

Abstract
Activation of the adenosine 2A receptor (A2AR) reduces inflammation in models of acute injury but contribution in development of chronic abdominal aortic aneurysms (AAAs) is unknown. Elastase perfusion to induce AAA formation in A2AR-knockout (A2ARKO) and C57BL6/J wild-type (WT) mice resulted in nearly 100% larger aneurysms in A2ARKO compared to WT at d 14 (P<0.05), with evidence of greater elastin fragmentation, more immune cell infiltration, and increased matrix metallatoproteinase (MMP) 9 expression (P<0.05). Separately, exogenous A2AR antagonism in elastase-perfused WT mice also resulted in larger aneurysms (P<0.05), while A2AR agonism limited aortic dilatation (P<0.05). Activated Thy-1.2+ T lymphocytes from WT mice treated in vitro with A2AR antagonist increased cytokine production, and treatment with A2AR agonist decreased cytokine production (P<0.05 for all). Primary activated CD4+ T lymphocytes from A2ARKO mice exhibited greater chemotaxis (P<0.05). A2AR antagonist increased chemotaxis of activated CD4+ cells from WT mice in vitro, and A2AR agonist reduced this effect (P<0.05). A2AR activation attenuates AAA formation partly by inhibiting immune cell recruitment and reducing elastin fragmentation. These findings support augmenting A2AR signaling as a putative target for limiting aneurysm formation.—Bhamidipati, C. M., Mehta, G. S., Moehle, C. W., Meher, A. K., Su, G., Vigneshwar, N. G., Barbery, C., Sharma, A. K., Kron, I. L., Laubach, V. E., Owens, G. K., Upchurch Jr., G. R., Ailawadi, G. Adenosine 2A receptor modulates inflammation and phenotype in experimental abdominal aortic aneurysms.
Keywords: immune modulation, CD4+ T lymphocytes, GPCR signaling, physiological regulation, inflammation
Abdominal aortic aneurysms (AAAs) are the 10th leading cause of death among men over age 65 and result in >15,000 surgical procedures annually in the United States (1). Despite advances in our knowledge of the mechanisms that regulate aneurysm formation (2, 3), the understanding of how aneurysms occur still remains unclear. It is known that aneurysm formation is accompanied by destruction of the medial layer, fewer smooth muscle cells (SMCs) due to apoptosis, mural thrombus deposition, and degradation of collagen and elastin, all processes that are modulated by inflammatory and immune cells (3–6). However, the precise mechanisms whereby immune cells influence aneurysm formation are complex and remain poorly understood (7). The high prevalence of circulating CD4+/CD28− T lymphocytes in patients with AAA suggests an important role of adaptive immune cells in AAA pathophysiology (8). Monocyte chemotactic protein-1 (MCP-1; also known as CCL2) stimulates adaptive immune cellular responses (9), including enhanced recruitment of CD4+ T lymphocytes (10). These CD4+ T lymphocytes contribute toward aneurysmal dilatation by promoting the release of IFN-γ (11).
The endogenous purine nucleoside adenosine is, in part, involved in numerous extracellular signaling circuits through its cognate receptor, adenosine A2A receptor (A2AR; ref. 12). The heterotrimeric stimulatory G protein α (Gsα) A2AR increases intracellular 3′-5′-cyclic adenosine monophosphate (cAMP), which, in part, inhibits proinflammatory transcriptional regulation by PI3K and Akt (12, 13). Activation of A2AR induces various cardiovascular effects by up-regulation of anti-inflammatory programs in immune cells in several models (12, 14, 15). These anti-inflammatory effects resulting from the activation of A2AR have been demonstrated to be, in part, consequent from bone marrow-derived (BMD) cells (i.e., neutrophils; refs. 16–18). While A2AR biology has been examined mostly in the context of ischemia-reperfusion injuries, its effects in diseases, such as aortic aneurysms, remain unclear. Further, A2AR-mediated adaptive cellular response in modulating inflammation, influencing extracellular matrix degradation by MMPs, and promoting SMC phenotypic switching in the context of aneurysm formation are also unknown.
Taken together, we hypothesized that anti-inflammatory signals from activated A2AR mediate cascades in T lymphocytes, playing an important role in regulating aneurysm formation, and sought to determine whether perturbations in A2AR expression or activity would result in altered AAA development.
MATERIALS AND METHODS
Murine elastase model
The elastase perfusion model of AAA formation in mice was utilized as described previously (19). Congenic age-, weight- and gender-matched mice were used for experiments. Briefly, 8- to 12-wk-old male C57BL/6J wild-type (WT) mice (stock no. 000664; Jackson Laboratories, Bar Harbor, ME, USA) or A2AR-knockout (A2ARKO) mice (development of A2ARKO strain described previously, ref. 20; provided by Dr. Victor E. Laubach, University of Virginia, backcrossed onto WT strain through 10 generations) that weighed between 20 and 28 g were randomly assigned to either experimental or control groups and underwent perfusion. There was an immediate mechanically induced aortic wall dilatation of 15–35% after perfusion with saline or elastase (0.47 U/ml porcine pancreatic elastase), which was then followed by either no aneurysmal dilatation (perfused with saline) or aneurysmal dilatation (perfused with elastase) over 14 d. Aneurysms are regarded as being 80–120% above baseline. Mice were housed individually throughout experiments, and maintained at 70°F, 50% humidity, with 12-h light-dark cycles per institutional animal protocols. All mice had ad libitum access to water and a standard maintenance diet (TD 8604; Teklad Diets, Madison, WI, USA) with no restrictions on movement. Video micrometry measurements of the aortic wall diameter were performed in situ before perfusion, following perfusion, and at the time of harvest using a Q-Color3 Optical Camera (Olympus Corp., Center Valley, PA, USA) attached to an operating microscope (Leica Microsystems, Bannockburn, IL, USA) using QCapture Pro Software version 6.0 (QImaging, Surrey, BC, Canada). At harvest following antegrade perfusion as described, the entire infrarenal aorta was explanted. The abdominal aortas (or aneurysms, when present) were then either snap-frozen in liquid nitrogen for analyses by real-time quantitative polymerase chain reaction (qPCR) or protein extraction, or processed for histology or immunohistochemistry. Animal care and use were in accordance with Institute for Laboratory Animal Research guidelines (21). The animal protocol was approved by the University of Virginia Institutional Animal Care and Use Committee (no. 3634) in compliance with the Office of Laboratory Animal Welfare guidelines (22).
Real-time PCR
Ribonucleic acid (RNA) from snap-frozen mouse aortic samples was isolated with TRIzol (Invitrogen, Carlsbad, CA, USA) after homogenization, according to the manufacturer's protocols. Total RNA quantification was determined using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Subsequently, 0.5 μg complimentary deoxyribonucleic acid (cDNA) was synthesized from RNA using RNase H+ iScript reverse transcriptase (Bio-Rad Laboratories, Hercules, CA, USA). Then cDNA was amplified by qPCR on a MyiQ color real-time PCR detection system (Bio-Rad Laboratories). The A2AR (NP_033760.2) and 18S (NR_003278.2) primer sequences provided by the National Center for Biotechnology Information (NCBI; Bethesda, MD, USA) were obtained by the Oligonucleotide Synthesis Service (Fisher Scientific, Pittsburgh, PA, USA). The MMP2 primer sequences were S: 5′-CATCGCTGCACCATCGCCCATCATC-′3 and A: 5′-CCCAGGGTCCACAGCTCATCATCATCAA-′3; MMP9 primer sequences were S: 5′-TGAATCAGCTGGCTTTTGTG-′3 and A: 5′-GTGGATAGCTCGGTGGTGTT-′3; and VCAM-1 primer sequences were S: 5′-AAGCAGAAGTGGAATTAGTTG-′3 and A: 5′-GGAGTCACAGCCAATAGC-′3. Amplicons were compared to the amplification of standards and then underwent relative quantification with normalization to 18S using iQ5 2.1 optical system software (Bio-Rad Laboratories).
Histology and immunohistochemistry
Aortas were harvested at euthanasia for histology analysis after undergoing left ventricular puncture and 4% paraformaldehyde (PFA) followed by PBS antegrade perfusion at physiological pressure. Further fixation was achieved by overnight incubation in 4% PFA at 4°C, followed by paraffin embedding and sectioning at 5 μm. After microwave antigen retrieval, antibodies were bound and detected using the VectaStain Elite Kit (Vector Laboratories, Burlingame, CA, USA). Hematoxylin and eosin (H&E) along with modified Russell-Movat pentachrome (Movat) for elastin layers, and immunohistochemical (IHC) staining were performed. Antibodies for IHC staining were anti-rat Mac2 for macrophages (Cedarlane Laboratories, Burlington, ON, Canada), anti-mouse anti-neutrophil (Ly 6B.2) for neutrophils (AbD Serotec, Oxford, UK), anti-mouse CD3ε (M-20) for T lymphocytes (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-goat MMP9 for matrix metalloproteinase (MMP) 9 (R&D Systems, Minneapolis, MN, USA), and anti-mouse SMαA (14A) for smooth muscle α-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Visualization color development was completed using diaminobenzidine (Dako Corp., Carpinteria, CA, USA) for Mac2, anti-neutrophil, CD3ε, and MMP9, and Cy3 (Sigma-Aldrich, St. Louis, MO, USA) for SMαA. Appropriate controls verified staining procedures. Images were acquired using AxioCam 4.6 software via ×10 and ×40 objectives and an AxioCam MRc camera (Carl Zeiss, Thornwood, NY, USA).
Immunohistochemistry quantification
The H&E images were used to calculate the circumference of the internal elastic lamina (IEL) by computing the traced IEL distance based on pixel scale. A circumferential area of interest (AOI) from IHC images was drawn to include the medial and adventitial layers of the aorta. Threshold-gated positive signal was detected within the AOI and quantified using Image-Pro Plus 7.0 (Media Cybernetics, Bethesda, MD, USA).
A2AR agonist and antagonist administration
CGS21680 (Sigma-Aldrich) is a commercially available A2AR agonist that has been shown to be efficacious in injury models (23, 24). The Ki values of CGS21680 at A1, A2A, A2B, and A3 receptors are >350 nM, 15 nM, >100 μM, and >1 mM, respectively (25). CGS21680 or vehicle was administered subcutaneously to mice (n=9/group) via Alzet 1002 osmotic minipumps (Durect Corp., Cupertino, CA, USA) 24 h prior to elastase perfusion (elution rate: 0.25 μl/h). Preliminary dose-response experiments with varying concentrations of CGS21680 solubilized in differing amounts of vehicle, administered via osmotic minipumps, in elastase-perfused WT mice were completed (data not shown). Mice were examined at d 14 by video micrometry for aneurysm phenotype. Separately, ZM241385 (Sigma-Aldrich), a known A2AR antagonist, was administered to mice in a similar fashion as CGS21680. All pumps were tunneled subcutaneously at the nape of the neck and positioned posteriorly on the backs of recipient mice, permitting uniform systemic administration by the time of perfusion.
Cell culture
All cell culture reagents were utilized as recommended by the manufacturer (Sigma-Aldrich). Mouse aortic SMCs (ASMCs) were isolated from 20–25-g male abdominal aortas of WT mice (n=8), plated on dishes coated with fibronectin, and cultured in DMEM containing nutrient mixture F-12 (DMEM/F12; Invitrogen), 100 U/ml penicillin/streptomycin, and 292 μg/ml glutamine (26). Cells were dispersed by treatment with trypsin (Invitrogen), and incubated in 5% CO2 at 37°C to confluence until passages 4 through 8. Postconfluent cultures were visually verified to have a hill and valley topography characteristic of ASMCs grown in vitro. Plated ASMCs (1.0×105 cells/well) were starved for 24 h in serum-free medium with DMEM/F12, 1.6 mM l-glutamine, 0.2 mM l-ascorbic acid, 5 μg/ml transferrin, 2.8 μg/ml insulin, 100 U/ml penicillin/streptomycin, and 6.25 ng/ml Na-selenate.
In vitro chemotaxis assays
In separate experiments, spleens were isolated from WT and A2ARKO mice and placed in RPMI 1640 with 10% FBS. Cells were released by blunt dissection using a sterile 3-ml syringe plunger and strained through a 40-μm nylon mesh (BD Biosciences, Bedford, MA, USA). Red blood cells were excluded by hypertonic lysis buffer (Sigma-Aldrich) for 5 min at room temperature and washed with RPMI 1640 and 10% FBS. To facilitate Thy-1.2 and CD4+ T-lymphocyte separation, commercially available magnetic bead-based cell isolation kits (Miltenyi Biotec, Auburn, CA, USA) were used serially to optimize enrichment. We achieved a relative enrichment of ≥90% of T lymphocytes determined by flow cytometry. The enriched cells were centrifuged at 1000 rpm for 10 min; resuspended in fluorescence-activated cell sorting (FACS) buffer (eBioscience, San Diego, CA, USA); and stained with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; eBioscience) to differentiate the live from dead cells, allophycocyanin Cy7 (APC Cy7)-conjugated CD45 (BioLegend, San Diego, CA, USA) and PerCP Cy5.5-conjugated CD3 (BioLegend) to identify CD3+ T lymphocytes, and APC-conjugated anti-TCRβ (eBioscience) and/or APC-conjugated anti-GK1.5 (eBioscience) to identify CD4+ T lymphocytes. All cells were examined by FACSCalibur (Becton Dickinson/Cytek Development, Fremont, CA, USA), and data were analyzed by FlowJo 8.8 software (Tree Star, Ashland, OR, USA).
CD4+ T lymphocytes were activated using Dynabeads (Invitrogen). Chemotaxis of WT and A2ARKO CD4+/CD28+/CD137+ cells (1.0×106 cells/ml) in DMEM with 0.5% BSA in the presence or absence of 10 ng/ml CCL2 for 4 h incubated at 37°C in 5% CO2 was determined by colorimetric QCM 5-μm chemotaxis assay (Millipore, Billerica, MA, USA) per manufacturer's protocols. Similarly, CD4+ T lymphocytes from WT mice treated with 0.125 μM CGS21680 or 150 nM ZM241385 underwent CCL2-induced chemotaxis. Cellular migration was assessed by absorbance spectrophotometry at 560 nm by iMark microplate absorbance reader (Life Science Research, Hercules, CA, USA).
In vitro cytokine/chemokine assays
Primary CD3+ T lymphocytes were isolated using a magnetic bead-based cell isolation kit (Miltenyi Biotech) and were pretreated with differing concentrations of CGS21680 or ZM241385 for 4 h and then activated by treatment with 2–10 μg/ml immobilized anti-CD3 mAb (BD Biosciences) at 37°C in 5% CO2 for 48 h. Cell cultures were centrifuged twice, and supernatants underwent quantification by mouse cytokine multiplex immunoassay (Bio-Rad Laboratories), according to manufacturer's protocols. MMP9 release was analyzed using ELISA (R&D Systems) per the manufacturer's protocol.
Human samples
Abdominal aortas from patients undergoing open elective aneurysmectomy for infrarenal AAA were obtained at the time of surgery. Control abdominal aorta samples were obtained from organ donors without clinical or macroscopic signs of aortic atherosclerosis at the University of Virginia Health System (Charlottesville, VA, USA). Approvals for these investigations, including patient consent waiver, was obtained through the Human Investigation Committee at the University of Virginia (HSR 13178).
Statistical analysis
Pair-wise comparisons were determined by unpaired nonparametric tests (Mann-Whitney U), with 2-tailed probabilities at a value of α = 0.05 being considered statistically significant. All calculations were performed using Statistical Package for Social Sciences 20 (SPSS, Armonk, NY, USA) and GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA). Data are presented as means ± se.
RESULTS
A2ARKO mice exhibited increased aortic aneurysm formation compared to WT mice following elastase perfusion
Initial experiments demonstrated that A2AR gene expression was lower at d 1 and 14 in abdominal aortas of WT mice perfused with elastase compared to saline, suggesting that attenuated A2AR signaling may contribute to aneurysm formation. Therefore, as an initial test of our hypothesis that A2AR plays an inhibitory role in aneurysm development, we compared AAA formation in WT and A2ARKO mice in the elastase perfusion model. At d 14, WT mice perfused with saline (negative controls) exhibited a 39 ± 6% increase in aortic diameter, whereas elastase-perfused WT mice developed a 107 ± 5% increase in aortic diameter (Fig. 1A). A2ARKO mice perfused with saline showed a 44 ± 4% increase in aortic diameter, while elastase-perfused mice exhibited a 190 ± 22% increase in aortic diameter (Fig. 1A). Notably, elastase-perfused A2ARKO mice had significantly larger aortic aneurysms vs. elastase perfused WT (positive control) mice (Fig. 1A). As a secondary marker of dilatation and to investigate whether the earlier findings translated to an increase in lumen diameter, circumferential measurements of the IEL from aneurysmal sections were compared. A2ARKO mice exhibited a greater IEL diameter following elastase perfusion compared to WT mice (Fig. 1B). These mice also showed increased medial layer fragmentation (Fig. 1C1) and loss of curvilinear morphology and increased elastin fiber fractures (Fig. 1C2, double-headed black arrow). These data indicate that the A2AR is important in limiting aortic aneurysm dilatation in experimental aneurysm formation.
Figure 1.
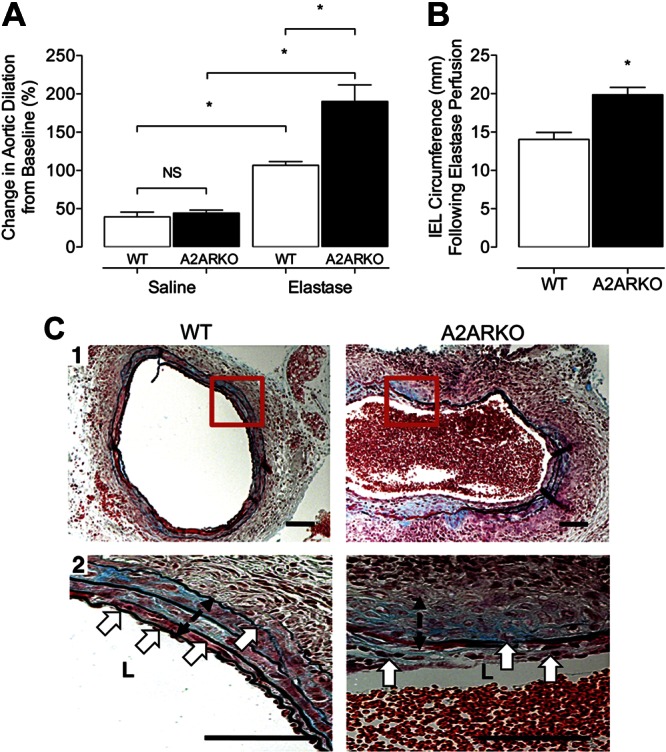
A2AR has a functional role in modulating abdominal aortic dilatation in experimental aneurysms. A) Video micrometry measured infrarenal AAA phenotype on d 14 following either saline or porcine pancreatic elastase perfusion. WT: saline, n = 11; elastase, n = 12. A2ARKO: saline, n = 6; elastase, n = 12. *P < 0.05. B) IEL circumference determined on d 14 among elastase-perfused mice. WT: n = 4; A2ARKO: n = 4. *P < 0.05. C) 1) Representative Movat cross-sectional photomicrographs at ×10 from WT and A2ARKO abdominal aneurysms at d 14 after elastase perfusion. 2) High-power (×40) views of boxed areas in panel 1. Black equilength double-headed arrows identify the medial layer of the aorta; white arrows depict fragmentation of the elastic lamina. Scale bars = 100 μm.
Aortas from A2ARKO mice had increased inflammatory cell infiltration vs. WT mice following elastase perfusion
Given the known anti-inflammatory influence of A2AR (27), we next tested whether enhanced AAA formation was associated with increased inflammatory cell infiltration. The infiltration by Ly-6B.2-positive neutrophils, Mac2-positive macrophages, and CD3ε (M-20)-positive T lymphocytes were markedly higher in aneurysms from A2ARKO mice compared to those from WT mice following elastase perfusion (Fig. 2A–C). In addition, MMP9 expression was also increased in aneurysms from A2ARKO mice (Fig. 2D). These data suggest that A2AR is functionally active in mediating immune cell recruitment and protease expression in the medial and adventitial layers of the aortic wall during AAA formation.
Figure 2.

A2AR gene deletion leads to increased inflammatory cell infiltration and greater matrix degradation. Representative cross-sectional photomicrographs from WT and A2ARKO abdominal aneurysms at d 14 after elastase perfusion. Immunohistochemical staining at ×40 magnification identifies neutrophils (A), macrophages (B), CD3ε T lymphocytes (C), and MMP9 (D) following elastase perfusion on d 14 in the medial and adventitial layers, indicated by arrows. Threshold-gated image quantification of DAB signal in AOI isolated to medial and adventitial layer of aorta. Immunostaining signal quantification was normalized to the total AOI background signal. WT: n = 4; A2ARKO: n = 4. L, lumen. *P < 0.05.
Treatment of WT mice with an A2AR antagonist (ZM241385) exacerbated, while treatment with A2AR agonist (CGS21680) attenuated, elastase perfusion-induced aneurysm formation
The preceding results provide evidence that A2ARKO mice develop larger aneurysms and exhibit greater recruitment of inflammatory cells. To determine whether these results are a direct consequence of genetic loss of the A2AR rather than being secondary to compensatory gene changes resulting from the absence of this receptor throughout development and maturation, we tested the effects of acute A2AR antagonism on AAA formation in WT mice. Elastase-perfused WT mice were dose dependently treated with a commercially available A2AR antagonist (ZM241385; Sigma-Aldrich) and examined by video micrometry 14 d following elastase perfusion. Mice treated with 50 or 200 mg/kg/min of ZM241385 developed larger aneurysms in situ vs. mice treated with vehicle (vehicle: 100±16%, 5 mg/kg/min: 125±30%, 50 mg/kg/min: 143±11%, 200 mg/kg/min: 151±12%; Fig. 3A). These findings indicate that the effects of acute A2AR antagonism mimic those of genetic knockout of A2AR.
Figure 3.

Treatment with exogenous A2AR antagonist and A2AR agonist influence phenotype of experimental aneurysm formation. A) Video micrometry measured infrarenal abdominal aortic aneurysm phenotype on d 14 in WT mice treated with exogenous A2AR antagonist (ZM241385). Vehicle: n = 6; 5 mg/kg: n = 5; 50 mg/kg: n = 6; 200 mg/kg: n = 6. B) Video micrometry measured infrarenal abdominal aortic aneurysm phenotype on d 14 in WT mice treated with either vehicle or CGS21680. Vehicle: n = 9; CGS21680: n = 9/group. *P < 0.05.
Next, we investigated whether acute agonism of the A2AR could mitigate aneurysm formation. To examine this, we used CGS21680, a commercially available selective A2AR agonist (Sigma-Aldrich). Elastase-perfused WT mice were treated with CGS21680 administered via osmotic minipumps initiated 24 h prior to elastase perfusion. CGS21680 dose dependently attenuated aortic aneurysm formation (vehicle: 104±0.1%, 300 ng/kg/min: 78±0.1%, 800 ng/kg/min: 64±0.1%; Fig. 3B). Pumps were explanted, and marginal residual volumes confirmed that the drug had been delivered (data not shown).
A2AR antagonism in vitro increased activated Thy-1.2 T-lymphocyte cytokine release, while agonism decreased cytokine release
Since we observed increased DAB-positive signal in CD3ε+ T-lymphocyte and MMP9-stained paraffin sections from A2ARKO compared to WT mice (Fig. 2C, D), we postulated that deletion of A2AR was associated with increased MMP9 release from activated CD3+ T lymphocytes. To examine this possibility, we measured MMP9 release by ELISA from activated Thy-1.2 T lymphocytes isolated from spleen of A2ARKO and WT mice. We observed a 7-fold increase in MMP9 release when A2AR was deleted (A2ARKO: 9.40±2.13 ng/ml, WT: 1.37±3.41 ng/ml, Fig. 4A). These data suggest that the increased MMP9 expression in the aortic wall of A2ARKO was, in part, likely due to an increased expression from activated CD3+ T lymphocytes.
Figure 4.

A2AR antagonism in vitro increases activated Thy-1.2 T-lymphocyte cytokine release, and agonism decreases cytokine release. A) MMP9 release measured by ELISA from activated CD3+ T lymphocytes normalized to unstimulated levels in WT and A2ARKO mice. WT: n = 4; A2ARKO: n = 4. B) Levels of IL-2, IFN-γ, TNF-α, IL-5, IL-4, IL-10, and CCL5 in cell culture medium of activated Thy-1.2+ lymphocytes treated with ZM241385. Vehicle: n = 4; ZM241385: n = 4/group. C) Levels of IL-2, IFN-γ, TNF-α, IL-5, IL-4, IL-10, and CCL5 released in cell culture medium of activated Thy-1.2+ lymphocytes treated with CGS21680 measured by multiplex ELISA. Vehicle: n = 4; CGS21680: n = 4/group. Open bars indicate vehicle-treated cells; solid bars indicate ZM241385-treated (B) or CGS21680-treated (C) cells. *P < 0.05.
Given the possibility that activated Thy-1.2 T lymphocytes at the aortic wall contribute to increased proteolytic fragmentation of elastin, we reasoned that influencing T-lymphocyte cytokine release, by moderating A2AR-mediated signaling, would change the cytokine/chemokine milieu and also several T-lymphocyte subset (i.e., CD4+) signaling cascades. To demonstrate this, we treated activated Thy-1.2 T lymphocytes with varying concentrations of ZM241385 (Fig. 4B) and CGS21680 (Fig. 4C). Treatment with A2AR antagonist-induced cytokine/chemokine release, while treatment with A2AR agonist attenuated cytokine/chemokine release. Canonical Th1 (i.e., IL-2, IFN-γ, and TNF-α) and Th2 (i.e., IL-4 and IL-10) cytokines were increased when ZM241385 blocked A2AR-mediated signaling in activated Th-1.2 lymphocytes. In contrast, these cytokines were reduced when CGS21680 augmented the A2AR pathway in activated Th-1.2 lymphocytes. Increased CCL5 release from Th-1.2 lymphocytes suggests that chemotactic signals for leukocyte and additional lymphocyte recruitment also increase with receptor antagonism.
CCL2-dependent chemotaxis is increased in activated CD4+ T lymphocytes of A2ARKO mice, increased in WT lymphocytes treated with A2AR antagonist, and reduced in WT lymphocytes treated with A2AR agonist
Data from human studies suggest that CD4+/CD28− lymphocytes are increased in patients with AAAs (8). We observed that Thy-1.2 lymphocytes were increased within the aneurysm wall of A2ARKO mice at d 14 after elastase perfusion (Fig. 2C). In addition, Th1 and Th2 cytokines and chemokines were influenced by A2AR signaling (Fig. 4B, C). Previous studies from our laboratory have shown a crucial role for CCL2 in aneurysm formation (28). Thus, we hypothesized that A2AR modulates chemotaxis of lymphocytes. To test this hypothesis, we compared CCL2-dependent chemotaxis in primary CD4+/CD28+/CD137+ lymphocytes from A2ARKO vs. WT mice. Activated CD4+ lymphocytes from A2ARKO mice had increased CCL2-dependent chemotaxis compared to WT lymphocytes (Fig. 5A). To examine whether chemotaxis of WT CD4+ lymphocytes is mediated by acute A2AR agonism, activated WT CD4+ lymphocytes were treated with CGS21680. Conversely, to examine whether chemotaxis is influenced by acute A2AR antagonism, similarly isolated lymphocytes were treated with ZM241385. We found reduced chemotaxis with exogenous A2AR agonism, and increased chemotaxis with A2AR antagonism (Fig. 5B). These data are consistent with the notion that the loss of integrity of the elastic lamina in elastase perfused A2ARKO mice, as well as in WT mice treated with the A2AR antagonist might be due, at least in part, to increased influx of CD4+/CD28+ lymphocytes and inflammatory cells secondary to loss of A2AR-dependent immunosuppression. Increased Th1-dependent cytokine and chemokine secretion influences the milieu that regulates chemotaxis and immune cell recruitment. Collectively, these data also suggest an important role for A2AR-mediated physiological responses in experimental aneurysm formation, and support the premise of therapeutic targeting of the A2AR as a novel approach to mitigating aneurysms.
Figure 5.
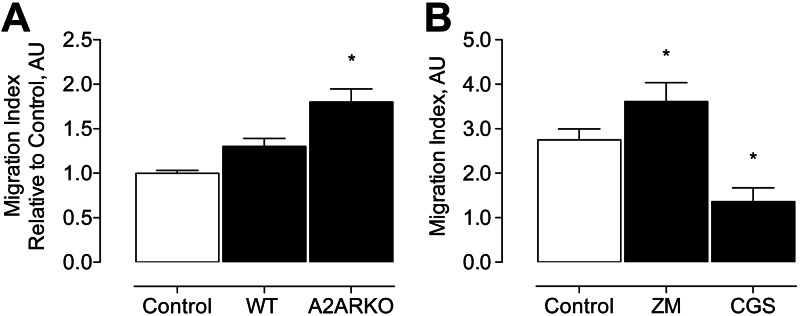
In vitro studies examining CCL2-dependent CD4+ T-lymphocyte chemotaxis. A) CCL2-induced chemotaxis of CD4+/CD28+/CD137+ T lymphocytes from WT and A2ARKO mice; n = 3/group. B) CCL2-induced chemotaxis of CD4+/CD28+/CD137+ T lymphocytes from WT mice treated with A2AR antagonist (ZM) or A2AR agonist (CGS); n = 3/group. Open bars indicate chemotaxis of untreated control WT cells; solid bars indicate chemotaxis of treated WT cells. *P < 0.05.
MMP2, MMP9, and VCAM1 gene expression in SMCs is increased by ZM241385 and decreased by CGS21680 treatment, and MMP9 transcripts are lowered by exogenous A2AR agonism
Given our observations that CCL2-mediated chemotaxis of CD4+/CD28+/CD137+ lymphocytes is influenced by A2AR receptor signaling (Fig. 5) and because SMCs are modulated in aneurysms (5), we postulated that SMC function is influenced in part by the A2AR receptor. We exhibit that MMP2, MMP9, and VCAM1 transcript levels in SMCs treated with ZM241385 were dose dependently increased, while CGS21680 treatments had the opposite effect (Fig. 6A). Moreover, MMP9 expression in CD4+/CD28+/CD137+ lymphocytes was reduced with CGS21680 treatment (Fig. 6B). Taken together, these data point toward an important signaling cascade between SMCs and T lymphocytes, which is, in part, modulated by the A2AR receptor and influences MMP2, MMP9, and VCAM1 gene expression.
Figure 6.

Gene expression of MMP2, MMP9, and VCAM1 is modulated by A2AR. A) Top panels: ZM241385-treated SMCs from WT mice. Bottom panels: CGS21680-treated SMCs from WT mice. Data are normalized to 18S mRNA transcript levels and represent relative fold change in treated WT cells. B) CGS21680-treated CD4+/CD28+/CD137+ T lymphocytes from WT mice. Open bars indicate vehicle-treated WT cells; solid bars indicate dose-dependent treatment of cells; n = 6/group. *P < 0.05.
A2AR gene expression is increased in human AAA tissue
Given evidence that A2AR-mediated experimental AAA formation is important to lesion development, we investigated the potential relevance of this pathway in humans. We examined abdominal aorta-specific gene expression of ADORA2A, the gene that encodes A2AR. A2AR gene expression was significantly elevated in aortic tissue samples from patients with known AAA compared to non-AAA control samples (Fig. 7).
Figure 7.
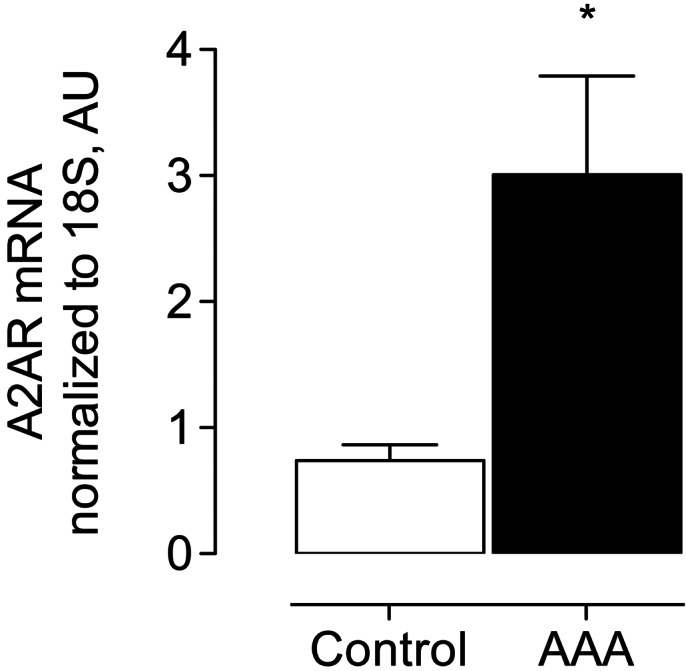
Gene expression of ADORA2A in human aneurysms. Normalized gene expression of ADORA2A in abdominal aorta specimen compared between humans with and without AAA. Control: n = 9; AAA: n = 7. *P < 0.05.
DISCUSSION
The present studies are the first to provide evidence that perturbation in A2AR activation regulates aortic aneurysms. We present multiple lines of evidence to support this assertion. First, A2ARKO mice and WT mice treated with an A2AR antagonist developed increased aneurysm formation. In addition, A2ARKO mice had more degradation of the elastic lamina, enhanced inflammatory cell infiltration, and greater MMP9 proteolytic activity in the aortic wall than WT mice. Second, we show that A2AR agonism attenuated AAA. Third, activated Thy-1.2 lymphocytes released more MMP9 when A2AR was deleted, and separately Thy-1.2 lymphocytes also demonstrated greater cytokine release when A2AR was antagonized, and less cytokine release when A2AR was agonized. Fourth, ASMCs from WT mice exogenously treated with A2AR agonist had reduced protease gene expression. Lastly, in response to CCL2, CD4+/CD28+/CD137+ lymphocytes from A2ARKO mice had greater chemotaxis, as did WT primary CD4+/CD28+/CD137+ lymphocytes treated with A2AR antagonist, while treatment of WT CD4+/CD28+/CD137+ lymphocytes with A2AR receptor agonist reduced chemotaxis. Taken together, results from the present study identify an important regulatory and anti-inflammatory function of A2AR in AAA. These results also suggest that A2AR influenced effects are, in part, combinatorial between ASMCS, and Thy-1.2 lymphocytes. To this end, the proteolytic events that lead to greater elastin fragmentation in AAA formation appear to be, at least in part, a result of increased A2AR activation on CD4+ lymphocytes.
Several groups have previously reported on the significance of adenosine and A2AR-mediated signaling in experimental models of asthma and chronic obstructive pulmonary disease, sepsis, ischemia-reperfusion, arthritis, inflammatory bowel disease, and wound healing (12, 27, 29, 30). Adenosine can activate A2AR, with EC50 values between 0.01 and 1 μM, and, since physiological adenosine concentrations are <1 μM, can readily activate A2AR (12). Genovese et al. (31) demonstrated that 0.1 mg/kg/min exogenous A2AR agonism with CGS21680 reduced JNK phosphorylation in oligodendrocytes in mouse spinal cord injury (31). Moreover, CGS 21680 administered after spinal cord injury reduced motor deficit for >2 wk after injury. Further evidence of physiological benefit of A2AR agonism was illustrated when intraperitoneal injection of CGS21680 administered on d 25 through 35 demonstrated decreased expression of inducible nitric oxide synthase and cyclooxygenase-2 in an experimental model of collagen-induced arthritis (32). Consistent with these data, we observed that acute exogenous A2AR antagonism increased, and agonism decreased aneurysm formation in experimental AAA. More recently, authors have demonstrated that MMP2 expression in SMCs is increased in smokers via nicotine-influenced phosphorylation of protein kinase α2 and aneurysm formation in mice (33). To this end, our data conceptually support a relationship between SMC-related MMP2 gene expression and cAMP pathways, in that agonism of A2AR leads to fewer transcript levels.
Immune cells are functionally important to aortic aneurysm formation. We have previously demonstrated that MCP-1 from BMD cells contributes to macrophage infiltration into AAAs and can act directly on SMCs to reduce contractile protein expression, while inducing MMP2 and MMP9 (28). Our group has previously demonstrated that antineutrophil antibody treatment and deletion of l-selectin in rodents reduces aneurysm formation (34, 35). Others have also reported similar reductions in aneurysm lesion development by global deletion of CD4+ T lymphocytes, IFN-γ, and TNF-α (11, 36). To this end, Baxter et al. (11) demonstrated that, in the absence of CD4+ T lymphocytes, mice were resistant to aneurysm formation. Although the mechanism of this phenomenon was attributed to IFN-γ release (11), the findings, when considered in the context of the current results, support a functional role of activated A2AR on CD4+ T lymphocytes. T lymphocytes initially stimulated in the presence of an A2AR agonist fail to proliferate, and produce IL-2 and IFN-γ when rechallenged without A2AR stimulation (37). Signaling through the T-lymphocyte receptor causes a rapid 5-fold increase in A2AR mRNA, which is correlated with a significant increase in the efficacy of A2AR-mediated cAMP accumulation in these cells (13). Collectively, these data support the hypothesis that aortic aneurysm pathogenesis is regulated partly by CD4+ T lymphocytes. Patients with small aortic aneurysms, who have higher levels of these T-lymphocyte subsets, could benefit from T-lymphocyte inhibition via A2AR-mediated mechanisms (8, 38). Our observations of elevated cytokine release by CD3+ T lymphocytes treated with an A2AR antagonist and reduced cytokine release when treated with an A2AR agonist support this hypothesis. These results combined with data from the chemotaxis assays, suggest that indeed activation of Th1 cells is potentially attenuated through A2AR-mediated signaling.
Recent data provide evidence that A2AR plays an important role in modulating inflammation through effects not only in immune cells but also by influencing secretion of inflammatory mediators by nonimmune cells, including platelets, vascular SMCs, and endothelial cells (39–41). Emaminia et al. (42) found lower tissue cytokine (IL-1β, IL-6, and IL-8) and IFN-γ levels in an ex vivo lung perfusion model following A2AR agonism. A2AR agonism has also been demonstrated to significantly reduce lung ischemia-reperfusion injury in mice (43). Moreover, expression of TNF-α, IL-17, KC, CCL2, MIP-1, and CCL5 were all reduced in CD4+ T-lymphocyte-depleted mice (16). A2AR activation on alveolar macrophages were a key immune signaling mechanism in these findings. Similarly Mazzon et al. (32) observed reduced MIP-1α, MIP-2, MPO activity, TNF-α, IL-1β, and IL-6 with A2AR agonism in a collagen-induced arthritis model. Together, these results suggest that augmenting A2AR signaling can also repress cytokine-mediated inflammation from nonimmune sources, and could have therapeutic implications in limiting aneurysm formation.
Future availability of novel A2AR agonists for humans with chronic disease is expected (12), as is the potential for identifying humans with single-nucleotide polymorphisms (SNPs) of A2AR as attempted by the NCT00253929 trial (44). An example that highlights the relevance of A2AR in cardiovascular disease is seen in patients with heart failure where 34C>T polymorphism in the adenosine monophosphate deaminase (AMPD1) gene is present, who benefit from improved clinical outcome mediated by increased endogenous adenosine (45). As detection of SNPs of A2AR increases across populations, the consequent increase in the risk of AAA formation among these individuals will evolve. In support of the potential importance of A2AR in human aneurysms, we observed increased transcriptional programming suggesting that A2AR may be functional in human aneurysms and that there may be an attempt to maintain homeostasis in response to increased proinflammatory signals. This examination of A2AR biology and T-lymphocyte crosstalk in aneurysms is in its infancy and requires corroborative mechanistic studies to better understand the contribution to nascent aneurysm development, as are feasibility and proof-of-concept preclinical studies to establish efficacy of A2AR agonists in mitigating aneurysm formation.
In summary, the current study is the first to our knowledge to demonstrate a pivotal role of A2AR in experimental aneurysm progression. Reduced inflammatory cell infiltration and proteolytic activity modulated by the A2AR is important to maintain vessel architecture, including the elastin layers in the aortic wall. As such, pharmacologic agonism of the A2AR may provide a novel therapeutic target for inhibiting inflammation and elastin loss in human aneurysms.
Acknowledgments
This work was supported by U.S. National Institutes of Health (NIH) grants to G.A. (K08HL098560) and I.L.K. (T32HL007849). The Research and Education Grant from the Thoracic Surgery Foundation also supported G.A.
The authors thank M. Bevard for her technical expertise and advice for histology. The authors also appreciate the administrative contributions of A. J. Herring and C. S. Dodson. The authors are grateful to Dr. M. Angulo (University of Virginia) and Dr. C. A. Whatling (AstraZeneca, Mölndal, Sweden) for critical review and editorial commentary. In addition, the authors thank Dr. M. Angulo (University of Virginia) for her technical expertise in the timely resubmission of revisions. The authors also acknowledge the initial work of Dr. Hong Pei (Joel Linden Laboratory, La Jolla Institute for Allergy and Immunology, La Jolla, CA, USA) in the area of A2AR and aortic aneurysms. The authors declare no competing financial interests. This work was presented at the Annual Scientific Sessions of the American Heart Association 2010 (Chicago, IL, USA).
Footnotes
- A2AR
- adenosine A2A receptor
- A2ARKO
- adenosine A2A receptor knockout
- AAA
- abdominal aortic aneurysm
- AOI
- area of interest
- APC
- allophycocyanin
- ASMC
- aortic smooth muscle cell
- BMD
- bone-marrow derived
- cAMP
- 3′-5′-cyclic adenosine monophosphate
- DAPI
- 4′,6-diamidino-2-phenylindole
- FACS
- fluorescence-activated cell sorting
- Gsα
- stimulatory G protein α
- H&E
- hematoxylin and eosin
- IEL
- internal elastic lamina
- IHC
- immunohistochemistry
- MCP-1
- monocyte chemotactic protein-1
- MMP
- matrix mellatoproteinase
- Movat
- modified Russell-Movat pentachrome
- qPCR
- quantitative polymerase chain reaction
- RNA
- ribonucleic acid
- SMC
- smooth muscle cell
- WT
- wild type
REFERENCES
- 1. Heron M. P., Hoyert D. L., Murphy S. L., Xu J. Q., Kochanek K. D., Tejada-Vera B. (2009) Deaths: final data for 2006. Natl. Vital Stat. Rep. 57, 14 http://www.cdc.gov/nchs/data/nvsr/nvsr57/nvsr57_14.pdf [PubMed] [Google Scholar]
- 2. Guo D. C., Papke C. L., He R., Milewicz D. M. (2006) Pathogenesis of thoracic and abdominal aortic aneurysms. Ann. N. Y. Acad. Sci. 1085, 339–352 [DOI] [PubMed] [Google Scholar]
- 3. Thompson R. W., Curci J. A., Ennis T. L., Mao D., Pagano M. B., Pham C. T. (2006) Pathophysiology of abdominal aortic aneurysms: insights from the elastase-induced model in mice with different genetic backgrounds. Ann. N. Y. Acad. Sci. 1085, 59–73 [DOI] [PubMed] [Google Scholar]
- 4. Curci J. A., Thompson R. W. (2004) Adaptive cellular immunity in aortic aneurysms: cause, consequence, or context? J. Clin. Invest. 114, 168–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ailawadi G., Moehle C. W., Pei H., Walton S. P., Yang Z., Kron I. L., Lau C. L., Owens G. K. (2009) Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J. Thorac. Cardiovasc. Surg. 138, 1392–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ailawadi G., Eliason J. L., Upchurch G. R., Jr. (2003) Current concepts in the pathogenesis of abdominal aortic aneurysm. J. Vasc. Surg. 38, 584–588 [DOI] [PubMed] [Google Scholar]
- 7. Rizas K. D., Ippagunta N., Tilson M. D., 3rd (2009) Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiol. Rev. 17, 201–210 [DOI] [PubMed] [Google Scholar]
- 8. Duftner C., Seiler R., Klein-Weigel P., Gobel H., Goldberger C., Ihling C., Fraedrich G., Schirmer M. (2005) High prevalence of circulating CD4+CD28- T-cells in patients with small abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 25, 1347–1352 [DOI] [PubMed] [Google Scholar]
- 9. Valente A. J., Rozek M. M., Sprague E. A., Schwartz C. J. (1992) Mechanisms in intimal monocyte-macrophage recruitment. A special role for monocyte chemotactic protein-1. Circulation 86, III20–25 [PubMed] [Google Scholar]
- 10. Taub D. D., Proost P., Murphy W. J., Anver M., Longo D. L., van Damme J., Oppenheim J. J. (1995) Monocyte chemotactic protein-1 (MCP-1), -2, and -3 are chemotactic for human T lymphocytes. J. Clin. Invest. 95, 1370–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiong W., Zhao Y., Prall A., Greiner T. C., Baxter B. T. (2004) Key roles of CD4+ T cells and IFN-γ in the development of abdominal aortic aneurysms in a murine model. J. Immunol. 172, 2607–2612 [DOI] [PubMed] [Google Scholar]
- 12. Haskó G., Linden J., Cronstein B., Pacher P. (2008) Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug. Discov. 7, 759–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lappas C. M., Sullivan G. W., Linden J. (2005) Adenosine A2A agonists in development for the treatment of inflammation. Expert Opin. Investig. Drugs 14, 797–806 [DOI] [PubMed] [Google Scholar]
- 14. Gazoni L. M., Walters D. M., Unger E. B., Linden J., Kron I. L., Laubach V. E. (2010) Activation of A1, A2A, or A3 adenosine receptors attenuates lung ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 140, 440–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haskó G., Pacher P., Deitch E. A., Vizi E. S. (2007) Shaping of monocyte and macrophage function by adenosine receptors. Pharmacol. Ther. 113, 264–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sharma A. K., Laubach V. E., Ramos S. I., Zhao Y., Stukenborg G., Linden J., Kron I. L., Yang Z. (2010) Adenosine A2A receptor activation on CD4+ T lymphocytes and neutrophils attenuates lung ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 139, 474–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laubach V. E., French B. A., Okusa M. D. (2011) Targeting of adenosine receptors in ischemia-reperfusion injury. Expert Opin. Ther. Targets 15, 103–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anvari F., Sharma A. K., Fernandez L. G., Hranjec T., Ravid K., Kron I. L., Laubach V. E. (2010) Tissue-derived proinflammatory effect of adenosine A2B receptor in lung ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 140, 871–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pyo R., Lee J. K., Shipley J. M., Curci J. A., Mao D., Ziporin S. J., Ennis T. L., Shapiro S. D., Senior R. M., Thompson R. W. (2000) Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J. Clin. Invest. 105, 1641–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ledent C., Vaugeois J. M., Schiffmann S. N., Pedrazzini T., El Yacoubi M., Vanderhaeghen J. J., Costentin J., Heath J. K., Vassart G., Parmentier M. (1997) Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature 388, 674–678 [DOI] [PubMed] [Google Scholar]
- 21. Institute for Laboratory Animal Research (2011) Guide for the Care and Use of Laboratory Animals, National Academies Press, Washington, DC [Google Scholar]
- 22. Applied Research Ethics National Organization/Office of Laboratory Animal Welfare (2002) Institutional Animal Care and Use Committee Guidebook, National Institutes of Health, Bethesda, MD, USA [Google Scholar]
- 23. Cassada D. C., Tribble C. G., Long S. M., Kaza A. K., Linden J., Rieger J. M., Rosin D., Kron I. L., Kern J. A. (2002) Adenosine A2A agonist reduces paralysis after spinal cord ischemia: correlation with A2A receptor expression on motor neurons. Ann. Thorac. Surg. 74, 846–849; discussion 849–850 [DOI] [PubMed] [Google Scholar]
- 24. Chehata V. J., Domeier P. P., Weilnau J. N., Lappas C. M. (2011) Adenosine A(2A) receptor activation limits chronic granulomatous disease-induced hyperinflammation. Cell. Immunol. 267, 39–49 [DOI] [PubMed] [Google Scholar]
- 25. Feoktistov I., Biaggioni I. (1997) Adenosine A2B receptors. Pharmacol. Rev. 49, 381–402 [PubMed] [Google Scholar]
- 26. Alexander M. R., Moehle C. W., Johnson J. L., Yang Z., Lee J. K., Jackson C. L., Owens G. K. (2012) Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J. Clin. Invest. 122, 70–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Linden J. (2011) Regulation of leukocyte function by adenosine receptors. Advances Pharmacol. 61, 95–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moehle C. W., Bhamidipati C. M., Alexander M. R., Mehta G. S., Irvine J. N., Salmon M., Upchurch G. R., Jr., Kron I. L., Owens G. K., Ailawadi G. (2011) Bone marrow-derived MCP1 required for experimental aortic aneurysm formation and smooth muscle phenotypic modulation. J. Thorac. Cardiovasc. Surg. 142, 1567–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Olah M. E., Stiles G. L. (1995) Adenosine receptor subtypes: characterization and therapeutic regulation. Annu. Rev. Pharmacol. Toxicol. 35, 581–606 [DOI] [PubMed] [Google Scholar]
- 30. Bruns R. F. (1990) Adenosine receptors. Roles and pharmacology. Ann. N. Y. Acad. Sci. 603, 211–225; discussion 225–216 [DOI] [PubMed] [Google Scholar]
- 31. Genovese T., Melani A., Esposito E., Mazzon E., Di Paola R., Bramanti P., Pedata F., Cuzzocrea S. (2009) The selective adenosine A2A receptor agonist CGS 21680 reduces JNK MAPK activation in oligodendrocytes in injured spinal cord. Shock 32, 578–585 [DOI] [PubMed] [Google Scholar]
- 32. Mazzon E., Esposito E., Impellizzeri D., Di Paola R., Melani A., Bramanti P., Pedata F., Cuzzocrea S. (2011) CGS 21680, an agonist of the adenosine (A2A) receptor, reduces progression of murine type II collagen-induced arthritis. J. Rheumatol. 38, 2119–2129 [DOI] [PubMed] [Google Scholar]
- 33. Wang S., Zhang C., Zhang M., Liang B., Zhu H., Lee J., Viollet B., Xia L., Zhang Y., Zou M. H. (2012) Activation of AMP-activated protein kinase alpha2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat. Med. 18, 902–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eliason J. L., Hannawa K. K., Ailawadi G., Sinha I., Ford J. W., Deogracias M. P., Roelofs K. J., Woodrum D. T., Ennis T. L., Henke P. K., Stanley J. C., Thompson R. W., Upchurch G. R., Jr. (2005) Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation 112, 232–240 [DOI] [PubMed] [Google Scholar]
- 35. Hannawa K. K., Eliason J. L., Woodrum D. T., Pearce C. G., Roelofs K. J., Grigoryants V., Eagleton M. J., Henke P. K., Wakefield T. W., Myers D. D., Stanley J. C., Upchurch G. R., Jr. (2005) L-selectin-mediated neutrophil recruitment in experimental rodent aneurysm formation. Circulation 112, 241–247 [DOI] [PubMed] [Google Scholar]
- 36. Xiong W., MacTaggart J., Knispel R., Worth J., Persidsky Y., Baxter B. T. (2009) Blocking TNF-α attenuates aneurysm formation in a murine model. J. Immunol. 183, 2741–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zarek P. E., Huang C. T., Lutz E. R., Kowalski J., Horton M. R., Linden J., Drake C. G., Powell J. D. (2008) A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 111, 251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yin M., Zhang J., Wang Y., Wang S., Bockler D., Duan Z., Xin S. (2010) Deficient CD4+CD25+ T regulatory cell function in patients with abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 30, 1825–1831 [DOI] [PubMed] [Google Scholar]
- 39. Shryock J. C., Belardinelli L. (1997) Adenosine and adenosine receptors in the cardiovascular system: biochemistry, physiology, and pharmacology. Am. J. Cardiol. 79, 2–10 [DOI] [PubMed] [Google Scholar]
- 40. Wagner D. R., Kubota T., Sanders V. J., McTiernan C. F., Feldman A. M. (1999) Differential regulation of cardiac expression of IL-6 and TNF-α by A2- and A3-adenosine receptors. Am. J. Physiol. Heart Circ. Physiol. 276, H2141–H2147 [DOI] [PubMed] [Google Scholar]
- 41. Ernens I., Rouy D., Velot E., Devaux Y., Wagner D. R. (2006) Adenosine inhibits matrix metalloproteinase-9 secretion by neutrophils: implication of A2a receptor and cAMP/PKA/Ca2+ pathway. Circ. Res. 99, 590–597 [DOI] [PubMed] [Google Scholar]
- 42. Emaminia A., Lapar D. J., Zhao Y., Steidle J. F., Harris D. A., Laubach V. E., Linden J., Kron I. L., Lau C. L. (2011) Adenosine AA agonist improves lung function during ex vivo lung perfusion. Ann. Thorac. Surg. 92, 1840–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. LaPar D. J., Laubach V. E., Emaminia A., Crosby I. K., Hajzus V. A., Sharma A. K., Sumner H. M., Webb D. V., Lau C. L., Kron I. L. (2011) Pretreatment strategy with adenosine A2A receptor agonist attenuates reperfusion injury in a preclinical porcine lung transplantation model. J. Thorac. Cardiovasc. Surg. 142, 887–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Riksen N. P., Franke B., van den Broek P., Smits P., Rongen G. A. (2007) The 1976C>T polymorphism in the adenosine A2A receptor gene does not affect the vasodilator response to adenosine in humans in vivo. Pharmacogenet. Genom. 17, 551–554 [DOI] [PubMed] [Google Scholar]
- 45. Riksen N. P., Franke B., Oyen W. J., Borm G. F., van den Broek P., Boerman O. C., Smits P., Rongen G. A. (2007) Augmented hyperaemia and reduced tissue injury in response to ischaemia in subjects with the 34C>T variant of the AMPD1 gene. Eur. Heart. J. 28, 1085–1091 [DOI] [PubMed] [Google Scholar]