

Abstract
Acute lung injury (ALI) is characterized by alveolar injury and uncontrolled inflammation. Since most cases of ALI resolve spontaneously, understanding the endogenous mechanisms that promote ALI resolution is important to developing effective therapies. Previous studies have implicated extracellular adenosine signaling in tissue adaptation and wound healing. Therefore, we hypothesized a functional contribution for the endogenous production of adenosine during ALI resolution. As a model, we administered intratracheal LPS and observed peak lung injury at 3 d, with resolution by d 14. Treatment with pegylated adenosine-deaminase to enhance extracellular adenosine breakdown revealed impaired ALI resolution. Similarly, genetic deletion of cd73, the pacemaker for extracellular adenosine generation, was associated with increased mortality (0% wild-type and 40% in cd73−/− mice; P<0.05) and failure to resolve ALI adequately. Studies of inflammatory cell trafficking into the lungs during ALI resolution revealed that regulatory T cells (Tregs) express the highest levels of CD73. While Treg numbers in cd73−/− mice were similar to controls, cd73-deficient Tregs had attenuated immunosuppressive functions. Moreover, adoptive transfer of cd73-deficient Tregs into Rag−/− mice emulated the observed phenotype in cd73−/− mice, while transfer of wild-type Tregs was associated with normal ALI resolution. Together, these studies implicate CD73-dependent adenosine generation in Tregs in promoting ALI resolution.—Ehrentraut, H., Clambey, E. T., McNamee, E. N., Brodsky, K. S., Ehrentraut, S. F., Poth, J. M., Riegel, A. K., Westrich, J. A., Colgan, S. P., Eltzschig, H. K. CD73+ regulatory T cells contribute to adenosine-mediated resolution of acute lung injury.
Keywords: ectonucleotidase, lipopolysaccharide, hypoxia, Adora2b, inflammation
Acute lung injury (ALI) is characterized by acute hypoxemic respiratory failure in the setting of noncardiogenic pulmonary edema with a significant contribution to morbidity and mortality of critically ill patients (1, 2). In fact, ALI mortality ranges between 35 and 60%, and ∼200,000 patients develop ALI annually in the United States, leading to 75,000 deaths (3). Among the hallmarks of ALI is massive accumulation of inflammatory cells into different compartments of the lungs in conjunction with cytokine release and inflammatory activation of recruited or resident cells (4). Other characteristics include epithelial injury, causing disruption of the alveolar-capillary barrier function, resulting in extensive pulmonary edema and attenuated gas exchange. Previous research studies have focused mainly on the onset phase of ALI and have proposed strategies to prevent ALI development (4–7). However, in many instances, ALI is not recognized in its early onset stage. Indeed, clinicians are more frequently faced with the challenge of having to treat patients that have fully developed ALI, including the presence of inflammatory cells within the lungs, exposure to toxins produced by microorganisms, epithelial injury, and pulmonary edema. At this stage, the inflamed mucosal tissues follow a decision pathway to either resolve or to proceed toward an uncontrolled inflammatory response that often leads to death. In the present studies, we hypothesize the existence of endogenous protective molecular pathways at the interface between innate and adaptive immunity that can be targeted to promote ALI resolution.
Extracellular adenosine is an endogenously produced signaling molecule (2) that has been implicated previously in attenuating inflammation during inflammatory diseases (5, 7–12), hypoxia (1, 13–17), or ischemia and reperfusion injury (16–20). Adenosine receptors 2a and 2b (A2aR and A2bR) are critical for the limitation of inflammatory responses. Genetic models and pharmacological inhibition have been shown to augment inflammation in A2aR- or A2bR-signaling-deficient mice (21, 22). However, their functional role during the resolution phase of ALI remains unclear. During inflammatory conditions, extracellular adenosine stems predominantly from the breakdown of precursor nucleotides, such as ATP, ADP, and AMP (2, 23). This process is controlled by a set of enzymes that regulate extracellular nucleotide phosphohydrolysis. The pacemaker enzyme for the extracellular generation of adenosine is 5′-ectonucleotidase (CD73), an ectoenzyme that converts extracellular AMP to adenosine and thereby controls the final step for the liberation of adenosine (24–27). In vitro evidence confirmed for the first time that regulatory T cells (Tregs) express high amounts of cd73 and generate adenosine (28), thereby acting inhibitory on activated A2aR-expressing effector T cells (29).
To address the functional role of extracellular adenosine during the resolution phase of ALI, we exposed mice to ALI induced by intratracheal installation of lipopolysaccharide (LPS) and allowed mice to recover over a time period of 14 d. Subsequently, we combined genetic and pharmacologic approaches to alter extracellular generation of adenosine and thereby its signaling effects. These studies pointed us toward a functional role of extracellular adenosine generation during ALI resolution and implicate CD73+ Tregs in adenosine-mediated ALI resolution.
MATERIALS AND METHODS
Experimental animals
Cd73+/+ (C57BL/6J) and cd73−/− mice were bred in house. Cd73−/− mice were generated on a BL6 strain as described previously (6, 7, 24, 30–32). B6.Rag1−/− mice (B6.129S7-Rag1tm1Mom) were obtained from Jackson Laboratory (Bar Harbor, ME, USA). Mice were bred and maintained in accordance with the recommendations of the U.S. National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Experimental protocols were approved by the Institutional Review Board at the University of Colorado Denver and were in accordance with the Protection of Animals and the NIH guidelines for the use of live animals.
ALI model
Age (8–12 wk old) and weight-matched male cd73+/+ and cd73−/− mice were anesthetized with pentobarbital (70 mg/kg) before the procedure. LPS (Escherichia coli 0111:B4, L4391; Sigma, St. Louis, MO, USA; 3.75 μg/g body weight unless otherwise indicated) or saline as control was administered intratracheally via a 22-gauge catheter (33). Within up to 14 d post-LPS exposure, weight loss and recovery were recorded.
Treatment with pegylated adenosine deaminase (PEG-ADA) to dampen extracellular adenosine production and signaling
BL6 mice were injected intraperitoneally with 10 U PEG-ADA 3 d before LPS or saline instillation and immediately after intratracheal treatment (see Fig. 1A). Polyethylene glycol-modified ADA possesses a prolonged circulatory time due to reduced renal clearance (34). One unit of PEG-ADA is defined as the amount of enzyme necessary to convert 1 μmol of adenosine to inosine per minute at 25°C (35). Animals incorporated in the weight loss recording groups received a third dose 7 d post-LPS application. A total volume of 100 μl was injected, and saline served as control treatment.
Figure 1.
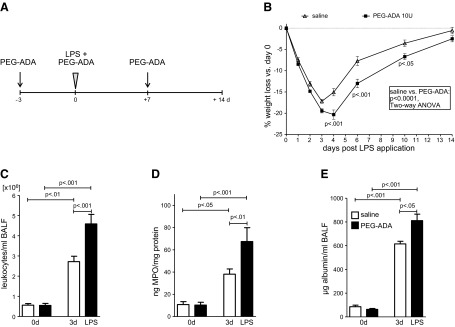
Enhanced adenosine turnover following PEG-ADA treatment aggravates weight loss and inflammation following LPS-induced lung injury. A) Mice received 10 U PEG-ADA or vehicle 3 d before and immediately after LPS instillation; tissue was harvested 3 d later. Mice included in the weight curve study were injected again after 7 d. B) Relative weight loss after LPS treatment (3.75 μg LPS/g body weight) in vehicle and PEG-ADA-treated wild-type mice. P values at indicated time points derived from Bonferroni post hoc testing (n=10–11/group). C) BALF derived from PEG-ADA group 3 d after LPS application contains higher leukocyte numbers (n=11–12/group). D) Pulmonary lysates from PEG-ADA-treated mice exhibit higher MPO activity (n=6–10/group). E) Increased albumin content in BALF from PEG-ADA mice harvested on d 3 post-LPS treatment (n=5–6/group).
5′-Nucleotidase (5′-NT) reconstitution
5′-NT from Crotalus atrox venom (Enzo Life Sciences, Farmingdale, NY, USA) was diluted in saline to a final concentration of 2 U 5′-NT/100 μl as done previously (10). Experimental animals received an intraperitoneal injection of 2 U 5′-NT or an equal volume of vehicle immediately after LPS treatment. Injection was repeated daily.
Tissue harvest
At indicated time points, mice were anesthetized and killed by exsanguination. Bronchoalveolar lavage fluid (BALF) was obtained after lavaging the lung with 3 × 1 ml saline. Cell-free BALF supernatant for ELISA studies was snap-frozen after centrifugation at 1000 g for 5 min at 4°C. Pulmonary tissue was flushed with 10 ml saline via the right ventricle, snap-frozen in liquid nitrogen, and stored at −80°C.
RNA isolation and real-time PCR
Total RNA was extracted from tissue by Trizol, followed by cDNA synthesis using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. Quantitative reverse transcriptase PCR (qPCR; ABI 7900HT; Applied Biosystems, Carlsbad, CA, USA) was performed to measure relative mRNA levels for various transcripts, with Power SYBR Green PCR Master Mix (Applied Biosystems), containing 1 μM sense and 1 μM antisense primers. Results were evaluated with the 2ΔΔCt method using β-actin as an internal control.
Primers for real-time RT-PCR
Real-time RT-PCR was performed with the following murine Quantitect Primer Assays (Qiagen, Valencia, CA, USA): β-actin (QT01136772), TNF-α (QT00104006), IL-1β (QT01048355), and IL-6 (QT00098875).
Leukocyte counts of BALF
BAL samples were mixed gently before diluting 50 μl of BALF with 50 μl of trypan blue (1:5 in 1× PBS) for viable cell counts. Then, 20 μl of diluted cell sample was pipetted on a cellometer cell counting chamber. Leukocytes were automatically counted using a Cellometer Auto T4 (Nexcelom Bioscience, Lawrence, MA, USA).
Measurement of BALF albumin content
Albumin content of BALF supernatants was measured with a mouse albumin ELISA Quantitation Set (Bethyl Laboratories, Montgomery, TX, USA) according to the manufacturer's instructions. Samples were diluted 1:10,000.
Myeloperoxidase (MPO) assay
MPO is rapidly released by activated polymorphonuclear neutrophils, monocytes and macrophages. Tissue was homogenized and lysed in ice-cold lysis buffer (200 mM NaCl, 5 mM EDTA, 10 mM Tris, 10% glycerol, 1 mM PMSF, 1 μg/ml leupeptide, and 28 μg/ml aprotinine, pH 7.4). Tissue samples were measured with a mouse MPO ELISA kit (Hycult Biotech, Plymouth Meeting, PA, USA). MPO levels were normalized to protein concentrations afterward.
Cytokine protein levels
Concentrations of TNF-α and IL-10 were measured in BALF or cell culture supernatant. All mediators were determined using commercially available ELISA kits for mouse (TNF-α DuoSet ELISA development system, R&D Systems, Minneapolis, MN, USA; IL-10 ELISA Ready-SET GO!, eBioscience, San Diego, CA, USA) according to the manufacturer's instructions.
Lung histology and lung injury scoring
To examine the amount of cellular infiltrates as well as the tissue integrity in histological slices of pulmonary tissue, mice were killed by a pentobarbital overdose, followed by exsanguination. Lungs were inflated with 0.7 ml of 10% formalin and fixed in 10% formalin solution overnight. After automated dehydration through a graded ethyl alcohol series, tissue was embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E). Two investigators blinded to group assignments analyzed the samples and determined the levels of lung injury according to semiquantitative scoring. All 25 fields of the left lung at ×20 view were examined for each sample. Assessment of histological lung injury was performed by grading as follows: infiltration or aggregation of inflammatory cells in air space or vessel wall: 1 = only wall, 2 = few cells (1–5 cells) in air space, 3 = intermediate, 4 = severe (air space congested); interstitial congestion and hyaline membrane formation: 1 = normal lung, 2 = moderate (<25% of lung section), 3 = intermediate (25–50% of lung section), 4 = severe (>50% of lung section); hemorrhage: 0 = absent, 1 = present.
Forkhead box protein 3 (FoxP3) immunohistology
For immunohistochemistry, sections were dewaxed in xylene and rehydrated in descending ethanol series. Antigen retrieval was performed in a decloaker in citrate-based antigen unmasking solution (Vector Laboratories, Burlingame, CA, USA) for 20 min at 125°C, followed by washing in TBS. Tissue was permeabilized with 0.05% Tween in TBS for 5 min and blocked in serum with 0.05% Tween for 60 min (Vectastain Elite ABC Kit; Vector Laboratories). Sections were incubated overnight at 4°C with primary antibody (Abcam 54501; Abcam, Cambridge, MA, USA; dilution 1:1000 in TBS with 1% serum). Slides were incubated in biotinylated secondary antibody with 1% serum (Vectastein Elite ABC Kit) and Vectastain Elite ABC reagent for 30 min, each. DAB was prepared according to manufacturer's instructions and applied for 45 s (DAB peroxidase substrate kit, Vector Laboratories). Counterstain was performed with methyl green for 30 s (Vector Methyl Green; Vector Laboratories). Sections were photographed with a Nikon DS-U2/L2 (Nikon Instruments, Melville, NY, USA). FoxP3+ cell counts were performed at ×20 view on 9 individual photographs per slide using NIS-Element BR (Nikon Instruments).
Flow cytometry analysis
The cell pellet from BALF was obtained after spinning the vials for 5 min at 400 g. The cell pellet, left lung, and spleen were kept in RPMI until staining. Tissue was mechanically disrupted, followed by 1 h incubation of tissue with collagenase D (Roche Diagnostics, Indianapolis, IN, USA) at a final concentration of 1 mg/ml at 37°C. The homogenate was forced through a 100-μm mesh using the plunger of a 3-ml syringe and washed. The following anti-mouse antibodies, including clone name and fluorophore, were purchased from eBioscience unless otherwise noted: CD73 (eBioTY/11.8, PE), CD45 (30-F11, APC-eFluor780), major histocompatibility complex II (MHCII; M5/114.15.2, PerCP; Biolegend, San Diego, CA, USA), Ly6C (HK1.4, PerCP-Cy5.5), Ly6G (1A8, PE-Cy7; BD Pharmingen, San Jose, CA, USA), CD4 (RM4-5, AlexaFluor700), T-cell receptor-β (TCRβ; H57-597, FITC), and CD11b (M1/70, eFluor450). Cell suspension or disrupted tissue was stained with a cocktail of antibodies containing Fc block (2.4G2) for 30 min at room temperature in the dark. For intracellular FoxP3 staining (FJK-16s, AF647; eBioscience), cells were treated with FoxP3 staining buffer according to manufacturer's instructions (FoxP3 fixation/permeabilization concentrate and diluent, and permeabilization buffer, 10×; eBioscience). Stains included a viability dye to identify viable cells (Live/Dead Fixable Aqua Dead Cell Stain Kit; Invitrogen, Life Technologies, Grand Island, NY, USA), and T cells were routinely identified as viable using this dye. Flow cytometry was performed on an LSRII (BD Biosciences, San Jose, CA, USA), with compensation done using FACSDiva software (BD Biosciences).
CD4+CD25+ T-cell purification and adoptive transfer
T cells were purified from spleen and mesenteric and popliteal lymph nodes of mice, mechanically disrupted over a 100-μm filter, and subjected to magnetic bead enrichment for CD4 T cells using a CD4+CD25+ Treg isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Enriched CD4+CD25+ T cells were then counted and resuspended in saline. A cell suspension containing 3.25 × 105 cells/50 μl was retroorbitally injected into B6.Rag1−/− mice anesthetized with pentobarbital (70 mg/kg). Intratracheal LPS application was performed 14 h post-T-cell injection. Animals were euthanized 7 d post-LPS treatment.
In vitro regulatory CD4+ T-cell differentiation assay
Naive CD4+ T cells (CD4+CD25−) were isolated from cd73+/+ or cd73−/− spleen and peripheral lymph nodes as described above and cultured in RPMI containing 10% fetal bovine serum (FBS), 1% pencillin-streptomycin, 1% l-glutamine, and β-mercaptoethanol (50 μM). Cells were treated with recombinant IL-2 (10 ng/ml; eBioscience) alone (TH0 condition) or were cultured in the presence of a Treg differentiating cytokine cocktail: rmIL-2 (10 ng/ml; eBioscience), rh-TGF-β1 (5 ng/ml; eBioscience), all-trans retinoic acid (100 μM), anti-mIFNγ (XMG1.2; 10 μg/ml, eBioscience), anti-mIL-4 (11B11; 10 μg/ml, BioXCell, West Lebanon, NH, USA), and anti-CD3/anti-CD28 microbeads (Dynabeads Mouse T-Activator CD3/CD28; Invitrogen). After 4 d, cells were harvested and incubated with fluorescently labeled antibodies against CD4 (RM4-5), CD25 (PC61.5), FoxP3 (FJK-16s), CLTA4 (UC10-4B9), and GITR (DTA-1, eBioscience). Live cells were discriminated with the use of Aqua fluorescence reactive dye (Invitrogen). Flow cytometry analysis was performed using a BD FACSCanto (BD Biosciences).
In vitro macrophage suppression assay
Cd73+/+ alveolar macrophages were harvested by flushing the airways with a total of 4 ml prewarmed PBS supplemented with 0.5 mM EDTA. BALF from 7 mice/strain was collected and centrifuged for 10 min at 400 g at 4°C. Cells were resuspended in 1 ml RPMI containing 10% FBS, 1% pencillin-streptomycin, and 1% glutamine. Alveolar macrophages and CD4+CD25+ T cells were seeded on a 96-well plate in a 2:1 ratio. Macrophages or CD4+CD25+ cells alone or in coculture were stimulated with 1 μg LPS/ml supernatant or medium as control. Supernatants were harvested after 20 h. TNF-α and IL-10 levels were determined with ELISA.
T-cell suppression assay
Measurement of Treg suppressive function was performed as described previously (36). Briefly, CD4+CD25+ Tregs were isolated by negative selection of CD4 T cells, followed by positive selection of CD25+ cells using the MACS Treg isolation kit (Miltenyi Biotec). CellTrace Violet-labeled (Invitrogen) CD4+CD25− effector T cells (50,000 cells/well) were stimulated with anti-CD3 (1 μg/ml) mAb in the presence of irradiated syngeneic CD90− antigen-presenting cells and varying ratios of purified CD4+CD25+ Tregs from cd73+/+ or cd73−/− mice. Percentage suppression of proliferation was determined by the CellTrace profile of dividing effector cells at 72 h.
Software and statistical analysis
Data analysis and plotting were done using Prism 4.0c (GraphPad Software, San Diego, CA, USA). Statistical analyses were performed with unpaired t test, 1-way ANOVA, or 2-way ANOVA and Bonferroni's multiple comparison posttest correction as indicated. All data are expressed as means ± se. Real-time PCR data were evaluated using SDS 2.3 (Applied Biosystems). Flow cytometric data were analyzed using FlowJo (TreeStar, Ashland, OR, USA).
RESULTS
Pharmacologic deprivation of adenosine by administration of PEG-ADA impairs ALI resolution
Previous studies had demonstrated a functional role of endogenously generated adenosine in attenuating acute inflammatory events (2). To address the functional role of extracellular adenosine in ALI resolution, we established a murine model of LPS-induced lung injury/resolution utilizing intratracheal LPS followed by 14 d recovery. To probe the functional role of extracellular adenosine in this ALI resolution mode, we first utilized treatment with PEG-ADA to degrade extracellular adenosine in the lung. ADA rapidly converts extracellular adenosine to inosine and thereby effectively terminates extracellular adenosine-signaling events (37). Based on previous studies examining different treatment regimens, and their effect on extracellular adenosine levels (18), we treated intraperitoneally with 10 U PEG-ADA 3 d before LPS instillation, immediately after intratracheal LPS treatment, and on d 7 post-LPS treatment (Fig. 1A). Indeed, LPS treatment was associated with a time-dependent weight loss (2-way ANOVA, P<0.0001). Consistent with our hypothesis that endogenous adenosine functions to promote ALI resolution, we observed a more dramatic weight loss and failure to recover in mice treated with PEG-ADA (2-way ANOVA, P<0.0001; Fig. 1B). Moreover, PEG-ADA-treated mice experienced prolonged and significantly increased lung inflammation during the resolution of ALI. As such, alveolar leukocyte infiltration was dramatically increased in the BALF at 3 d following LPS treatment in the PEG-ADA LPS group (P<0.01; Fig. 1C). Similarly, pulmonary MPO activity as a measure of phagocyte influx and activation was increased significantly in pulmonary tissue from PEG-ADA LPS mice (P<0.01; Fig. 1D). In addition, PEG-ADA administration was associated with prolonged and increased pulmonary barrier dysfunction. Albumin content of the BALF was considerably increased at 3 d following LPS treatment (P<0.05; Fig. 1E).
Mice with a genetic defect in extracellular adenosine generation (cd73−/− mice) fail to resolve LPS-induced ALI
After demonstrating that endogenous adenosine production contributes to ALI resolution, we next utilized genetic models to investigate the functional role of endogenous adenosine production and signaling during ALI resolution. We have previously demonstrated that extracellular adenosine production during ALI is controlled by CD73, the terminal enzyme catalyzing extracellular conversion of AMP to adenosine (6, 7, 24). For example, adenosine measurements in wild-type mice exposed to ventilator-induced lung injury showed dramatic increases in their adenosine levels. This endogenous increase of adenosine with ALI was completely blunted in cd73−/− mice (7). Therefore, we felt confident that we could utilize cd73−/− mice as a genetic model to address the functional role of extracellular adenosine production during ALI. We compared the response of cd73+/+ and cd73−/− mice to LPS-induced lung injury resolution. We found that cd73−/− mice experienced dramatic problems to resolve LPS-induced inflammation. As depicted in Fig. 2A, an LPS concentration of 1.875 μg/g body weight induced a transient weight loss in both genotypes, as confirmed by 2-way ANOVA (P<0.0001). However, cd73−/− mice exhibited higher weight loss compared with cd73+/+ mice between d 2 and 14 (2-way ANOVA, P<0.0001). Moreover, treatment with a higher LPS dose was associated with increased mortality of cd73−/− mice. The cd73−/− mice treated with a high LPS dosing regimen (3.75 μg/g body weight) demonstrated significantly aggravated weight loss (2-way ANOVA, P<0.0001; data not shown) with 40% of the mice succumbing within the observation period. None of the cd73+/+ mice died (log-rank Mantel-Cox test, P<0.05 vs. cd73+/+ mice; Fig. 2B). Similarly to the above studies in mice treated with PEG-ADA, cd73−/− mice failed to control lung inflammation and to adequately resolve ALI. At 3 d after intratracheal administration of LPS, leukocyte numbers in BALF from cd73−/− mice significantly exceeded those determined in cd73+/+ mice (P<0.01; Fig. 2C). Furthermore, lung injury scores in histological sections of cd73−/− mice compared with controls revealed significantly increased ALI at d 3 and 14, indicating a failure of cd73−/− mice to resolve ALI (Fig. 2D). This is also displayed in H&E staining from lung tissue depicting increased abundance of inflammatory cells in cd73−/−-deficient lungs. At 14 d after the LPS insult, tissue sections from cd73+/+ mice resembled those from previously untreated mice (0 d), whereas in sections from cd73−/− mice lung injury was not resolved, still demonstrating signs of injury with interstitial infiltrates and cellular infiltration into the air space. Also, genetic deletion of cd73 was associated with increased mRNA expression of proinflammatory mediators (Fig. 3) at 3 d after LPS administration, as shown for TNF-α (P<0.001), IL-1β (P<0.01), and IL-6 (P<0.001). Taken together, these genetic studies demonstrate that CD73-dependent production of extracellular adenosine plays a functional role for promoting ALI resolution.
Figure 2.

Lack of CD73 increases weight loss and enhances inflammation following intratracheal LPS instillation. A) Relative weight loss (percentage of initial individual weight) after LPS treatment (1.875 μg LPS/g body weight) in cd73+/+ and cd73−/− mice (n=6/group). P values indicate significant differences at indicated time points between genotypes, calculated by Bonferroni posttest. B) Survival curve after high-dose LPS treatment (3.75 μg/g body weight; n=8–12). C) BALF derived from cd73−/− mice 3 d after LPS application contains higher leukocyte numbers (1.875 μg LPS/g body weight; n=6–8/group). D) Lung injury scores (1.875 μg LPS/g body weight; n=4–5/group/time point). E) Representative H&E-stained tissue sections of pulmonary tissue at d 0, 3, and 14 post-LPS application, ×20 view (1.875 μg LPS/g body weight; n=4–5/group/time point).
Figure 3.

Inflammatory mediators are increased in cd73−/− mice 3 d post-LPS treatment (1.875 μg LPS/g body weight). Genetic ablation of CD73 elevates transcript levels of TNF-α (A), IL-1β (B), and IL-6 (C) 3 d post-LPS treatment in pulmonary tissue (n=5–8/group).
5′-NT reconstitution reduces pulmonary injury in cd73−/− mice
To affirm that the observed phenotype of cd73−/− mice was caused by a lack in extracellular adenosine generation from precursor nucleotides, we pursued reconstitution in these cd73−/− mice. As we have done in previous studies, we utilized soluble nucleotidase (5′-NT) derived from snake venom for this purpose (7, 24, 30–32, 38). In the line of these studies, we provided cd73−/− mice with daily injections of 5′-NT after LPS administration. 5′-NT augments adenosine levels by catalyzing the dephosphorylation of 5′-AMP to adenosine. In accordance with the previous weight curve, weight reduction in cd73−/− mice was more pronounced compared with cd73+/+ mice (2-way ANOVA, P<0.0001; Fig. 4A). Comparison of LPS cd73−/− groups revealed a significant acceleration of weight recovery in animals that received 5′-NT reconstitution compared with vehicle control (2-way ANOVA, P<0.0001). Determination of cellular infiltrates and albumin leakage into BALF on d 3 post-LPS treatment demonstrated less barrier disruption in 5′-NT-treated mice (Fig. 4B, C). Only cd73−/− vehicle mice exhibited significantly increased albumin levels compared with baseline values (P<0.05) but not cd73+/+ or cd73−/− 5′-NT groups. In line with this observation, leukocyte numbers in the BALF were highest in cd73−/− vehicle mice (P<0.05 vs. non-LPS group) whereas cell counts in 5′-NT-treated cd73−/− were not different from wild-type LPS mice and did not reach the level of significance compared with non-LPS-treated mice. Also, we observed reconstitution of a wild-type response when assessing lung injury scores (Fig. 5B). This is also displayed in representative tissue section of the different treatment groups (Fig. 5A). Taken together, these studies demonstrate that impaired resolution of ALI observed in cd73−/− mice can be rescued by treatment with soluble nucleotidase, thereby highlighting the functional role of extracellular adenosine production in promoting ALI resolution.
Figure 4.
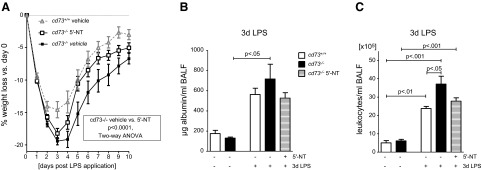
5′-NT therapy accelerates the recovery of cd73−/− mice after intratracheal LPS instillation (3.75 μg/g body weight). A) 5′-NT-treated cd73−/− mice recover faster than vehicle-treated cd73−/− mice (n=12–15/group). B) Albumin content in BALF is highest in vehicle-treated cd73−/− mice (n=5/group). C) BALF derived from vehicle-treated cd73−/− mice contains significantly more leukocytes compared with cd73+/+ mice, whereas cell counts from 5′-NT-treated cd73−/− mice show intermediate numbers (n=5/group).
Figure 5.
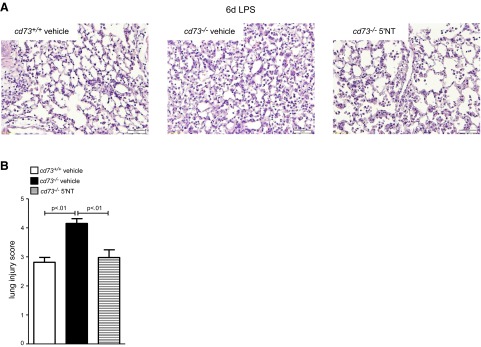
Resolution of lung injury is impaired on d 6 in cd73−/− mice. A) H&E staining displays most prominent interstitial wall thickening and cellular infiltrates in alveolae from vehicle-treated cd73−/− (n=5/group). B) Lung injury score is highest in cd73−/− vehicle group (n=5/group).
CD73high Tregs promote ALI resolution
Previous studies had shown that Tregs play a key role in ALI resolution (33), while other studies demonstrate that Treg-dependent immune functions are linked to CD73 expression and CD73-dependent adenosine production (29). To examine the functional role of CD73 in the immunological response to ALI, we determined CD73 expression on BAL cells 6 d following ALI initiation. Interestingly, we observed that Tregs possess exceptionally high CD73 levels (P<0.001; Fig. 6A). We and others detected Tregs in the alveolar compartment as early as 24–48 h after exposure to intratracheal LPS (33, 39). However, we found the maximum number of Tregs after 7 d following the LPS insult. To exclude the assumption that cd73−/− mice possess less Tregs, we performed fluorescence activated cell sorting which did not show a difference in Treg numbers between the strains (Fig. 6B). In contrast, anti-inflammatory IL-10 secretion into BALF was significantly attenuated in cd73−/− mice on d 6 (P<0.05; Fig. 6C). Next, we harvested CD4+CD25+ T cells from spleen and lymph nodes and characterized the cd73+/+ and cd73−/− Treg populations. Based on our previous observations in BALF, we confirmed that splenic CD4+ Tregs are the highest expressers of CD73 (Fig. 7A). Next, a Treg differentiation assay of cd73+/+ and cd73−/− CD4+CD25− T cells was performed, and the abundance of FoxP3+ Tregs was detected (Fig. 7B). In line with the comparable Treg frequencies measured in vivo, CD73 expression did not have any developmental influence on the amount of differentiated Tregs. Glucocorticoid-induced TNFR-related (GITR), cytotoxic T-lymphocyte antigen 4 (CTLA4), CD25, and FoxP3 are proteins characteristically expressed by Tregs. The cd73+/+ and cd73−/− Tregs showed equal expression levels of these (Fig. 7C). Due to the fact that Tregs are known to express TLR4 and directly respond to LPS (40, 41), we examined cd73+/+ and cd73−/− CD4+CD25+ cells in culture, stimulated them with LPS for 24 h, and measured TNF-α and IL-10 in the supernatant (Fig. 7D). Interestingly, cd73−/− cells secreted significantly more TNF-α (P<0.001) but less IL-10 (P<0.001) into the medium, suggesting an anti-inflammatory phenotype dependent on functional CD73. In further experiments, we tested whether CD73 expression on Tregs influences TNF secretion in coculture with cd73+/+ macrophages. Here we observed that the presence of cd73−/− Tregs significantly failed to limit the expression of TNF-α in the medium (P<0.001; Fig. 7E). Furthermore, lack of cd73 decreased the suppressive function of Tregs on the proliferation of effector T cells (Fig. 7F). Suppressor function differed significantly at 1:1 to 1:4 ratios of Tregs to cd73+/+ responding T cells. Studies were additionally performed using cd73−/− responder T cells with comparable results. Together, these studies demonstrate that while cd73−/− mice have similar numbers of Tregs, their immune-suppressive function is significantly attenuated.
Figure 6.
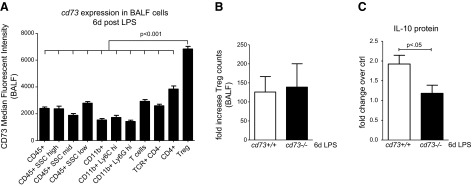
CD73 expression levels were defined in multiple cell populations by flow cytometric analysis. All cells were defined as CD45+, hematopoietically derived cells. Cells were analyzed for CD73 expression in various CD45+ populations according to their differential side-scatter profile. CD11b+ cells were identified as CD45+CD11b+ cells by sequential gating. T cells were identified as viable, CD45+TCR+ events (total T cells) that were either CD4− or CD4+, with Tregs defined as CD4+FoxP3+ events. A) Median fluorescent intensity of CD73 is highest on Tregs detected in the BALF 6 d after LPS (n=5/group). B) Treg numbers increase in both genotypes 6 d after LPS-induced lung injury (fold change of untreated naive control mice; n=5/group). C) At 6 d after LPS application, cd73+/+ but not cd73−/− mice exhibit increased IL-10 protein secretion into BALF (fold change of untreated naive control mice; n=5/group).
Figure 7.

CD73 expression has no influence on the abundance of Tregs but alters function. A) Tregs derived from spleen exhibit highest CD73 mean fluorescent intensity (histogram overlay from n=3). Analysis of CD73 protein expression levels in multiple splenocyte populations, as measured by flow cytometric analysis, with populations defined as in Fig. 6. B) Treg abundance following in vitro differentiation assay is not dependent on CD73 expression. C) In vitro Treg differentiation assay of CD4+CD25− naive T cells has no influence on CD25, FoxP3, CTLA4, and GITR expression. D) At 24 h of in vitro LPS stimulation (1 μg/ml medium) cd73+/+ Tregs secreted less TNF-α but more IL-10 into medium when compared with cd73−/− Tregs (n=4/group, representative of 2 independent experiments). E) Coculture of cd73+/+ macrophages and cd73+/+ Tregs (2:1 ratio) suppresses TNF-α secretion (n=4/group, representative of 2 independent experiments). F) cd73−/− Tregs less effectively suppress proliferation of T effector cells (n=4 samples, representative of 2 independent studies with similar results).
Adoptive transfer of cd73+/+ but not cd73−/− Tregs mediates the resolution of lung injury in B6.Rag1−/− mice
To address the functional role of Treg-dependent CD73 during ALI, we next performed adoptive transfer studies. For this purpose, CD4+CD25+ T cells separated from spleen and lymph node suspensions enriched for Tregs were retroorbitally injected into B6.Rag1−/− mice at equal cell numbers 14 h before intratracheal LPS stimulation (Fig. 8A). BALF was collected 7 d later, and leukocyte numbers were counted (Fig. 8B). Lavage from mice receiving cd73−/− Tregs contained significantly more leukocytes (P<0.05; Fig. 8B). In line with the above-mentioned in vitro studies, IL-10 protein secretion was maximal in those mice injected with cd73+/+ Tregs (P<0.05; Fig. 8C). Lung injury scoring reflected these results, and histology revealed a progressive resolution in mice receiving cd73+/+ Tregs compared with cd73−/− Tregs (P<0.05; Fig. 8D, E). To exclude any suspicion that these results were based on differing survival of Tregs, we stained for FoxP3 and counted the positive cells. Evaluation demonstrated no differences in the recovery of cells 7 d after adoptive transfer and LPS treatment (cd73+/+ vs. cd73−/− Tregs: 65.44±7.697 vs. 72.98±2.109 cells per photograph; 9 photographs/slide from n=5 mice/group; Fig. 8F). Taken together, these studies implicate a crucial role for Treg-dependent CD73 in ALI resolution.
Figure 8.

Adoptive transfer (AT) of cd73+/+ or cd73−/− Tregs to B6.Rag1−/− mice. A) cd73+/+ or cd73−/− Tregs (3.25×105 cells/mouse) isolated from spleen and lymph nodes (LN) by CD4+CD25+ selection are retroorbitally injected into B6.Rag1−/− mice 14 h before LPS instillation (3.75 μg/g body weight). B) At 7 d after LPS insult, BALF from mice receiving cd73−/− Tregs contained higher leukocyte numbers (n=7–8/group). C) IL-10 secretion into BALF increased in the cd73+/+ Treg group (n=7–8/group). D) Lung injury score demonstrates accelerated lung injury resolution after AT of cd73+/+ Tregs (n=5/group). E) Representative H&E tissue sections, ×20 view (n=5/group). F) FoxP3 immunohistological staining showed no differences in FoxP3+ cell numbers between slides from cd73+/+ and cd73−/− mice (n=5 slides/group). Representative slide for FoxP3 staining and rabbit IgG control only.
DISCUSSION
During the course of an acute inflammatory event, endogenous signaling pathways determine the outcome and progression of the disease process. Activation of these pathways can result in the development of chronicity, fibrosis, and organ dysfunction. However, more commonly, an inflammatory insult will resolve toward its resolution, promote healing, and establish normal organ function (2, 42). Here, we hypothesized that the endogenous generation of adenosine is central to inflammatory resolution of lung inflammation following ALI. In support of this hypothesis, our studies demonstrated that recovery from LPS-induced lung injury was significantly impaired in mice treated with PEG-ADA at levels well above those that have been shown to lower adenosine concentrations in the lung following injury (43–45). The association between pharmacologic inactivation of extracellular adenosine and diminished recovery from ALI provides the first evidence for a functional role of extracellular adenosine in ALI resolution. As a second step, we performed studies in cd73−/− gene-targeted mice, which are characterized by their inability to generate extracellular adenosine from precursor nucleotides. These studies provide the first genetic evidence that cd73 deficiency is associated with a profound failure to adequately resolve ALI. Expressional studies of CD73 on inflammatory cells trafficking to the lung during ALI resolution revealed high levels of CD73 expression on Tregs. Indeed, a combination of in vitro studies examining cd73-deficient Tregs and in vivo studies of adoptive transfer of wild-type or cd73−/− Tregs revealed a functional role of Treg-dependent CD73 in ALI resolution.
The present studies implicating CD73-dependent adenosine production in ALI are consistent with previous studies showing a functional role of CD73 during the acute phase of ALI. For example, we previously demonstrated that cd73 deficiency is associated with an enhanced acute inflammatory response during bleomycin-induced lung injury (46). Similarly, studies of ventilator-induced lung injury or lung injury induced by LPS inhalation indicated a more profound early inflammatory phase in cd73−/− mice compared with control animals (2, 7). However, during chronic disease states, extracellular adenosine signaling can become detrimental (47–50). For example, mice with genetic deletion of ADA and concomitant elevations of extracellular adenosine suffer from a chronic form of lung disease (43). Extensions of these studies revealed that while extracellular adenosine signaling is lung protective during the early onset of inflammation, adenosine signaling can become detrimental once chronicity has developed (49). However, the present studies suggest that during inflammatory lung disease, as opposed to established pulmonary fibrosis or chronic obstructive lung disease, adenosine signaling can prevent the development of disease chronicity by enhancing Treg functions and Treg-dependent ALI resolution.
Previous studies have implicated extracellular adenosine signaling in modulating immune functions of inflammatory cells from the innate or the adoptive immune system. For example, the first pharmacologic studies to implicate adenosine signaling in attenuating inflammatory cell activation come from the laboratory of Cronstein (51). A landmark study from Ohta and Sitkovsky (21) provided genetic evidence that an individual adenosine receptor, the A2aR, plays a critical part of the physiological negative feedback mechanism for limitation and termination of both tissue-specific and systemic inflammatory responses. The A2aR-mediated lung tissue protection was further approved by Thiel et al. (52) who demonstrated that deleterious side effects of high oxygen treatment in acute respiratory distress syndrome were overcome by selective A2aR agonist application. Other studies implicate the adenosine signaling in attenuating TNF release from neutrophils during inflammatory disease (53) or on dendritic cells during ischemia and reperfusion injury (54). Also, adenosine generation and signaling have been implicated in T-cell functions, for example, by inhibiting inflammatory activation of CD4+-T cells (55, 56). A recent study elucidated that CD4+CD73+ T cells may contribute to HIV-1 pathogenesis since this T-cell subset was depleted in HIV-1-infected patients, correlating inversely with T-cell activation (57). Furthermore, adenosine enhances the immunosuppressive functions of Tregs (29, 39). These later findings are consistent with the results from our present studies implicating adenosine generation on Tregs in promoting ALI resolution. Indeed, ALI resolution has been closely linked to functional Tregs (33).
Previous studies have demonstrated a transcriptionally regulated pathway for CD73 that is under the control of the transcription factor hypoxia-inducible factor (HIF). Inflammatory diseases of the mucosa, such as inflammatory bowel disease or during ALI, are characterized by an alteration of the balance between oxygen demand and supply, thereby resulting in profound tissue hypoxia (1, 12). Indeed, the interdependent relationship between hypoxia and inflammation during inflammatory diseases of the mucosa results in the activation of hypoxia-dependent transcriptional pathways, including HIF (51–55). As such, previous studies utilizing ambient hypoxia exposure of epithelial cells demonstrated robust induction of CD73 transcript, protein and function with hypoxia. Moreover, studies with CD73 promoter constructs, HIF loss of function, and transcription factor binding assays demonstrated that HIF controls the hypoxic induction of CD73 (27). In addition, cd73−/− mice are more prone to hypoxia-associated vascular dysfunction (24) and hypoxia-driven inflammation (25). Hypoxia within inflamed areas of the lungs could represent a stimulus for CD73 induction and enhanced adenosine production and previous studies from the laboratory of Sitkovsky (52, 58) have implicated hypoxia in enhancing adenosine responses during ALI, and concomitant protection from bacterial ALI. Furthermore, recent studies indicate that hypoxia can directly impact Treg differentiation via HIF-dependent induction of FoxP3 and concomitant Treg expansion (59).
Taken together, the present studies define a fundamental role for CD73-dependent adenosine production in ALI resolution. These studies implicate Treg expression of CD73 as a central contributor to endogenous lung protection. Ultimately, these observations provide a number of therapeutic opportunities. Future challenges will include hurdles to translating these experimental findings from the laboratory into novel therapeutic treatment modalities, for example, by defining clinical strategies to enhance Treg functions during ALI or to target adenosine signaling to promote ALI resolution in patients. Prospective studies may rule out time-dependent activation and contribution of A2aR and A2bR to adenosine-mediated lung protection and resolution of injury, thereby providing further therapeutic applications through selective agonist treatment.
Acknowledgments
The authors acknowledge Kristann Magee and Sheema Rabbaig for technical assistance. The cd73−/− mice were kindly provided by Linda Thompson (OK Medical Research Foundation, Okalahoma City, OK, USA). In addition, the authors acknowledge Yang Xia and Michael R. Blackburn (University of Texas Medical School, Houston, TX, USA) and Edwin F de Zoeten and Colm B Collins (University of Colorado School of Medicine, Aurora, CO, USA) for help with addressing questions that came up in the review process of this study.
The present research work was supported by U.S. National Institutes of Health grants R01-HL-092188, R01-DK-083385, and R01-HL-098294 (to H.K.E.) and HL-560569 and DK-095491 (to S.P.C.); Crohn's and Colitis Foundation of America (CCFA) grants (to H.K.E.); German Academic Exchange Service [Deutscher Akademischer Austausch Dienst (DAAD)] grant D/10/52531 (to J.M.P.); and German Research Foundation [Deutsche Forschungsgemeinschaft (DFG)] grants EH401/1-1 (to H.E.) and ES 371/1-1 (to S.F.E.).
Footnotes
- 5′-NT
- 5′-nucleotidase
- A2aR
- adenosine receptor 2a
- A2bR
- adenosine receptor 2b
- ADA
- adenosine deaminase
- ALI
- acute lung injury
- BALF
- bronchoalveolar lavage fluid
- CD73
- 5′-ectonucleotidase
- CTLA4
- cytotoxic T-lymphocyte antigen 4
- FBS
- fetal bovine serum
- FoxP3
- forkhead box protein 3
- GITR
- glucocorticoid-induced TNFR related
- H&E
- hematoxylin and eosin
- HIF
- hypoxia inducible factor
- LPS
- lipopolysaccharide
- MPO
- myeloperoxidase
- PEG-ADA
- pegylated adenosine deaminase
- TCRβ
- T-cell receptor-β
- Treg
- regulatory T cell
REFERENCES
- 1. Eltzschig H. K., Carmeliet P. (2011) Hypoxia and inflammation. N. Engl. J. Med. 364, 656–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eltzschig H. K., Sitkovsky M. V., Robson S. C. (2012) Purinergic signaling during inflammation. N. Engl. J. Med. 367, 2322–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rubenfeld G. D., Caldwell E., Peabody E., Weaver J., Martin D. P., Neff M., Stern E. J., Hudson L. D. (2005) Incidence and outcomes of acute lung injury. N. Engl. J. Med. 353, 1685–1693 [DOI] [PubMed] [Google Scholar]
- 4. Eckle T., Koeppen M., Eltzschig H. K. (2009) Role of extracellular adenosine in acute lung injury. Physiology (Bethesda) 24, 298–306 [DOI] [PubMed] [Google Scholar]
- 5. Schingnitz U., Hartmann K., Macmanus C. F., Eckle T., Zug S., Colgan S. P., Eltzschig H. K. (2010) Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J. Immunol. 184, 5271–5279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reutershan J., Vollmer I., Stark S., Wagner R., Ngamsri K. C., Eltzschig H. K. (2009) Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. FASEB J. 23, 473–482 [DOI] [PubMed] [Google Scholar]
- 7. Eckle T., Fullbier L., Wehrmann M., Khoury J., Mittelbronn M., Ibla J., Rosenberger P., Eltzschig H. K. (2007) Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J. Immunol. 178, 8127–8137 [DOI] [PubMed] [Google Scholar]
- 8. Eckle T., Grenz A., Laucher S., Eltzschig H. K. (2008) A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J. Clin. Invest. 118, 3301–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aherne C. M., Collins C. B., Masterson J. C., Tizzano M., Boyle T. A., Westrich J. A., Parnes J. A., Furuta G. T., Rivera-Nieves J., Eltzschig H. K. (2012) Neuronal guidance molecule netrin-1 attenuates inflammatory cell trafficking during acute experimental colitis. Gut 61, 695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hart M. L., Grenz A., Gorzolla I. C., Schittenhelm J., Dalton J. H., Eltzschig H. K. (2011) Hypoxia-inducible factor-1alpha-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J. Immunol. 186, 4367–4374 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11. Frick J. S., Macmanus C. F., Scully M., Glover L. E., Eltzschig H. K., Colgan S. P. (2009) Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J. Immunol. 182, 4957–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Colgan S. P., Eltzschig H. K. (2012) Adenosine and hypoxia-inducible factor signaling in intestinal injury and recovery. Annu. Rev. Physiol. 74, 153–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rosenberger P., Schwab J. M., Mirakaj V., Masekowsky E., Mager A., Morote-Garcia J. C., Unertl K., Eltzschig H. K. (2009) Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat. Immunol. 10, 195–202 [DOI] [PubMed] [Google Scholar]
- 14. Morote-Garcia J. C., Rosenberger P., Nivillac N. M., Coe I. R., Eltzschig H. K. (2009) Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology 136, 607–618 [DOI] [PubMed] [Google Scholar]
- 15. Morote-Garcia J. C., Rosenberger P., Kuhlicke J., Eltzschig H. K. (2008) HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood 111, 5571–5580 [DOI] [PubMed] [Google Scholar]
- 16. Hart M. L., Gorzolla I. C., Schittenhelm J., Robson S. C., Eltzschig H. K. (2010) SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J. Immunol. 184, 4017–4024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eltzschig H. K., Kohler D., Eckle T., Kong T., Robson S. C., Colgan S. P. (2009) Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood 113, 224–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grenz A., Bauerle J. D., Dalton J. H., Ridyard D., Badulak A., Tak E., McNamee E. N., Clambey E., Moldovan R., Reyes G., Klawitter J., Ambler K., Magee K., Christians U., Brodsky K. S., Ravid K., Choi D. S., Wen J., Lukashev D., Blackburn M. R., Osswald H., Coe I. R., Nurnberg B., Haase V. H., Xia Y., Sitkovsky M., Eltzschig H. K. (2012) Equilibrative nucleoside transporter 1 (ENT1) regulates postischemic blood flow during acute kidney injury in mice. J. Clin. Invest. 122, 693–710 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19. Eckle T., Hartmann K., Bonney S., Reithel S., Mittelbronn M., Walker L. A., Lowes B. D., Han J., Borchers C. H., Buttrick P. M., Kominsky D. J., Colgan S. P., Eltzschig H. K. (2012) Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat. Med. 18, 774–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eltzschig H. K., Eckle T. (2011) Ischemia and reperfusion–from mechanism to translation. Nat. Med. 17, 1391–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ohta A., Sitkovsky M. (2001) Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414, 916–920 [DOI] [PubMed] [Google Scholar]
- 22. Yang D., Zhang Y., Nguyen H. G., Koupenova M., Chauhan A. K., Makitalo M., Jones M. R., St H. C., Seldin D. C., Toselli P., Lamperti E., Schreiber B. M., Gavras H., Wagner D. D., Ravid K. (2006) The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J. Clin. Invest. 116, 1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eltzschig H. K. (2009) Adenosine: an old drug newly discovered. Anesthesiology 111, 904–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thompson L. F., Eltzschig H. K., Ibla J. C., Van De Wiele C. J., Resta R., Morote-Garcia J. C., Colgan S. P. (2004) Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J. Exp. Med. 200, 1395–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eltzschig H. K., Thompson L. F., Karhausen J., Cotta R. J., Ibla J. C., Robson S. C., Colgan S. P. (2004) Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood 104, 3986–3992 [DOI] [PubMed] [Google Scholar]
- 26. Eltzschig H. K., Ibla J. C., Furuta G. T., Leonard M. O., Jacobson K. A., Enjyoji K., Robson S. C., Colgan S. P. (2003) Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J. Exp. Med. 198, 783–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Synnestvedt K., Furuta G. T., Comerford K. M., Louis N., Karhausen J., Eltzschig H. K., Hansen K. R., Thompson L. F., Colgan S. P. (2002) Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Invest. 110, 993–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kobie J. J., Shah P. R., Yang L., Rebhahn J. A., Fowell D. J., Mosmann T. R. (2006) T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′-adenosine monophosphate to adenosine. J. Immunol. 177, 6780–6786 [DOI] [PubMed] [Google Scholar]
- 29. Deaglio S., Dwyer K. M., Gao W., Friedman D., Usheva A., Erat A., Chen J. F., Enjyoji K., Linden J., Oukka M., Kuchroo V. K., Strom T. B., Robson S. C. (2007) Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hart M. L., Much C., Gorzolla I. C., Schittenhelm J., Kloor D., Stahl G. L., Eltzschig H. K. (2008) Extracellular adenosine production by ecto-5′-nucleotidase protects during murine hepatic ischemic preconditioning. Gastroenterology 135, 1739–1750 [DOI] [PubMed] [Google Scholar]
- 31. Eckle T., Krahn T., Grenz A., Kohler D., Mittelbronn M., Ledent C., Jacobson M. A., Osswald H., Thompson L. F., Unertl K., Eltzschig H. K. (2007) Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation 115, 1581–1590 [DOI] [PubMed] [Google Scholar]
- 32. Grenz A., Zhang H., Eckle T., Mittelbronn M., Wehrmann M., Kohle C., Kloor D., Thompson L. F., Osswald H., Eltzschig H. K. (2007) Protective role of ecto-5′-nucleotidase (CD73) in renal ischemia. J. Am. Soc. Nephrol. 18, 833–845 [DOI] [PubMed] [Google Scholar]
- 33. D'Alessio F. R., Tsushima K., Aggarwal N. R., West E. E., Willett M. H., Britos M. F., Pipeling M. R., Brower R. G., Tuder R. M., McDyer J. F., King L. S. (2009) CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J. Clin. Invest. 119, 2898–2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Veronese F. M., Pasut G. (2005) PEGylation, successful approach to drug delivery. Drug Discov. Today 10, 1451–1458 [DOI] [PubMed] [Google Scholar]
- 35. Blackburn M. R., Aldrich M., Volmer J. B., Chen W., Zhong H., Kelly S., Hershfield M. S., Datta S. K., Kellems R. E. (2000) The use of enzyme therapy to regulate the metabolic and phenotypic consequences of adenosine deaminase deficiency in mice. Differential impact on pulmonary and immunologic abnormalities. J. Biol. Chem. 275, 32114–32121 [DOI] [PubMed] [Google Scholar]
- 36. Collins C. B., Aherne C. M., Yeckes A., Pound K., Eltzschig H. K., Jedlicka P., de Zoeten E. F. (2013) Inhibition of N-terminal ATPase on HSP90 attenuates colitis through enhanced Treg function. [E-pub ahead of print] Mucosal. Immunol. doi: 10.1038/mi.2012.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eltzschig H. K., Faigle M., Knapp S., Karhausen J., Ibla J., Rosenberger P., Odegard K. C., Laussen P. C., Thompson L. F., Colgan S. P. (2006) Endothelial catabolism of extracellular adenosine during hypoxia: the role of surface adenosine deaminase and CD26. Blood 108, 1602–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hart M. L., Henn M., Kohler D., Kloor D., Mittelbronn M., Gorzolla I. C., Stahl G. L., Eltzschig H. K. (2008) Role of extracellular nucleotide phosphohydrolysis in intestinal ischemia-reperfusion injury. FASEB J. 22, 2784–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ehrentraut H., Westrich J. A., Eltzschig H. K., Clambey E. T. (2012) Adora2b adenosine receptor engagement enhances regulatory T cell abundance during endotoxin-induced pulmonary inflammation. PLoS ONE 7, e32416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sutmuller R. P., Morgan M. E., Netea M. G., Grauer O., Adema G. J. (2006) Toll-like receptors on regulatory T cells: expanding immune regulation. Trends Immunol. 27, 387–393 [DOI] [PubMed] [Google Scholar]
- 41. Caramalho I., Lopes-Carvalho T., Ostler D., Zelenay S., Haury M., Demengeot J. (2003) Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J. Exp. Med. 197, 403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Serhan C. N., Savill J. (2005) Resolution of inflammation: the beginning programs the end. Nat. Immunol. 6, 1191–1197 [DOI] [PubMed] [Google Scholar]
- 43. Blackburn M. R., Volmer J. B., Thrasher J. L., Zhong H., Crosby J. R., Lee J. J., Kellems R. E. (2000) Metabolic consequences of adenosine deaminase deficiency in mice are associated with defects in alveogenesis, pulmonary inflammation, and airway obstruction. J. Exp. Med. 192, 159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blackburn M. R., Lee C. G., Young H. W., Zhu Z., Chunn J. L., Kang M. J., Banerjee S. K., Elias J. A. (2003) Adenosine mediates IL-13-induced inflammation and remodeling in the lung and interacts in an IL-13-adenosine amplification pathway. J. Clin. Invest. 112, 332–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chunn J. L., Molina J. G., Mi T., Xia Y., Kellems R. E., Blackburn M. R. (2005) Adenosine-dependent pulmonary fibrosis in adenosine deaminase-deficient mice. J. Immunol. 175, 1937–1946 [DOI] [PubMed] [Google Scholar]
- 46. Volmer J. B., Thompson L. F., Blackburn M. R. (2006) Ecto-5′-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J. Immunol. 176, 4449–4458 [DOI] [PubMed] [Google Scholar]
- 47. Zhang Y., Dai Y., Wen J., Zhang W., Grenz A., Sun H., Tao L., Lu G., Alexander D. C., Milburn M. V., Carter-Dawson L., Lewis D. E., Zhang W., Eltzschig H. K., Kellems R. E., Blackburn M. R., Juneja H. S., Xia Y. (2011) Detrimental effects of adenosine signaling in sickle cell disease. Nat. Med. 17, 79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Karmouty-Quintana H., Zhong H., Acero L., Weng T., Melicoff E., West J. D., Hemnes A., Grenz A., Eltzschig H. K., Blackwell T. S., Xia Y., Johnston R. A., Zeng D., Belardinelli L., Blackburn M. R. (2012) The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J. 26, 2546–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou Y., Schneider D. J., Morschl E., Song L., Pedroza M., Karmouty-Quintana H., Le T., Sun C. X., Blackburn M. R. (2011) Distinct roles for the A2B adenosine receptor in acute and chronic stages of bleomycin-induced lung injury. J. Immunol. 186, 1097–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peng Z., Borea P. A., Varani K., Wilder T., Yee H., Chiriboga L., Blackburn M. R., Azzena G., Resta G., Cronstein B. N. (2009) Adenosine signaling contributes to ethanol-induced fatty liver in mice. J. Clin. Invest. 119, 582–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cronstein B. N., Daguma L., Nichols D., Hutchison A. J., Williams M. (1990) The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J. Clin. Invest. 85, 1150–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thiel M., Chouker A., Ohta A., Jackson E., Caldwell C., Smith P., Lukashev D., Bittmann I., Sitkovsky M. V. (2005) Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 3, e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Grenz A., Kim J. H., Bauerle J. D., Tak E., Eltzschig H. K., Clambey E. T. (2012) Adora2b adenosine receptor signaling protects during acute kidney injury via inhibition of neutrophil-dependent TNF-alpha release. J. Immunol. 189, 4566–4573 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54. Li L., Huang L., Ye H., Song S. P., Bajwa A., Lee S. J., Moser E. K., Jaworska K., Kinsey G. R., Day Y. J., Linden J., Lobo P. I., Rosin D. L., Okusa M. D. (2012) Dendritic cells tolerized with adenosine A(2)AR agonist attenuate acute kidney injury. J. Clin. Invest. 122, 3931–3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Day Y. J., Huang L., Ye H., Li L., Linden J., Okusa M. D. (2006) Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: the role of CD4+ T cells and IFN-gamma. J. Immunol. 176, 3108–3114 [DOI] [PubMed] [Google Scholar]
- 56. Day Y. J., Huang L., McDuffie M. J., Rosin D. L., Ye H., Chen J. F., Schwarzschild M. A., Fink J. S., Linden J., Okusa M. D. (2003) Renal protection from ischemia mediated by A2A adenosine receptors on bone marrow-derived cells. J. Clin. Invest. 112, 883–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Macatangay B. J., Schuler P. J., Buchanan W. G., Riddler S. A., Jackson E. K., Whiteside T. L., Rinaldo C. R. (2012) CD4+CD73+ T cells regulate T cell immune activation and plasma CRP levels and are depleted in HIV-1 infection regardless of viral suppression. 19th International AIDS Conference Abstract No. MOPE009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sitkovsky M. V., Lukashev D., Apasov S., Kojima H., Koshiba M., Caldwell C., Ohta A., Thiel M. (2004) Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu. Rev. Immunol. 22, 657–682 [DOI] [PubMed] [Google Scholar]
- 59. Clambey E. T., McNamee E. N., Westrich J. A., Glover L. E., Campbell E. L., Jedlicka P., de Zoeten E. F., Cambier J. C., Stenmark K. R., Colgan S. P., Eltzschig H. K. (2012) Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. U. S. A. 109, E2784–E2793 [DOI] [PMC free article] [PubMed] [Google Scholar]