, n = 10 |
Vehicle, n = 9 |
TH-848, n = 10 |
||||
|---|---|---|---|---|---|---|---|---|
| d 1 | d 8 | d 1 | d 8 | d 1 | d 8 | d 1 | d 8 | |
| G-CSF | 17 ± 12 | 12 ± 6 | 9 ± 9 | 8 ± 4 | 11 ± 8 | 7 ± 4 | 11 ± 12 | 10 ± 6 |
| GM-CSF | 88 ± 62 | 81 ± 52 | 38 ± 18 | 37 ± 22 | 52 ± 35 | 29 ± 13 | 56 ± 63 | 50 ± 29 |
| IFN-γ | 92 ± 71 | 70 ± 32 | 61 ± 65 | 50 ± 36 | 65 ± 54 | 43 ± 36 | 75 ± 65 | 73 ± 73 |
| IL-1β | 178 ± 103 | 188 ± 75 | 157 ± 116 | 149 ± 109 | 116 ± 51 | 93 ± 27 | 152 ± 73 | 166 ± 56 |
| IL-4 | 156 ± 118 | 96 ± 59 | 59 ± 29 | 54 ± 33 | 72 ± 50 | 38 ± 19 | 90 ± 76 | 61 ± 35 |
| IL-6 | 1436 ± 1165 | 1060 ± 554 | 717 ± 530 | 593 ± 390 | 896 ± 626 | 591 ± 402 | 1489 ± 2170 | 1019 ± 805 |
| IL-7 | 217 ± 163 | 194 ± 110 | 105 ± 64 | 143 ± 115 | 139 ± 106 | 98 ± 72 | 117 ± 130 | 137 ± 61 |
| IL-10 | 1733 ± 1195 | 1358 ± 681 | 771 ± 249 | 884 ± 370 | 897 ± 537 | 685 ± 149 | 1153 ± 840 | 1011 ± 284 |
| IL-12p70 | 57 ± 50 | 50 ± 37 | 24 ± 11 | 25 ± 18 | 32 ± 26 | 19 ± 13 | 33 ± 41 | 37 ± 13 |
| IL-13 | 90 ± 68 | 90 ± 46 | 52 ± 20 | 50 ± 22 | 64 ± 35 | 43 ± 15 | 58 ± 48 | 58 ± 20 |
| IL-18 | 698 ± 252 | 589 ± 261 | 1009 ± 868 | 678 ± 394 | 673 ± 371 | 798 ± 422 | 692 ± 679 | 671 ± 453 |
| M-CSF | 259 ± 27 | 300 ± 46 | 207 ± 48 | 196 ± 40 | 191 ± 35 | 203 ± 34 | 212 ± 27 | 201 ± 71 |
| MIP-3α | 76 ± 58 | 56 ± 17 | 40 ± 17 | 40 ± 13 | 43 ± 21 | 31 ± 8 | 50 ± 35 | 39 ± 19 |
| RANTES | 620 ± 136 | 561 ± 135 | 416 ± 104 | 491 ± 97 | 591 ± 259 | 447 ± 112 | 551 ± 304 | 545 ± 225 |
| TNFα | 96 ± 55 | 93 ± 44 | 47 ± 19 | 49 ± 24 | 56 ± 30 | 48 ± 12 | 70 ± 58 | 59 ± 30 |
Levels of cytokines (pg/ml) were analyzed in rat blood plasma from tail blood of nonligated (NL) control rats, rats with induced experimental periodontitis receiving only ligatures (OL), vehicle treatment, or TH-848 treatment. Values are means ± sd. No significant changes in cytokine levels were found within or between treatment groups.
TH-848 decreased PGE2 production in rat gingival fibroblasts
To investigate further the mechanisms behind the capacity of TH-848 to reduce alveolar bone loss in our experimental periodontitis rat model, gingival fibroblasts were also isolated from healthy rat gingiva for in vitro studies. We stimulated the primary rat gingival fibroblasts with the cytokine IL-1β alone or in combination with TH-848, and the production of PGE2 was analyzed by EIA. IL-1β (0.3 ng/ml) stimulated the PGE2 production in rat gingival fibroblasts, and treatment of the cells with TH-848 (2 μM) reduced the IL-1β-stimulated PGE2 production with no cytotoxic effects, as measured by MTT assay (Fig. 8). This finding is in line with our results on human gingival fibroblasts, confirming that TH-848 can inhibit the rat ortholog of mPGES-1, which differs slightly from human mPGES-1 (53).
Figure 8.
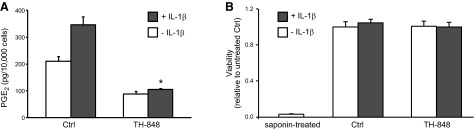
Effect of TH-848 on PGE2 production and cell viability in rat gingival fibroblasts stimulated with IL-1β. Cells were treated with IL-1β (0.3 ng/ml, A; 0.5 ng/ml, B), alone or in combination with TH-848 (2 μM). Control (Ctrl) cells were not treated with TH-848. PGE2 production was measured by EIA (A), and viability was assessed by MTT assay (B). Results are presented as means ± sd (n=3). *P < 0.05 vs. IL-1β-stimulated cells for PGE2 production. Results shown represent ≥3 independent experiments.
DISCUSSION
Thiazoles and their derivatives exhibit a range of biological activities, including anti-inflammatory properties (43, 44), although their mechanism of action is not well known. Here we show that aminothiazoles inhibited PGE2 production in gingival fibroblasts by targeting mPGES-1, the terminal enzyme regulating inflammation-induced PGE2 synthesis. This study also demonstrates, for the first time, that the aminothiazole TH-848 decreased alveolar bone destruction in experimental periodontitis in a rat model.
Increased levels of the inflammatory mediator PGE2 have been reported in gingival tissues, as well as in gingival fluid of patients with periodontitis, indicating an essential role for this mediator in the pathogenesis of the chronic inflammatory disease periodontitis (3, 4, 8). In addition, our group has recently reported that PGE2-synthesizing enzymes PGES and COX-2 were expressed in the gingival tissues of patients with periodontitis (14). In the current study, gingival fibroblasts, the dominant cell type in the gingival connective tissue, were used as in vitro model to evaluate the effect of two aminothiazole derivatives on mPGES-1 expression and its end product PGE2. In human gingival fibroblasts, the aminothiazoles TH-848 and TH-644, identified as potential mPGES-1 inhibitors by docking studies, decreased the IL-1β-stimulated PGE2 production in a dose-dependent manner. In a cell-free mPGES-1 enzyme activity assay, these compounds also decreased mPGES-1 activity but not COX-2 activity. Neither TH-848 nor TH-644 affected the IL-1β-induced COX-2 protein expression or the basal expression of COX-2 in gingival fibroblasts. Moreover, the aminothiazoles did not affect mPGES-1 or COX-2 mRNA expression in 6 h cultures, indicating that the inhibitory effect of TH-848 and TH-644 on the regulation of PGE2 was mainly mediated by decreased mPGES-1 activity. In addition, treatment with TH-848 or TH-644 did not affect the basal expression of the isoenzymes mPGES-2 or cPGES, which, in agreement with our previous findings, did not increase in response to cytokine stimulation (14, 36). Collectively, these results suggest a selective inhibition of the mPGES-1 enzyme by aminothiazoles TH-848 and TH-644. The aminothiazole TH-848 slightly reduced the cytokine-stimulated mPGES-1 protein expression, in contrast to TH-644, similar to mPGES-1 inhibitors MK-886 and imidazole derivatives, which have been shown to reduce mPGES-1 expression (36, 54). This modest reduction in mPGES-1 protein expression may not be biologically relevant as compared to the pronounced effect of TH-848 on mPGES-1 activity. Several novel inhibitors potentially targeting mPGES-1 have been identified in structural computer models and have not been further studied in relevant biological systems (55). The mechanisms of action of the well-characterized COX inhibitors that are used in clinical therapies to reduce PGE2 synthesis include effects on both expression and activity of the COX enzymes (56, 57).
The inhibitory effect of aminothiazoles TH-848 and TH-644 on PGE2 was compared with different substances known to inhibit PGE2 production and used in the clinical management of inflammatory conditions. The glucocorticoid dexamethasone, acting on a broad spectrum of inflammatory mechanisms (58), the selective COX-2 inhibitor celecoxib, and the antibacterial and anti-inflammatory agent triclosan all reduced PGE2 production in gingival fibroblasts in a similar manner as the two aminothiazoles. In contrast to the widely used triclosan, no antibacterial effects were observed for TH-848 or TH644, indicating that these aminothiazoles may be used without concern for bacterial resistance.
One of the aminothiazole derivatives, TH-848, was further investigated in vivo for its potential efficacy on bone resorption using experimental periodontitis. We here used a ligature-induced experimental periodontitis rat model, which is commonly used for studying the pathogenesis of periodontitis, as well as for testing of anti-inflammatory and antibacterial agents for treatment of periodontal disease (34, 59, 60). Our in vivo studies demonstrated a reduced alveolar bone resorption of experimental periodontitis after treatment with the aminothiazole TH-848. This reduction may be partly due to the decreased PGE2 production in the gingiva in response to treatment with the potential mPGES-1 inhibitor TH-848 with the capacity to inhibit PGE2 synthesis. This assumption was further supported by the in vitro studies using rat gingival fibroblasts that also demonstrated a reduction of cytokine-induced PGE2 production in the presence of TH-848. We did not observe any changes in systemic PGE2 or cytokine levels in the rat model, which may be due to the fact that the treatment was local rather than systemic.
Substances affecting the eicosanoid pathways, such as resolvin E1, have been shown to have a beneficial effect on periodontitis (29, 30). Similarly, the selective COX-2 inhibitor celecoxib has previously been reported to have a beneficial effect on the progression of periodontal disease, as well as other diseases associated with inflammatory bone destruction, such as rheumatoid arthritis (20, 32). It has been suggested that these COX-2 inhibitors should be used with caution, since there are several associated side-effects, such as gastrointestinal toxicity and disturbed homeostasis leading to cardiovascular disease (22–24). Our results showed no effect of the aminothiazole inhibitors on COX-2 activity. Furthermore, systemic LTB4 levels were unaffected, as were the levels of 6-keto PGF1α, the stable breakdown product of prostacyclin. These results suggest that the locally applied aminothiazoles may avoid the side effects related to COX inhibition, and may therefore be possible potential candidates for selective inhibition of PGE2 production.
Despite intensive research, there are currently no selective mPGES-1 inhibitors available for clinical use, although a number of potential mPGES-1 inhibitors have been identified and investigated in vitro (55, 61–63). Our previous findings show that triclosan inhibits cytokine-induced mPGES-1 mRNA and protein expression, accompanied by inhibition of PGE2 production in gingival fibroblasts, without affecting COX-2 expression (18). This agent has been reported to reduce gingival inflammation in patients and alveolar bone loss in experimental periodontitis in rats (33, 34). However, triclosan has other properties, including environmental toxicity and a potentially increased risk of bacterial resistance, indicating that this agent should be avoided in commercial use (64, 65). The substance curcumin was reported to reduce IL-1β-stimulated mPGES-1 expression in vitro and cytokine expression in gingival tissues of experimental periodontitis in rats (37–39), but in a recent study, curcumin failed to decrease alveolar bone resorption in ligature-induced experimental periodontitis after systemic administration by oral gavage (60). In this study, TH-848 was administrated topically to the ligature-equipped tooth, which we believe was necessary to obtain accurate treatment outcomes, in this case a decrease in alveolar bone resorption.
The potential anti-inflammatory activities of thiazoles have previously been attributed to the ability of these compounds to inhibit 5-lipoxygenase enzyme activity and LTB4 production, and to inhibit the synthesis of the inflammatory mediator nitric oxide (43, 44). Studies have also demonstrated that thiazoles prevent inflammatory cell infiltration and bone destruction in induced arthritis mouse models (42, 66). During the preparation of this article, findings that thiazoles bind to mPGES-1 protein and decrease IL-1β-induced PGE2 production in cancer cell lines were published (67), which are in line with our results. In this study we show for the first time that aminothiazoles inhibit cytokine-induced PGE2 production in gingival fibroblasts by targeting the enzyme mPGES-1, and that the aminothiazole TH-848 reduces the inflammation-associated alveolar bone loss in experimental periodontitis in rats. We conclude that aminothiazole derivatives represent novel mPGES-1 inhibitors that may be used to complement future treatment strategies of the chronic inflammatory disease periodontitis.
Acknowledgments
This work was supported by grants from the Swedish Patent Revenue Fund (T.Y.-L.); the Swedish Research Council, project number 73XD-15005 (T.Y.-L.); the Swedish Dental Society (A.K.); and Karolinska Institutet (A.K., T.Y.-L.).
The authors thank Ms. Ann-Britt Lundberg and Professor Dowen Birkhed (Institute of Odontology, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden) for disc diffusion tests; Ms. Haleh Davanian and Ms. Kaja Eriksson (Department of Dental Medicine, Karolinska Institutet) for assistance with laboratory work; and Dr. Rachael Sugars (Department of Dental Medicine, Karolinska Institutet) for editing the English language.
Footnotes
- BHI
- brain heart infusion
- COX
- cyclooxygenase
- cPGES
- cytosolic prostaglandin E synthase
- DMEM
- Dulbecco's modified Eagle medium
- DuP-697
- 5-bromo-2-(4-fluorophenyl)-3-[4-(methylsulfonyl)phenyl]-thiophene
- EIA
- enzyme immunoassay
- FCS
- fetal calf serum
- GSH
- glutathione
- HBA
- horse blood agar
- IL-1β
- interleukin-1β
- LDH
- lactate dehydrogenase
- mPGES
- microsomal prostaglandin E synthase
- LTB4
- leukotriene B4
- MK-886
- 1-[(4-chlorophenyl)methyl]-3-[(1,1-dimethylethyl)thio]-α,α-dimethyl-5-(1-methylethyl)-1H-indole-2-propanoic acid
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NSAID
- nonsteroid anti-inflammatory drug
- PBS
- phosphate-buffered saline
- PE
- phycoerythrin
- PGE2
- prostaglandin E2
- PGES
- prostaglandin E synthase
- PGF1α
- prostaglandin F1α
- PGH2
- prostaglandin H2
- TNF-α
- tumor necrosis factor α
- TH-644
- 4-(3-fluoro-4-methoxyphenyl)-N-(4-phenoxyphenyl)-1,3-thiazol-2-amine
- TH-848
- 4-([4-(2-naphthyl)-1,3-thiazol-2-yl]amino)phenol
REFERENCES
- 1. Armitage G. C. (2004) Periodontal diagnoses and classification of periodontal diseases. Periodontol. 2000 34, 9–21 [DOI] [PubMed] [Google Scholar]
- 2. Kinney J. S., Ramseier C. A., Giannobile W. V. (2007) Oral fluid-based biomarkers of alveolar bone loss in periodontitis. Ann. N. Y. Acad. Sci. 1098, 230–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Page R. C. (1991) The role of inflammatory mediators in the pathogenesis of periodontal disease. J. Periodontal Res. 26, 230–242 [DOI] [PubMed] [Google Scholar]
- 4. Inada M., Matsumoto C., Uematsu S., Akira S., Miyaura C. (2006) Membrane-bound prostaglandin E synthase-1-mediated prostaglandin E2 production by osteoblast plays a critical role in lipopolysaccharide-induced bone loss associated with inflammation. J. Immunol. 177, 1879–1885 [DOI] [PubMed] [Google Scholar]
- 5. Blackwell K. A., Raisz L. G., Pilbeam C. C. (2010) Prostaglandins in bone: bad cop, good cop? Trends Endocrinol. Metab. 21, 294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang M., Song W. L., Cheng Y., Fitzgerald G. A. (2008) Microsomal prostaglandin E synthase-1 inhibition in cardiovascular inflammatory disease. J. Intern. Med. 263, 500–505 [DOI] [PubMed] [Google Scholar]
- 7. Akaogi J., Nozaki T., Satoh M., Yamada H. (2006) Role of PGE2 and EP receptors in the pathogenesis of rheumatoid arthritis and as a novel therapeutic strategy. Endocr. Metab. Immune Disord. Drug Targets 6, 383–394 [DOI] [PubMed] [Google Scholar]
- 8. Preshaw P. M., Heasman P. A. (2002) Prostaglandin E2 concentrations in gingival crevicular fluid: observations in untreated chronic periodontitis. J. Clin. Periodontol. 29, 15–20 [DOI] [PubMed] [Google Scholar]
- 9. Murakami M., Kudo I. (2004) Recent advances in molecular biology and physiology of the prostaglandin E2-biosynthetic pathway. Prog. Lipid Res. 43, 3–35 [DOI] [PubMed] [Google Scholar]
- 10. Jakobsson P. J., Thoren S., Morgenstern R., Samuelsson B. (1999) Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. U. S. A. 96, 7220–7225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tanikawa N., Ohmiya Y., Ohkubo H., Hashimoto K., Kangawa K., Kojima M., Ito S., Watanabe K. (2002) Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochem. Biophys. Res. Commun. 291, 884–889 [DOI] [PubMed] [Google Scholar]
- 12. Tanioka T., Nakatani Y., Semmyo N., Murakami M., Kudo I. (2000) Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J. Biol. Chem. 275, 32775–32782 [DOI] [PubMed] [Google Scholar]
- 13. Murakami M., Naraba H., Tanioka T., Semmyo N., Nakatani Y., Kojima F., Ikeda T., Fueki M., Ueno A., Oh S., Kudo I. (2000) Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J. Biol. Chem. 275, 32783–32792 [DOI] [PubMed] [Google Scholar]
- 14. Båge T., Kats A., Lopez B. S., Morgan G., Nilsson G., Burt I., Korotkova M., Corbett L., Knox A. J., Pino L., Jakobsson P. J., Modeer T., Yucel-Lindberg T. (2011) Expression of prostaglandin E synthases in periodontitis immunolocalization and cellular regulation. Am. J. Pathol. 178, 1676–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gudis K., Tatsuguchi A., Wada K., Futagami S., Nagata K., Hiratsuka T., Shinji Y., Miyake K., Tsukui T., Fukuda Y., Sakamoto C. (2005) Microsomal prostaglandin E synthase (mPGES)-1, mPGES-2 and cytosolic PGES expression in human gastritis and gastric ulcer tissue. Lab. Invest. 85, 225–236 [DOI] [PubMed] [Google Scholar]
- 16. Westman M., Korotkova M., af Klint E., Stark A., Audoly L. P., Klareskog L., Ulfgren A. K., Jakobsson P. J. (2004) Expression of microsomal prostaglandin E synthase 1 in rheumatoid arthritis synovium. Arthritis Rheum. 50, 1774–1780 [DOI] [PubMed] [Google Scholar]
- 17. Yucel-Lindberg T., Hallström T., Kats A., Mustafa M., Modeer T. (2004) Induction of microsomal prostaglandin E synthase-1 in human gingival fibroblasts. Inflammation 28, 89–95 [DOI] [PubMed] [Google Scholar]
- 18. Mustafa M., Wondimu B., Yucel-Lindberg T., Kats A., Hallström T., Jonsson A. S., Modeer T. (2005) Triclosan reduces microsomal prostaglandin E synthase-1 expression in human gingival fibroblasts. J. Clin. Periodontol. 32, 6–11 [DOI] [PubMed] [Google Scholar]
- 19. Turini M. E., DuBois R. N. (2002) Cyclooxygenase-2: a therapeutic target. Annu. Rev. Med. 53, 35–57 [DOI] [PubMed] [Google Scholar]
- 20. Chen Y. F., Jobanputra P., Barton P., Bryan S., Fry-Smith A., Harris G., Taylor R. S. (2008) Cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs (etodolac, meloxicam, celecoxib, rofecoxib, etoricoxib, valdecoxib and lumiracoxib) for osteoarthritis and rheumatoid arthritis: a systematic review and economic evaluation. Health Technol. Assess. 12, 1–278, iii [DOI] [PubMed] [Google Scholar]
- 21. Anderson G. D., Keys K. L., De Ciechi P. A., Masferrer J. L. (2009) Combination therapies that inhibit cyclooxygenase-2 and leukotriene synthesis prevent disease in murine collagen induced arthritis. Inflamm. Res. 58, 109–117 [DOI] [PubMed] [Google Scholar]
- 22. Koeberle A., Werz O. (2009) Inhibitors of the microsomal prostaglandin E(2) synthase-1 as alternative to non steroidal anti-inflammatory drugs (NSAIDs)–a critical review. Curr. Med. Chem. 16, 4274–4296 [DOI] [PubMed] [Google Scholar]
- 23. Ng S. C., Chan F. K. (2010) NSAID-induced gastrointestinal and cardiovascular injury. Curr. Opin. Gastroenterol. 26, 611–617 [DOI] [PubMed] [Google Scholar]
- 24. Sanghi S., MacLaughlin E. J., Jewell C. W., Chaffer S., Naus P. J., Watson L. E., Dostal D. E. (2006) Cyclooxygenase-2 inhibitors: a painful lesson. Cardiovasc. Hematol. Disord. Drug Targets 6, 85–100 [DOI] [PubMed] [Google Scholar]
- 25. Cannon C. P., Cannon P. J. (2012) Physiology. COX-2 inhibitors and cardiovascular risk. Science 336, 1386–1387 [DOI] [PubMed] [Google Scholar]
- 26. Offenbacher S., Heasman P. A., Collins J. G. (1993) Modulation of host PGE2 secretion as a determinant of periodontal disease expression. J. Periodontol. 64, 432–444 [DOI] [PubMed] [Google Scholar]
- 27. Pouliot M., Clish C. B., Petasis N. A., Van Dyke T. E., Serhan C. N. (2000) Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: a role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry 39, 4761–4768 [DOI] [PubMed] [Google Scholar]
- 28. Cavanaugh P. F., Jr., Meredith M. P., Buchanan W., Doyle M. J., Reddy M. S., Jeffcoat M. K. (1998) Coordinate production of PGE2 and IL-1 beta in the gingival crevicular fluid of adults with periodontitis: its relationship to alveolar bone loss and disruption by twice daily treatment with ketorolac tromethamine oral rinse. J. Periodontal. Res. 33, 75–82 [DOI] [PubMed] [Google Scholar]
- 29. Hasturk H., Kantarci A., Goguet-Surmenian E., Blackwood A., Andry C., Serhan C. N., Van Dyke T. E. (2007) Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J. Immunol. 179, 7021–7029 [DOI] [PubMed] [Google Scholar]
- 30. Hasturk H., Kantarci A., Ohira T., Arita M., Ebrahimi N., Chiang N., Petasis N. A., Levy B. D., Serhan C. N., Van Dyke T. E. (2006) RvE1 protects from local inflammation and osteoclast- mediated bone destruction in periodontitis. FASEB J. 20, 401–403 [DOI] [PubMed] [Google Scholar]
- 31. Salvi G. E., Lang N. P. (2005) The effects of non-steroidal anti-inflammatory drugs (selective and non-selective) on the treatment of periodontal diseases. Curr. Pharm. Des. 11, 1757–1769 [DOI] [PubMed] [Google Scholar]
- 32. Yen C. A., Damoulis P. D., Stark P. C., Hibberd P. L., Singh M., Papas A. S. (2008) The effect of a selective cyclooxygenase-2 inhibitor (celecoxib) on chronic periodontitis. J. Periodontol. 79, 104–113 [DOI] [PubMed] [Google Scholar]
- 33. Blinkhorn A., Bartold P. M., Cullinan M. P., Madden T. E., Marshall R. I., Raphael S. L., Seymour G. J. (2009) Is there a role for Triclosan/copolymer toothpaste in the management of periodontal disease? Br. Dent. J. 207, 117–125 [DOI] [PubMed] [Google Scholar]
- 34. Luan Q., Desta T., Chehab L., Sanders V. J., Plattner J., Graves D. T. (2008) Inhibition of experimental periodontitis by a topical boron-based antimicrobial. J. Dent. Res. 87, 148–152 [DOI] [PubMed] [Google Scholar]
- 35. Mancini J. A., Blood K., Guay J., Gordon R., Claveau D., Chan C. C., Riendeau D. (2001) Cloning, expression, and up-regulation of inducible rat prostaglandin e synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J. Biol. Chem. 276, 4469–4475 [DOI] [PubMed] [Google Scholar]
- 36. Båge T., Modeer T., Kawakami T., Quezada H. C., Yucel-Lindberg T. (2007) Regulation of prostaglandin E synthases: effects of siRNA-mediated inhibition of microsomal prostaglandin E synthase-1. Biochim. Biophys. Acta 1773, 1589–1598 [DOI] [PubMed] [Google Scholar]
- 37. Koeberle A., Northoff H., Werz O. (2009) Curcumin blocks prostaglandin E2 biosynthesis through direct inhibition of the microsomal prostaglandin E2 synthase-1. Mol. Cancer Ther. 8, 2348–2355 [DOI] [PubMed] [Google Scholar]
- 38. Moon Y., Glasgow W. C., Eling T. E. (2005) Curcumin suppresses interleukin 1beta-mediated microsomal prostaglandin E synthase 1 by altering early growth response gene 1 and other signaling pathways. J. Pharmacol. Exp. Ther. 315, 788–795 [DOI] [PubMed] [Google Scholar]
- 39. Guimaraes M. R., de Aquino S. G., Coimbra L. S., Spolidorio L. C., Kirkwood K. L., Rossa C., Jr. (2012) Curcumin modulates the immune response associated with LPS-induced periodontal disease in rats. Innate Immun. 18, 155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tonetti M. S., Chapple I. L. (2011) Biological approaches to the development of novel periodontal therapies–consensus of the Seventh European Workshop on Periodontology. J. Clin. Periodontol. 38(Suppl. 11), 114–118 [DOI] [PubMed] [Google Scholar]
- 41. Nishikaku F., Koga Y. (1993) Suppression of murine collagen-induced arthritis by treatment with a novel thiazole derivative, SM-8849. Immunopharmacology 25, 65–74 [DOI] [PubMed] [Google Scholar]
- 42. Ohno H., Uemura Y., Murooka H., Takanashi H., Tokieda T., Ohzeki Y., Kubo K., Serizawa I. (2008) The orally-active and selective c-Fms tyrosine kinase inhibitor Ki20227 inhibits disease progression in a collagen-induced arthritis mouse model. Eur. J. Immunol. 38, 283–291 [DOI] [PubMed] [Google Scholar]
- 43. Cho Y. S., Kim C. H., Surh J. H., Kang N. S., Yoo S. E., Cheon H. G. (2010) Identification of 4-[4-(4-fluoro-phenyl)-thiazol-2-ylamino]-2,6-dimethyl-phenol (KR-33749) as an inhibitor of 5-lipoxygenase with potent antiinflammatory activity. Pharmacology 86, 65–72 [DOI] [PubMed] [Google Scholar]
- 44. Moldovan C. M., Oniga O., Parvu A., Tiperciuc B., Verite P., Pirnau A., Crisan O., Bojita M., Pop R. (2011) Synthesis and anti-inflammatory evaluation of some new acyl-hydrazones bearing 2-aryl-thiazole. Eur. J. Med. Chem. 46, 526–534 [DOI] [PubMed] [Google Scholar]
- 45. Trott O., and Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jegerschold C., Pawelzik S. C., Purhonen P., Bhakat P., Gheorghe K. R., Gyobu N., Mitsuoka K., Morgenstern R., Jakobsson P. J., Hebert H. (2008) Structural basis for induced formation of the inflammatory mediator prostaglandin E2. Proc. Natl. Acad. Sci. U. S. A. 105, 11110–11115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yucel-Lindberg T., Olsson T., Kawakami T. (2006) Signal pathways involved in the regulation of prostaglandin E synthase-1 in human gingival fibroblasts. Cell. Signal. 18, 2131–2142 [DOI] [PubMed] [Google Scholar]
- 48. Friesen R. W., Mancini J. A. (2008) Microsomal prostaglandin E2 synthase-1 (mPGES-1): a novel anti-inflammatory therapeutic target. J. Med. Chem. 51, 4059–4067 [DOI] [PubMed] [Google Scholar]
- 49. Båge T., Lindberg J., Lundeberg J., Modeer T., Yucel-Lindberg T. (2010) Signal pathways JNK and NF-kappaB, identified by global gene expression profiling, are involved in regulation of TNFalpha-induced mPGES-1 and COX-2 expression in gingival fibroblasts. BMC Genomics 11, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Koeberle A., Siemoneit U., Buhring U., Northoff H., Laufer S., Albrecht W., Werz O. (2008) Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J. Pharmacol. Exp. Ther. 326, 975–982 [DOI] [PubMed] [Google Scholar]
- 51. Rosenstock M., Danon A., Rimon G. (1999) PGHS-2 inhibitors, NS-398 and DuP-697, attenuate the inhibition of PGHS-1 by aspirin and indomethacin without altering its activity. Biochim. Biophys. Acta 1440, 127–137 [DOI] [PubMed] [Google Scholar]
- 52. Di Paola R., Mazzon E., Maiere D., Zito D., Britti D., De Majo M., Genovese T., Cuzzocrea S. (2006) Rosiglitazone reduces the evolution of experimental periodontitis in the rat. J. Dent. Res. 85, 156–161 [DOI] [PubMed] [Google Scholar]
- 53. Pawelzik S. C., Uda N. R., Spahiu L., Jegerschold C., Stenberg P., Hebert H., Morgenstern R., Jakobsson P. J. (2010) Identification of key residues determining species differences in inhibitor binding of microsomal prostaglandin E synthase-1. J. Biol. Chem. 285, 29254–29261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tseng C. H., Tzeng C. C., Shih P. K., Yang C. N., Chuang Y. C., Peng S. I., Lin C. S., Wang J. P., Cheng C. M., Chen Y. L. (2012) Identification of furo[3′, 2′:3,4]naphtho[1,2-d]imidazole derivatives as orally active and selective inhibitors of microsomal prostaglandin E(2) synthase-1 (mPGES-1). Mol. Divers. 16, 215–229 [DOI] [PubMed] [Google Scholar]
- 55. Chang H. H., Meuillet E. J. (2011) Identification and development of mPGES-1 inhibitors: where we are at? Future Med. Chem. 3, 1909–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Capone M. L., Tacconelli S., Di Francesco L., Sacchetti A., Sciulli M. G., Patrignani P. (2007) Pharmacodynamic of cyclooxygenase inhibitors in humans. Prostaglandins Other Lipid Mediat. 82, 85–94 [DOI] [PubMed] [Google Scholar]
- 57. El-Rayes B. F., Ali S., Sarkar F. H., Philip P. A. (2004) Cyclooxygenase-2-dependent and -independent effects of celecoxib in pancreatic cancer cell lines. Mol. Cancer Ther. 3, 1421–1426 [PubMed] [Google Scholar]
- 58. Barnes P. J. (2005) Molecular mechanisms and cellular effects of glucocorticosteroids. Immunol. Allergy Clin. North Am. 25, 451–468 [DOI] [PubMed] [Google Scholar]
- 59. Cheng W. C., Huang R. Y., Chiang C. Y., Chen J. K., Liu C. H., Chu C. L., Fu E. (2010) Ameliorative effect of quercetin on the destruction caused by experimental periodontitis in rats. J. Periodontal Res. 45, 788–795 [DOI] [PubMed] [Google Scholar]
- 60. Guimaraes M. R., Coimbra L. S., de Aquino S. G., Spolidorio L. C., Kirkwood K. L., Rossa C., Jr. (2011) Potent anti-inflammatory effects of systemically administered curcumin modulate periodontal disease in vivo. J. Periodontal Res. 46, 269–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bruno A., Di Francesco L., Coletta I., Mangano G., Alisi M. A., Polenzani L., Milanese C., Anzellotti P., Ricciotti E., Dovizio M., Di Francesco A., Tacconelli S., Capone M. L., Patrignani P. (2010) Effects of AF3442 [N-(9-ethyl-9H-carbazol-3-yl)-2-(trifluoromethyl)benzamide], a novel inhibitor of human microsomal prostaglandin E synthase-1, on prostanoid biosynthesis in human monocytes in vitro. Biochem. Pharmacol. 79, 974–981 [DOI] [PubMed] [Google Scholar]
- 62. Giroux A., Boulet L., Brideau C., Chau A., Claveau D., Cote B., Ethier D., Frenette R., Gagnon M., Guay J., Guiral S., Mancini J., Martins E., Masse F., Methot N., Riendeau D., Rubin J., Xu D., Yu H., Ducharme Y., Friesen R. W. (2009) Discovery of disubstituted phenanthrene imidazoles as potent, selective and orally active mPGES-1 inhibitors. Bioorg. Med. Chem. Lett. 19, 5837–5841 [DOI] [PubMed] [Google Scholar]
- 63. Mbalaviele G., Pauley A. M., Shaffer A. F., Zweifel B. S., Mathialagan S., Mnich S. J., Nemirovskiy O. V., Carter J., Gierse J. K., Wang J. L., Vazquez M. L., Moore W. M., Masferrer J. L. (2010) Distinction of microsomal prostaglandin E synthase-1 (mPGES-1) inhibition from cyclooxygenase-2 inhibition in cells using a novel, selective mPGES-1 inhibitor. Biochem. Pharmacol. 79, 1445–1454 [DOI] [PubMed] [Google Scholar]
- 64. Ricart M., Guasch H., Alberch M., Barcelo D., Bonnineau C., Geiszinger A., Farre M., Ferrer J., Ricciardi F., Romani A. M., Morin S., Proia L., Sala L., Sureda D., Sabater S. (2010) Triclosan persistence through wastewater treatment plants and its potential toxic effects on river biofilms. Aquat. Toxicol. 100, 346–353 [DOI] [PubMed] [Google Scholar]
- 65. Saleh S., Haddadin R. N., Baillie S., Collier P. J. (2011) Triclosan - an update. Lett. Appl. Microbiol. 52, 87–95 [DOI] [PubMed] [Google Scholar]
- 66. Nishikaku F., Aono S., Koga Y. (1994) Protective effects of D-penicillamine and a thiazole derivative, SM-8849, on pristane-induced arthritis in mice. Int. J. Immunopharmacol. 16, 91–100 [DOI] [PubMed] [Google Scholar]
- 67. Chang H. H., Song Z., Wisner L., Tripp T., Gokhale V., Meuillet E. J. (2011) Identification of a novel class of anti-inflammatory compounds with anti-tumor activity in colorectal and lung cancers. Invest. New Drugs 30, 1865–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]