

Abstract
Until recently, the apicomplexan parasites, Cryptosporidium hominis and C. parvum, were considered the same species. However, the two parasites, now considered distinct species, exhibit significant differences in host range, infectivity, and pathogenicity, and their sequenced genomes exhibit only 95–97% identity. The availability of the complete genome sequences of these organisms provides the potential to identify the genetic variations that are responsible for the phenotypic differences between the two parasites. We compared the genome organization and structure, gene composition, the metabolic and other pathways, and the local sequence identity between the genes of these two Cryptosporidium species. Our observations show that the phenotypic differences between C. hominis and C. parvum are not due to gross genome rearrangements, structural alterations, gene deletions or insertions, metabolic capabilities, or other obvious genomic alterations. Rather, the results indicate that these genomes exhibit a remarkable structural and compositional conservation and suggest that the phenotypic differences observed are due to subtle variations in the sequences of proteins that act at the interface between the parasite and its host.
1. Introduction
Organisms of the genus Cryptosporidium are protozoa of the phylum Apicomplexa. These obligatory intracellular organisms parasitize animals of all vertebrate classes [1]. Although mostly ignored as a pathogen until relatively late in the 20th century, diarrhea caused by Cryptosporidium species is debilitating for adults and children and can be life threatening for immunocompromised individuals such as those with AIDS. Cryptosporidiosis is also a significant factor in animal husbandry practices and represents a significant challenge to agriculture, for example, the beef industry [2]. Development of molecular tools now permits efficient differentiation of morphologically indistinguishable isolates of these parasites, and this new capability has led to important new insights into their epidemiology and pathogenicity. Although several Cryptosporidium species can cause human disease, two species, C. hominis and C. parvum, are responsible for the majority of the human impact. C. parvum infects ruminants as primary hosts and humans as incidental hosts. C. hominis, in contrast, is highly infectious to humans but generally does not infect other species [3]. Until very recently, these two species were considered genotypes of C. parvum [4]: genotype 1 (or type H) found nearly exclusively in humans; and genotype 2 (or type C) found naturally infecting cattle and other animals as well as humans [5]. Later investigations established that these genotypes are sufficiently distinct in host range, genetics, pathogenicity, intensity of infection, and other growth characteristics to be considered separate species [6]. Recently, it has been shown that these two parasites use different mechanisms for host cell invasion, a significant finding considering their differential host preferences [7].
Cryptosporidiosis is a zoonotic, primarily water-borne disease that is transmitted by the oral-fecal route. Cryptosporidium has a simpler life cycle than other apicomplexans. The only life stage found outside the host is the oocyst, a resistant spore-like form that is largely quiescent until ingested by a new host. Although the disease is usually self-limiting, it has been suggested that cryptosporidiosis can be a significant component in malnutrition, impaired growth, and intellectual acuity in developing countries, where children are exposed to repeated infections during a critical time of their development [8]. In developed countries, drinking and recreational water-borne outbreaks and their economical consequences, as well as agricultural and veterinary impacts are the major concerns. Immunocompromised individuals and the elderly are also at risk of complications of cryptosporidiosis, since immunoprophylactics for the disease are unavailable, and treatment is often late and targeted at symptoms.
With the completion of the genome sequences of C. hominis and C. parvum [9, 10], it is now possible to carefully and accurately compare their genetic architectures and compositions with the goal of identifying the root causes of their phenotypic differences. Herein, comparisons were performed at three levels. First, the genome sequences were compared, focusing on general features of genome organization, for example, rearrangements and insertions or deletions. Second, gene level comparisons were performed with two goals: to evaluate the gene complements and compositions of these organisms and to search for specific genes undergoing noticeable selective pressure, as determined by nonsynonymous to synonymous substitution ratios in protein evolution. Finally, comparisons at the level of inferred pathways were performed to investigate how eventual differences in gene composition could impact the organization of metabolic and other networks in these organisms. Thus, genomes of the two species of Cryptosporidium were carefully compared to each other and simultaneously to the genomes of other apicomplexans for which the genomes are available.
2. Results and Discussion
2.1. Genome Synteny and Collinearity
To analyze the genomic organization in these two organisms, we used the published C. parvum sequence [10], updated with additional data, and an updated assembly of the published C. hominis genome [9]. In the new C. hominis assembly (deposited at DDBJ/EMBL/GenBank under the whole genome shotgun project accession number AAEL00000000, the version used in this paper being AAEL02000000), additional directed sequencing was used to close most of the sequence gaps and reduce the number of contigs to ~330 with an N50 of 49.2 kb. Alignment of the C. hominis and C. parvum contigs revealed no significant differences; that is, despite the 3–5% sequence variation observed between the two genomes, we detected no significant insertions, deletions, or rearrangements.
This observation was confirmed by generation of genome wide optical maps of both species. Thus, optical maps of the C. hominis genome generated using the SacI restriction enzyme were compared to the virtual SacI restriction maps of the genome sequences of C. parvum (see Section 4). Discrepancies were found in the ordering and orientation of contigs of the two species. Each of these apparent discrepancies was examined by attempting to amplify the DNA from both C. hominis and C. parvum in the apparent gaps using primers targeting the ends of the contigs in question in polymerase chain reaction (PCR) experiments (see Section 4). These experiments confirmed that apparent organizational discrepancies, for example, rearrangements, between the two genomes were due to incorrect assemblies (data not shown). For example, the orientations of fragment no. 1 of chromosome 2 and fragment no. 2 of chromosome 4 in the published C. parvum sequence [10] are reversed. No insertions or deletions of significant size were detected, and most differences in chromosome length were due to remaining gaps in the C. hominis sequence. In summary, despite the fairly high level of sequence divergence observed between C. hominis and C. parvum, the genomes of these two parasites are essentially collinear. Since both parasites exhibit the same gene composition (see below), we conclude they are syntenic at all sites in all eight of their chromosomes.
The synteny between the C. parvum genome and those of three other apicomplexans, selected for the quality of their genome annotation (Plasmodium falciparum, P. knowlesi, and P. vivax), was evaluated by comparing both chromosome composition and gene order. Thus, orthologs were identified and each chromosome of the three plasmodia species was evaluated for its enrichment in orthologs from given chromosomes of C. parvum (see Section 4). As shown in Table S1 in Supplementary Material available online at http://dx.doi.org/10.1155/2013/832756, orthologs are not uniformly spread into the plasmodia chromosomes. Thus, although the genes from each Cryptosporidium chromosome are dispersed among the Plasmodium chromosomes, a significant fraction of them are colocalized on a given Plasmodium chromosome. This observation suggests that chromosome composition is significantly conserved across the apicomplexans. However, despite this conservation of gene composition, the order of the genes on the chromosomes (i.e., the synteny) shows little evidence of conservation between the Cryptosporidium and plasmodia (see Supplementary Figure 1). These results strongly suggest that genomes of these parasites evolved from a common ancestor through extensive cis-rearrangements (insertion, deletion, and reordering of genome fragments or genes within chromosomes) rather than through trans-rearrangements (exchange of genome fragments or genes among chromosomes).
We have previously reported that the genome of Cryptosporidium is heavily tailored and apparently has maintained only the fraction of genes required for its survival in the specific environments it inhabits [9, 10]. In contrast, the genomes of other apicomplexans, including the Plasmodium species, are quite robust. Thus, we assume that the genome of the apicomplexan progenitor was more robust and that the Cryptosporidium genome is the product of broad-scale gene deletion. Although the selective pressures for this phenomena are apparent, the genetic mechanisms remain obscure. It is interesting therefore to note that the mechanism that led to broad scale deletions in Cryptosporidium largely preserved the gene compositions of specific chromosome fragments while simultaneously not conserving the gene order.
2.2. Gene Complement and Composition
We compared the gene complements of C. hominis and C. parvum in order to investigate potential differences in the repertoires of proteins encoded. We found that effectively all genes from one organism have a corresponding putative ortholog in the other. The only apparent exceptions were 67 genes initially found only in C. parvum and 246 genes initially present only in C. hominis. However, more careful alignment of these genes to the corresponding genome [11] showed that all putative C. parvum-specific genes are also present in C. hominis but were not predicted due to very small gaps remaining in the sequence. Conversely, all but one of the genes apparently specific to C. hominis were similarly found in the C. parvum genome. The remaining apparently C. hominis-specific gene (Chro.00003) is truly absent in C. parvum. This gene is in a contig that is unlinked to the remainder of the C. hominis genome, and its sequence most closely resembles related genes of alphaproteobacterial origin (data not shown). Since C. hominis is purified from the feces of an infected mammal, this single remaining gene is most likely derived from an alphaproteobacterial contaminant of the initial C. hominis stock used to generate shotgun sequence for the C. hominis genome. Therefore, we conclude that C. hominis and C. parvum have identical gene complements.
We also examined the frequency of putative paralogs per gene across all Apicomplexa for which the genomes are available (Supplementary Figure 2). This analysis showed that Cryptosporidium has less than 0.05 paralogs per gene, while the next most compact genomes (Plasmodium berghei and Toxoplasma gondii) have levels of gene duplication about one order of magnitude higher (0.43 and 0.53, resp.). Species of Theileria exhibit 0.8–1.4 paralogs per gene, despite having genomes about 1 Mb smaller than Cryptosporidium and a similar number of genes (~3,800 to ~4,000) [12]. In short, our analysis suggested that Cryptosporidium species, in spite of having neither the smallest genomes nor the lowest numbers of proteins, have the most compact proteomes of these apicomplexans, exhibiting the lowest level of redundancy. Clearly, such a low level of protein redundancy is convenient for the investigation of potential drug targets and vaccine candidates for treating and preventing cryptosporidiosis.
2.3. Metabolic and Signaling Capabilities
Metabolic and signaling pathways were identified in the two Cryptosporidium species and other apicomplexans for which sequences are available, using sequence similarity approaches (see Section 4). In order to evaluate any potential impairment of these pathways due to the minor gene content discrepancies identified above, we evaluated the pathways using three scores: completeness, connectedness, and support (Figure 1). These scores, described in Section 4, reflect how complete the enzymatic equipment of a given pathway is, as well as the ability of the pathway to process key metabolites shared with other pathways [13, 14]. A high completeness score for a pathway indicates that most of the genes that are traditionally associated with this pathway are present in the genome. A high connectedness score indicates that the actual set of enzymes present ensures the interconversion of the metabolites, both input and output, that the pathway exchanges with other pathways. Finally, the support score reflects the number of reference species from the pathway databases that have been used to assess the presence of this pathway in Cryptosporidium. Therefore, a high support score reflects a high level of confidence that the annotation of the pathway is correct.
Figure 1.

Pathway scores (excerpt). Completeness, connectedness, and support scores of the inferred metabolic pathways of C. hominis and C. parvum, along with nine other apicomplexans and an external reference (Saccharomyces cerevisiae). The three scores are represented as follows. The color of the circles reflects the support, from white (support of 0) to blue (maximal support). The size of each circle is proportional to the completeness; that is, the larger the circle, the more complete the pathway (see text). The color of the wedge—available for metabolic pathways only—reflects the connectedness, from red (connectedness of 0) to green (connectedness of 100%). Species are clustered according to their completeness score. Pathways are ranked by decreasing power to discriminate between the Cryptosporidium and the other apicomplexans. Only the 25 most discriminative pathways are shown; a complete figure is available as Supplementary Figure 3. All score values are available in Supplementary Table 2.
Figure 1—an excerpt of Supplementary Figure 3—provides a comparative analysis of Cryptosporidium and nine other apicomplexans for which genomic data is available (T. parva, T. annulata, Plasmodium chabaudi, P. berghei, P. yoelii, P. falciparum, P. vivax, P. knowlesi, and Toxoplasma gondii). The figure is a composite graphical representation of the three scores—completeness, connectedness, and support—for the two species of Cryptosporidium, the nine other apicomplexans, and an external reference (S. cerevisiae). The 12 species were clustered according to the completeness of their pathways. All scores are available in Supplementary Table 2.
As expected from the comparative analyses outlined above, these results confirmed that the two Cryptosporidium species have highly similar pathways and other functions. In contrast, their metabolic potential is markedly distinct from the other Apicomplexa. Despite the fact that some of the other species in the phylum have smaller genomes (e.g., Theileria spp. genomes are about 1 Mb smaller than the ~9.2 Mb genomes of the Cryptosporidium spp.), Cryptosporidium has the most highly reduced metabolic capabilities. As previously reported [9], and in contrast to most other apicomplexans, our observations further confirm that Cryptosporidium lacks most mitochondrial and apicoplast functions. Biosynthetic capabilities (amino acid, nucleic acid, carbohydrate, etc.) are limited, and energy generation machinery is largely dependent on glycolysis as the TCA cycle, oxidative phosphorylation, pentose phosphate pathway, and so forth are largely absent. This diminished metabolic capability is likely related to the relatively simple life cycle of the parasite; that is, in contrast to most other apicomplexan parasites, Cryptosporidium has only a single host and no vector. Clearly, the parasite is highly evolved to take advantage of the host functions and capabilities. For example, the C. hominis genome encodes more than 80 predicted transporters [9], while the similarly sized genome of T. parva seems to encode only about 60 transporters [15]. Another important difference is the giant enzyme, bacterial derived type I fatty acid biosynthesis mechanism present in Cryptosporidium [16], in contrast with the multienzyme type II mechanism functional in the apicoplast of all other Apicomplexa.
2.4. Gene Sequence Comparison
The high degree of identity of the gene compositions and pathways of C. hominis and C. parvum begs the question of the root of their clear phenotypic differences. Lack of differences in gene composition suggests a more subtle cause, for example, missense mutations in protein coding sequences, for the differences in their characteristics. The ratio of nonsynonymous missense and synonymous substitution rates (dN/dS ratio, see Section 4) is often used to identify genes subject to positive or negative selection within a given genome. Nonsynonymous changes in a DNA sequence, that is, mutations that change the amino acid sequence, are more likely to alter protein function and would therefore be expected to be rapidly eliminated from essential or important functional genes in microbial genomes. Genes with a dN/dS ratio greater than 1.0 are generally considered to be selected for sequence change (positive selection), while genes with a ratio below 1.0 are considered to be selected for sequence conservation (negative selection). We calculated dN/dS ratios for the pairs of orthologs in C. hominis and C. parvum as identified above to assess which genes were being selected for change or driven to evolve and those that are highly conserved in an attempt to begin identifying the source of the phenotypic divergence between the two species. This analysis identified 37 genes with dN/dS ratios greater than 1.1, indicating positive selective pressure for divergence (Supplementary Table 3).
Positively selected proteins have previously been shown to be associated with the surface of diverging pathogens, probably due to the necessity of the pathogen to alter its surface antigenicity to avoid the host immune response. [12] demonstrated this phenomenon in comparison of two related apicomplexan parasites, Theileria annulata and T. parva. Therefore, we used publicly available sequence analysis tools (see Section 4) to identify putative surface-associated or secreted proteins, that is, those with transmembrane domains, glycosylphosphatidylinositol anchors, signal sequences, or signal peptides (Supplementary Table 4). We used this information to evaluate the association between protein localization and the dN/dS ratio. Thus, we selected subsets of proteins having a high dN/dS ratio when considering different cutoff values. The enrichment of these subsets in proteins of given cellular localization was calculated as the fraction of proteins in a subset having a given localization divided by the fraction of proteins in the genome having this same localization. The statistical significance of this enrichment was evaluated by calculating a P value (the probability that this enrichment, or a better one, would be obtained by chance alone) using Fisher's exact test. The results (Figure 2) showed that positively selected proteins (dN/dS ratio equal or greater to 1.1) were significantly enriched in putatively secreted products; that is, 21.62% of the 37 positively selected proteins share this localization. This represents an enrichment of 4.16 and a P value of 9.48 × 10−5. A similar trend was observed for membrane-bound proteins (enrichment of 1.5, P value of 0.057). In contrast, typical cytoplasmic proteins showed strongly conserved dN/dS ratios; for example, glycolytic enzymes exhibit a mean dN/dS ratio of only 0.06, and enzymes of pyruvate metabolism exhibit a ratio of only 0.07 (Supplementary Table 4). These observations suggest that surface-associated and secreted proteins are strongly selected for divergence in Cryptosporidium.
Figure 2.
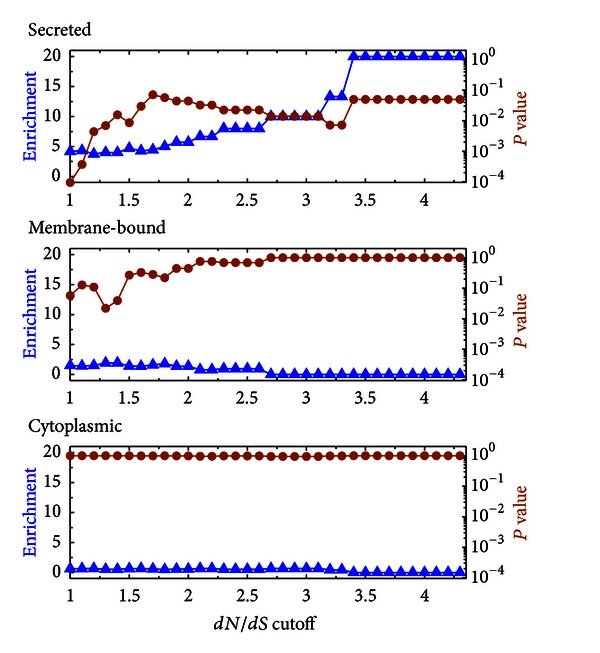
Protein localization and selective pressure. Association between protein localization and selective pressure. Abscissa: cutoffs used for the identification of positively selected proteins (are considered as positively selected proteins with dN/dS ratios higher than or equal to the cutoff). Ordinate: enrichment (triangles) and P value (circles) of the set of positively selected proteins for a given localization—either cytoplasmic, membrane-bound, or secreted.
We expected to observe a similar trend when examining the sequence identities of orthologous proteins in C. hominis and C. parvum; that is, surface-associated proteins should exhibit the greatest sequence divergence. Thus, we measured the identities of each protein orthologous pair in these two organisms and sorted them according to their cellular compartment. As it might be expected, the results of this analysis reinforced those of the dN/dS comparison. Thus, of the 145 protein pairs with identity lower than 90% (the average identity of all C. hominis and C. parvum genes being ~97%), 33 (22.76%) are predicted to be secreted proteins and 51 (35.17%) are predicted to be membrane-bound. This is 4.76-fold (P value of 8.21 × 10−15) and 1.8-fold (P value of 8.50 × 10−6) enrichment, respectively, over that expected by chance alone.
These results indicate that the two Cryptosporidium species have diverged most rapidly at the level of the proteins interacting with their environments, that is, the environment, the host, and the host immune system. This observation likely explains the significant host range and pathogenicity differences exhibited by the two pathogens.
3. Conclusions
Herein, we have compared the genomes of C. hominis and C. parvum to each other and to the genomes of related apicomplexan parasites. Our results show that despite the rather large sequence divergence of 3–5% between the two Cryptosporidium genomes, similar to that observed between humans and chimpanzees, the structure and composition of these two genomes are largely conserved. Not only is the number of chromosomes conserved at eight, but the two genomes are apparently completely collinear and each of the chromosomes in the two parasites is completely syntenic. Despite the synteny between C. hominis and C. parvum, almost no synteny was observed between the Cryptosporidium and Plasmodium. In contrast, gene composition of a chromosome fragments was highly conserved between these two genera, suggesting that the mechanisms by which Cryptosporidium evolved from its more genetically robust progenitors involved large scale gene deletion and more extensive cis- than trans-rearrangements. We also found no genes that are unique to either of the parasites, that is, present in only one of the two genomes. Comparison of the gene and pathway composition of the two parasites to those of other apicomplexans showed that the Cryptosporidium genome and metabolism are remarkably reduced, even more so than the Theileria spp. which have a smaller genome size. Finally, a comparison of the mutation rates of Cryptosporidium genes showed that those proteins associated with the surface of the parasite are being selected for rapid divergence, in stark contrast to the typical cytoplasmic proteins, which show a much higher level of conservation across the two species. This observation suggests that the phenotypic differences exhibited by C. hominis and C. parvum are due to selective forces exerted by the host parasite interaction largely upon the surface of the parasite.
4. Methods
4.1. Genome Comparison
Chromosome numbers, size, and sequences, as well as genome annotations (i.e., identification of the open reading frames) were obtained from the sequencing projects of C. hominis [9] and C. parvum [10]. In order to minimize the influence of annotation methodology in the accuracy of comparisons, C. parvum proteins were predicted de novo for this work, as previously described [9]. This was necessary since the versions of C. parvum proteins available in the general databases at the time of these analyses—GenBank and CryptoDB [17]—were missing around 400 proteins when compared to C. hominis or to our version of C. parvum gene predictions. Comparison of the organization of these two genomes was done first by obtaining restriction optical maps [18–26] for the SacI restriction enzyme, experimentally for C. hominis and in silico for C. parvum. The restriction fragment patterns generated by the optical mapping procedure were aligned for direct comparison using the Map Assembler algorithm [27–29]. For each given chromosome, discrepancies found in the alignment of the optical maps from the two species were investigated by the polymerase chain reaction (PCR). In brief, sequences ~3 kb upstream and downstream from the putative sites of discrepancy were retrieved from the C. parvum sequences. Primers directed towards the sites of discrepancy were designed and used in PCR reactions to assess and validate the presumed orientations of the contigs. Reactions were performed in volumes of 15 μL containing 250 μM dNTP, 2 ng/μL of each oligonucleotide primer, 1.5 units of Taq DNA Polymerase (HotMaster, 5 Prime Gaithersburg, MD, USA), 15 ng of C. parvum (IA) DNA, and the appropriate buffer as provided by the manufacturer. The following PCR conditions were used: 98°C for 3 min, 35 cycles of 95°C for 30 sec, 49°C for 30 sec, and 68°C for 5 min. DNA amplification was assessed by agarose gel electrophoresis. Amplification products were taken as verification that the contigs from which the two primers in a reaction were selected were colocalized in the genome of C. hominis.
Genome annotations of the three plasmodia, P. falciparum, P. knowlesi, and P. vivax, were retrieved from the release 5.5 of the PlasmoDB database [30]. Orthologs with C. parvum were identified using OrthoMCL [31]. The enrichment of each chromosome of the three plasmodia genomes in orthologs identified in given chromosomes of C. parvum was statistically evaluated using Fisher's exact test. The ordering of the orthologs within chromosomes was evaluated for each pair of species by drawing dot plots.
4.2. Gene Complement Comparison
Gene content comparison was performed by identifying putative orthologs between the two parasites, as well as genes in each organism having no counterpart in the other one. Ortholog and paralog identification was performed using InParanoid [32], a tool that performed an automated bidirectional BLAST search [11] involving two proteomes. Genes found to be apparently unique in each of the two parasites were manually examined to identify and remove artifacts due to remaining gaps in the two sequences. Putative ortholog and paralog assignment involving all Apicomplexa were performed using OrthoMCL [31], and the results were further filtered and summarized by locally developed software tools.
4.3. Pathway Comparison
Comparison of the metabolic pathways in C. hominis and C. parvum was performed in two steps. First, the two genomes were annotated for the presence of the pathways defined in the KEGG [33] and BioCarta (http://www.biocarta.com/) databases by sequence similarity, as previously described [9]. To ensure results were completely comparable between the two species, we reannotated the proteins from C. parvum using the same methods employed for C. hominis. Second, the potential impairments of each pathway for each organism were determined by calculation of a completeness and a connectedness score [13, 14]. Each pathway putatively present in the query organism was compared to a canonical pathway that includes all the enzymatic reactions known to be present in any of the organisms available in KEGG for this pathway. Completeness is defined as the fraction of reactions present in the query organism when compared to the reactions of the canonical pathway. Connectedness is the fraction of intact paths between ports (input and output metabolites for a pathway) in the pathway according to the inferred set of reactions when compared to all the possible paths between the canonical ports in that pathway. Ports are defined as metabolites exchanged between the considered pathway and at least one other pathway; that is, a port is a metabolite that is used either as reactant or product in at least two pathways. To this list of ports, we added metabolites produced but never consumed by any known reaction, as well as metabolites consumed but never produced by any known reaction. These were considered artifacts due to either a missing reaction or the presence of a transporter. In both cases, these metabolites are eligible to be considered as ports for the pathways to which they belong. Finally, we defined the support score as the number of species in KEGG that contains information about the pathway being evaluated.
4.4. Selective Pressure Analysis
For the identification of genes under positive, negative, or neutral selective pressure, pairs of orthologs were aligned globally using the Smith-Waterman algorithm implemented in the FASTA package [34]. Nonsynonymous to synonymous substitution ratios (dN/dS ratio) were calculated using the Yang and Nielson method [35] as implemented in the PAML package [36]. A software pipeline, MUTATION HUNTER, available at http://github.com/ajmazurie/MutationsHunter, was written to automate this task. Automated annotation of the protein sequences was performed on C. hominis sequences to predict the presence of transmembrane domains using tmhmm 2.0 [37], glycosylphosphatidylinositol anchors using GPI-SOM [38], and signal sequences and peptides using SIGNALP 3.0 [39].
Supplementary Material
Supplementary Figure 1: Synteny of Plasmodia and Cryptosporidia genus Position of the orthologs identified in the genomes of C. parvum, P. falciparum, P. knowlesi and P. vivax. For each pair of species, a green cross denotes an ortholog present in the same strand in both genomes, while a red cross denotes an inversion.
Supplementary Figure 2: Genome Compactness Comparison of the genome compactness of well-studied apicomplexans, as the average number of putative paralogs per genes.
Supplementary Figure 3: Pathway Scores See Figure 1.
Supplementary Table 1: Chromosomes Composition Results of the statistical evaluation performed using the Fisher's exact test of the enrichment of the Plasmodia chromosomes in orthologs identified in C. parvum. Values of enrichment are given as the log2 of the ratio between the fraction of orthologs in a given Plasmodium chromosome coming from a same chromosome of C. parvum, and the fraction of orthologs in the whole Plasmodium genome coming from this C. parvum chromosome. A value of 0 (ratio of 1) means that the Plasmodium chromosome contains proportionally as many orthologs from the C. parvum chromosome considered as in the whole genome. The values obtained show that the Plasmodium chromosomes significantly retain the composition of the C. parvum chromosomes. P-values below 0.05 were discarded.
Supplementary Table 2: Pathway Scores Completeness, connectedness and support scores of the inferred metabolic pathways of C. hominis and C. parvum, along with nine other apicomplexans and an external reference, S. cerevisiae (see text).
Supplementary Table 3: Orthologs Sequence Comparison Results of the pairs of putative orthologs sequences comparisons. Identity: percentage of identity between the two protein sequences. dN/dS: non-synonymous on synonymous substitution rate ratio, or None if undefined (when dS equals to zero). Ti/Tv: transition on transversion ratio.
Supplementary Table 4: Protein Localizations Predicted protein localization, based on the putative presence of transmembrane domains, GPI anchor and/or signal peptides (see text). For each pair of orthologs, the electronic annotation was done on the C. hominis sequence (identifier in the first column). P(signal peptide) and P(signal anchor): probability of a signal peptide and signal anchor as calculated by SignalP. # GPI anchors: putative number of GPI anchors as predicted by gpi-som. # TM domains: putative number of transmembrane helices as predicted by tmhmm.
Acknowledgments
This work was supported by Grants NIH U01 AI046418, NIH R01 AI050425, NIH RO1 AI50196, NIH U34 AI57168, and NIH 1R01 AI55347 from the National Institutes of Health (Buck, PI). The authors also wish to thank all members of the Buck Laboratory for helpful discussions and input. Oligonucleotide synthesis, sequencing, and qRT PCR analyses were performed in the Nucleic Acids Research Facilities at VCU.
References
- 1.Fayer R. Cryptosporidium: a water-borne zoonotic parasite. Veterinary Parasitology. 2004;126(1-2):37–56. doi: 10.1016/j.vetpar.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Thompson RCA, Palmer CS, O’Handley R. The public health and clinical significance of giardia and cryptosporidium in domestic animals. The Veterinary Journal. 2008;177(1):18–25. doi: 10.1016/j.tvjl.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leoni F, Amar C, Nichols G, Pedraza Díaz S, McLauchlin J. Genetic analysis of Cryptosporidium from 2414 humans with diarrhoea in England between 1985 and 2000. Journal of Medical Microbiology. 2006;55, part 6:703–707. doi: 10.1099/jmm.0.46251-0. [DOI] [PubMed] [Google Scholar]
- 4.Peng MM, Xiao L, Freeman AR, et al. Genetic polymorphism among Cryptosporidium parvum isolates: evidence of two distinct human transmission cycles. Emerging Infectious Diseases. 1997;3(4):567–573. doi: 10.3201/eid0304.970423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunter PR, Thompson RCA. The zoonotic transmission of Giardia and Cryptosporidium . International Journal for Parasitology. 2005;35(11-12):1181–1190. doi: 10.1016/j.ijpara.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Morgan-Ryan UM, Fall A, Ward LA, et al. Cryptosporidium hominis n. sp. (Apicomplexa: Cryptosporidiidae) from Homo sapiens . Journal of Eukaryotic Microbiology. 2002;49(6):433–440. doi: 10.1111/j.1550-7408.2002.tb00224.x. [DOI] [PubMed] [Google Scholar]
- 7.Hashim A, Mulcahy G, Bourke B, Clyne M. Interaction of Cryptosporidium hominis and Cryptosporidium parvum with primary human and bovine intestinal cells. Infection and Immunity. 2006;74(1):99–107. doi: 10.1128/IAI.74.1.99-107.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao L, Fayer R, Ryan U, Upton SJ. Cryptosporidium taxonomy: recent advances and implications for public health. Clinical Microbiology Reviews. 2004;17(1):72–97. doi: 10.1128/CMR.17.1.72-97.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu P, Widmer G, Wang Y, et al. The genome of Cryptosporidium hominis . Nature. 2004;431(7012):1107–1112. doi: 10.1038/nature02977. [DOI] [PubMed] [Google Scholar]
- 10.Abrahamsen MS, Templeton TJ, Enomoto S, et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum . Science. 2004;304(5669):441–445. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- 11.Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pain A, Renauld H, Berriman M, et al. Genome of the host-cell transforming parasite Theileria annulata compared with T. parva . Science. 2005;309(5731):131–133. doi: 10.1126/science.1110418. [DOI] [PubMed] [Google Scholar]
- 13.Arasappan D, Mazurie A, Alves J, Bonchev D, Buck GA. Path-a-way: a strategy for network analysis of microarray data. Proceedings of the BioMedical Engineering and Informatics: New Development and the Future (BMEI '08); May 2008; pp. 432–436. [Google Scholar]
- 14.Buck G, Mazurie A, Roberts S, et al. Genomes to Networks, Pathways and Function. Vol. 7. Brill Academic; 2006. Recent progress in computational sciences and engineering; pp. 758–763. (Lecture Series on Computer and Computational Sciences). [Google Scholar]
- 15.Gardner MJ, Bishop R, Shah T, et al. Genome sequence of Theileria parva, a bovine pathogen that transforms lymphocytes. Science. 2005;309(5731):134–137. doi: 10.1126/science.1110439. [DOI] [PubMed] [Google Scholar]
- 16.Zhu G, Li Y, Cai X, Millership JJ, Marchewka MJ, Keithly JS. Expression and functional characterization of a giant Type I fatty acid synthase (CpFAS1) gene from Cryptosporidium parvum . Molecular and Biochemical Parasitology. 2004;134(1):127–135. doi: 10.1016/j.molbiopara.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 17.Puiu D, Enomoto S, Buck GA, Abrahamsen MS, Kissinger JC. CryptoDB: the Cryptosporidium genome resource. Nucleic Acids Research. 2004;32(supplement 1):D329–D331. doi: 10.1093/nar/gkh050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aston C, Mishra B, Schwartz DC. Optical mapping and its potential for large-scale sequencing projects. Trends in Biotechnology. 1999;17(7):297–302. doi: 10.1016/s0167-7799(99)01326-8. [DOI] [PubMed] [Google Scholar]
- 19.Zhou S, Deng W, Anantharaman TS, et al. A whole-genome shotgun optical map of Yersinia pestis strain KIM. Applied and Environmental Microbiology. 2002;68(12):6321–6331. doi: 10.1128/AEM.68.12.6321-6331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou S, Kile A, Bechner M, et al. Single-molecule approach to bacterial genomic comparisons via optical mapping. Journal of Bacteriology. 2004;186(22):7773–7782. doi: 10.1128/JB.186.22.7773-7782.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou S, Kile A, Kvikstad E, et al. Shotgun optical mapping of the entire Leishmania major Friedlin genome. Molecular and Biochemical Parasitology. 2004;138(1):97–106. doi: 10.1016/j.molbiopara.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 22.Dimalanta ET, Lim A, Runnheim R, et al. A microfluidic system for large DNA molecule arrays. Analytical Chemistry. 2004;76(18):5293–5301. doi: 10.1021/ac0496401. [DOI] [PubMed] [Google Scholar]
- 23.Reslewic S, Zhou S, Place M, et al. Whole-genome shotgun optical mapping of Rhodospirillum rubrum. Applied and Environmental Microbiology. 2005;71(9):5511–5522. doi: 10.1128/AEM.71.9.5511-5522.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valouev B, Schwartz DC, Zhou S, Waterman MS. An algorithm for assembly of ordered restriction maps from single dna molecules. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(43):15770–15775. doi: 10.1073/pnas.0604040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou S, Bechner MC, Place M, et al. Validation of rice genome sequence by optical mapping. BMC Genomics. 2007;8, article 278 doi: 10.1186/1471-2164-8-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou S, Herschleb J, Schwartz D. New High Throughput Technologies for DNA Sequencing and Genomics. Vol. 2. Elsevier Scientific; 2007. A single molecule system for whole genome analysis; pp. 265–300. [Google Scholar]
- 27.Anantharaman TS, Mishra B, Schwartz DC. Genomics via optical mapping II: ordered restriction maps. Journal of Computational Biology. 1997;4(2):91–118. doi: 10.1089/cmb.1997.4.91. [DOI] [PubMed] [Google Scholar]
- 28.Anantharaman T, Mishra B, Schwartz D. Genomics via optical mapping. III: contiging genomic DNA. Proceedings of the International Conference on Intelligent Systems for Molecular Biology; 1999; pp. 18–27. [PubMed] [Google Scholar]
- 29.Valouev A, Li L, Liu Y-C, et al. Alignment of optical maps. Journal of Computational Biology. 2006;13(2):442–462. doi: 10.1089/cmb.2006.13.442. [DOI] [PubMed] [Google Scholar]
- 30.Aurrecoechea C, Brestelli J, Brunk BP, et al. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Research. 2009;37(1):D539–D543. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Research. 2003;13(9):2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Remm M, Storm CE, Sonnhammer EL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. Journal of Molecular Biology. 2001;314(5):1041–1052. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- 33.Kanehisa M, Goto S, Hattori M, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Research. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(8):2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Molecular Biology and Evolution. 2000;17(1):32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- 36.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Computer Applications in the Biosciences. 1997;13(5):555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 37.Krogh A, Larsson B, Von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. Journal of Molecular Biology. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 38.Fankhauser N, Mäser P. Identification of GPI anchor attachment signals by a Kohonen self-organizing map. Bioinformatics. 2005;21(9):1846–1852. doi: 10.1093/bioinformatics/bti299. [DOI] [PubMed] [Google Scholar]
- 39.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: signalP 3.0. Journal of Molecular Biology. 2004;340(4):783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Synteny of Plasmodia and Cryptosporidia genus Position of the orthologs identified in the genomes of C. parvum, P. falciparum, P. knowlesi and P. vivax. For each pair of species, a green cross denotes an ortholog present in the same strand in both genomes, while a red cross denotes an inversion.
Supplementary Figure 2: Genome Compactness Comparison of the genome compactness of well-studied apicomplexans, as the average number of putative paralogs per genes.
Supplementary Figure 3: Pathway Scores See Figure 1.
Supplementary Table 1: Chromosomes Composition Results of the statistical evaluation performed using the Fisher's exact test of the enrichment of the Plasmodia chromosomes in orthologs identified in C. parvum. Values of enrichment are given as the log2 of the ratio between the fraction of orthologs in a given Plasmodium chromosome coming from a same chromosome of C. parvum, and the fraction of orthologs in the whole Plasmodium genome coming from this C. parvum chromosome. A value of 0 (ratio of 1) means that the Plasmodium chromosome contains proportionally as many orthologs from the C. parvum chromosome considered as in the whole genome. The values obtained show that the Plasmodium chromosomes significantly retain the composition of the C. parvum chromosomes. P-values below 0.05 were discarded.
Supplementary Table 2: Pathway Scores Completeness, connectedness and support scores of the inferred metabolic pathways of C. hominis and C. parvum, along with nine other apicomplexans and an external reference, S. cerevisiae (see text).
Supplementary Table 3: Orthologs Sequence Comparison Results of the pairs of putative orthologs sequences comparisons. Identity: percentage of identity between the two protein sequences. dN/dS: non-synonymous on synonymous substitution rate ratio, or None if undefined (when dS equals to zero). Ti/Tv: transition on transversion ratio.
Supplementary Table 4: Protein Localizations Predicted protein localization, based on the putative presence of transmembrane domains, GPI anchor and/or signal peptides (see text). For each pair of orthologs, the electronic annotation was done on the C. hominis sequence (identifier in the first column). P(signal peptide) and P(signal anchor): probability of a signal peptide and signal anchor as calculated by SignalP. # GPI anchors: putative number of GPI anchors as predicted by gpi-som. # TM domains: putative number of transmembrane helices as predicted by tmhmm.