

Abstract
This study focused on the enzymatic biotransformation of the major ginsenoside Rb1 into Rd for the mass production of minor ginsenosides using a novel recombinant β-glucosidase from Flavobacterium johnsoniae. The gene (bglF3) consisting of 2,235 bp (744 amino acid residues) was cloned and the recombinant enzyme overexpressed in Escherichia coli BL21(DE3) was characterized. This enzyme could transform ginsenoside Rb1 and gypenoside XVII to the ginsenosides Rd and F2, respectively. The glutathione S-transferase (GST) fused BglF3 was purified with GST-bind agarose resin and characterized. The kinetic parameters for β-glucosidase had apparent Km values of 0.91±0.02 and 2.84±0.05 mM and Vmax values of 5.75±0.12 and 0.71±0.01 μmol·min-1·mg of protein-1 against p-nitrophenyl-β-D-glucopyranoside and Rb1, respectively. At optimal conditions of pH 6.0 and 37℃, BglF3 could only hydrolyze the outer glucose moiety of ginsenoside Rb1 and gypenoside XVII at the C-20 position of aglycon into ginsenosides Rd and F2, respectively. These results indicate that the recombinant BglF3 could be useful for the mass production of ginsenosides Rd and F2 in the pharmaceutical or cosmetic industry.
Keywords: Panax ginseng, Biotransformation, β-glucosidase, Ginsenoside F2, Flavobacterium johnsoniae
INTRODUCTION
Ginseng (Panax ginseng Meyer) has been used as a celebrated traditional herbal medicine to cure diseases and promote health in the Orient for thousands of years [1,2]. Ginsenosides can be categorized into the tetracyclic triterpenoid saponins, including protopanaxadiol (PPD) and protopanaxatriol, and the pentacyclic triterpenoid saponins. The PPD-type ginsenosides are further classified by the position and number of sugar moieties attached by a glycosidic bond to the aglycon at positions C-3 and C-20 [3]. After oral intake of ginseng, the major ginsenosides are hydrolyzed through human intestinal digestion into the more active minor ginsenosides, which are easily absorbed. For instance, ginsenoside Rb1 is converted to ginsenosides Rd, F2, compound K, and aglycon by intestinal microflora [4-6]. Therefore , converting the major ginsenosides, which account for more than 80% of the total ginsenosides, to highly active minor ginsenosides has much significance for the pharmaceutical industry.
Ginsenosides, the major active constituents of ginseng, have various biological and pharmacological activities, including anti-cancer effects [7], anti-inflammatory activity [8], and neuro-protective effects [9]. Ginsenosides Rd and F2 have several pharmaceutical functions such as anti-tumor and anti-cancer effects, curing atherosclerosis, neuro-protective effects [10-13], and so on. The minor ginsenosides can be produced by hydrolyzing sugar moieties from the major ginsenosides [14]. To date, several methods to produce pure ginsenosides such as heating, acid treatment, and enzymatic methods have been developed. The enzymatic methods are considered as the most promising approach with advantages of fewer byproducts, superior environmental protection, and better stereo-specificity [15]. In particular, the purified recombinant enzymes exhibit higher selectivity and efficiency than those isolated and purified from cultured microorganisms [16].
In this study, we report the cloning of a new gene encoding ginsenoside-hydrolyzing β-glucosidase (BglF3) from Flavobacterium johnsoniae, followed by expression in Escherichia coli and characterization of β-glucosidase (BglF3). BglF3 belongs to glycoside hydrolase family 3, and the recombinant enzyme hydrolyzed only the outer glucose moiety at the C-20 position of ginsenoside Rb1 and gypenoside XVII, which are effectively converted to ginsenosides Rd and F2, respectively.
MATERIALS AND METHODS
Chemicals
Ginsenosides Rb1, Rb2, Rc, Rd, Re, Rg1, Rg3(S), F2, and C-K were purchased from Nanjing Zelang Medical Technology Co., Ltd. (Nanjing, China) and gypenoside XVII was obtained by our group as described by An et al. [17]. All the chemic als used in this study were at least analytical reagent grade, and the sources are noted individually in the methods section.
Bacterial strains, vectors, and media
F. johnsoniae KACC 11414T, used as a source for cloning of β-glucosidase gene, was cultured on R2A agar (BD, Sparks, MD, USA) under an aerobic condition at 30℃. E. coli BL21 (DE3) and pGEX 4T-1 plasmid (GE Healthcare, Waukesha, WI, USA) for gene cloning and expression were cultured in a Luria–Bertani (LB) medium with ampicillin (100 mg/L).
Cloning, expression, and purification of recombinant BglF3
Genomic DNA of F. johnsoniae KACC 11414T was extracted by using a genomic DNA extraction kit (Solgent, Daejeon, Korea). The gene, termed bglF3, encoding β-glucosidase (GenBank accession number ABQ03809) was amplified by polymerase chain reaction (PCR) with Pfu DNA polymerase (Solgent) using the following primers (with BamHI and XhoI restriction sites in boldface): bglF3F (5’-CGGGATCCAAAAACAAAATGATATACCTTTCTGC- 3’) and bglF3R (5’-CCCCTCGAGTTATTTAATTGTGAAGTTAATTTCC- 3’). The amplified fragment was digested with BamHI and XhoI and then inserted to the pGEX 4T-1 vector to generate a glutathione S-transferase (GST)-bglF3 gene fusion. E. coli BL21 (DE3), transformed with recombinant pGEX-bglF3, was grown in LB-ampicillin medium at 37℃ until the culture reached an OD600 of 0.6, at which point protein expression was induced by adding 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG). Bacteria were incubated additionally for 24 h at 18℃ and then harvested by centrifuging at 10,000× rpm for 20 min at 4℃. The cells were washed twice with 50 mM sodium phosphate buffer (pH 7.0, 5 mM ethylenediaminetetraacetic acid [EDTA], and 1% Triton X-100) and then suspended in 50 mM sodium phosphate buffer (pH 7.0). The cells were disrupted by ultrasonication (Vibracell; Sonics & Materials, Newtown, CT, USA) and the intact cells and debris were removed by centrifugation at 12,000× rpm for 20 min at 4℃. The GST-tagged fusion protein was purified by GST-bind agarose resin (Elpisbiotech Co., Daejeon, Korea). The GST tag was removed from the GST-bind agarose resin after being incubated with thrombin. The homogeneity of the protein was assessed by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and EZ-Gel staining solution (Daeillab Co., Seoul, Korea).
Enzyme characterization and determination of kinetic parameters
The specific activity of purified BglF3 was determined by using p-nitrophenyl-β-D-glucopyranoside (PNPG) as substrate in 50 mM sodium phosphate buffer (pH 6.0) at 37℃. The release of p-nitrophenol was immediately measured using a microplate reader at 405 nm (Model 680; Bio-Rad, Hercules, CA, USA). One unit of activity was defined as the amount of protein required to generate 1 μmol of p-nitrophenol per minute. To check the optimum condition for the specific enzyme activity, pH, temperature, metals and chemical reagents were investigated as previously described [17]. Substrate preference was examined by using chromogenic o-nitrophenyl (ONP) and p-nitrophenyl (PNP). Kinetic studies were performed with freshly purified enzyme using PNPG and Rb1 at concentrations ranging from 0.1 mM to 5 mM. All enzyme assays were performed with triplicate. The parameters (Km and Vmax) were determined as described by Wang et al. [3].
Enzymatic hydrolysis of ginsenosides
To investigate the biotransformation ability of recombinant β-glucosidase, BglF3, 9 different ginsenosides (Rb1, Rb2, Rc, Rd, Gypenoside XVII, Rg3(S), Re, Rg1, and F2) were evaluated as substrates. The initial biotransformation experiments using ginsenoside Rb1 as the substrate revealed that GST fused with BglF3 did not affect the activiti es of BglF3. Each ginsenoside was reacted with fused protein solution (0.2 mg/mL in 50 mM sodium phosphate buffer, pH 6.0) with same ratio (1:1, v/ v) at 37℃. Also, the hydrolyzing capacity of BglF3 (0.2 mg/mL) was determined by using 1.0, 2.5, and 5.0 mg/ mL Rb1 as a substrate in 50 mM sodium phosphate buffer (pH 6.0) at 37℃. Samples were withdrawn at regular intervals. An equal volume of water-saturated n-butanol was added to stop the reaction, and the reactant present in the n-butanol fraction was analyzed by TLC after pretreatment.
Analysis of ginsenosides by thin-layer chromatography
TLC was performed using 60F254 silica gel plates (Merck, Darmstadt, Germany) with CHCl3-CH3OH-H2O (65:35:10, v/v, lower phase) in solvent system. The spots on the TLC plates were detected through spraying with 10% (vol/vol) H2SO4, followed by heating at 110℃ for 5 min.
RESULTS AND DISCUSSION
Cloning, expression, and purification of recombinant BglF3
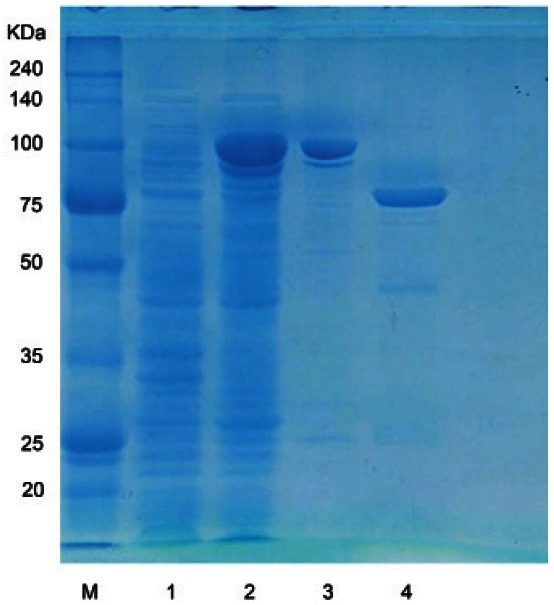
The β-glucosidase gene consisting of 2, 235 bp encoding 744 amino acids, which have homology to the protein domain of glycoside hydrolase family 3, was amplified by PCR and then inserted into the pGEX 4T-1 vector. The GST-BglF3 fusion gene was expressed in E. coli BL21 (DE3) followed by the induction of 0.1 mM IPTG and incubated at 18℃ for 24 h. The GSTBglF3 fusion protein was purified by GST-bind agarose resin and then the GST tag was removed by thrombin at room temperature during a 12 h incubation period. The predictive molecular mass (81.8 KDa) of the BglF3 was determined by SDS-PAGE (Fig. 1).
Fig. 1. Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis of purified BglF3. 1, uninduced crude extract; 2, induced crude extract; 3, glutathione S-transferase (GST)-BglF3 enzyme fraction after purification by GST-bind agarose resin; 4, cleavage of GSTBglF3 by thrombin. KDa, kilodalton; M, marker.
Enzyme characterization
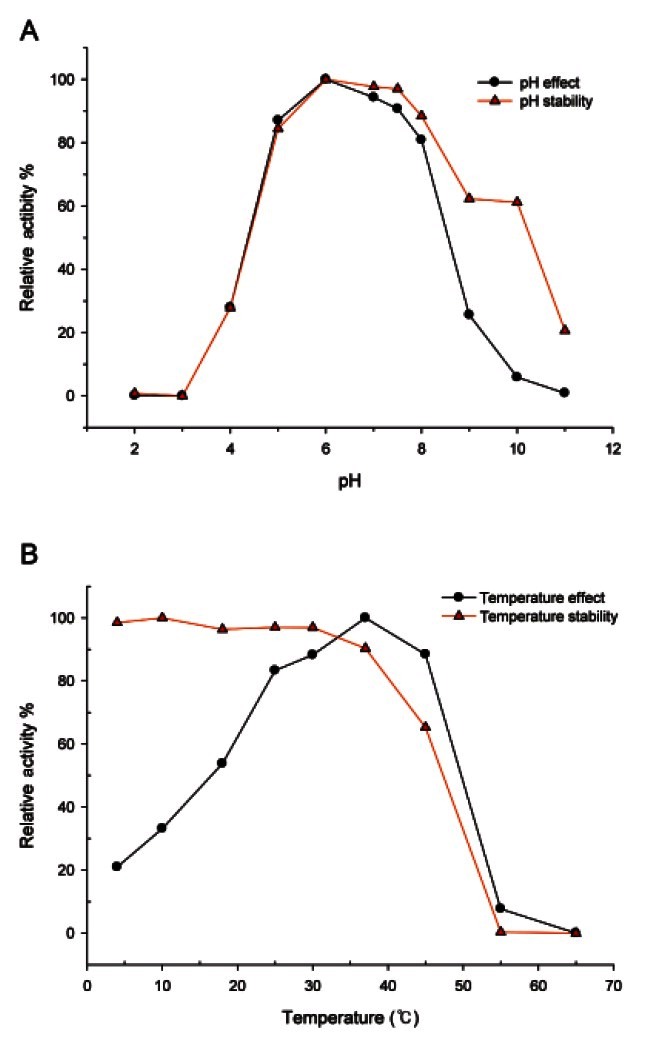
BglF3 was active over a broad pH range (pH 4.0 to 9.0) at 37℃. The optimum pH was pH 6.0 in sodium phosphate buffer (Fig. 2A). The enzyme activity retained more than 80% of its optimal activity from pH 5.0 to 8.0, while above pH 10.0 enzyme activity decreased upto 95% and at pH 4.0 the enzyme activity decreased to 30%. The β-glucosidase from Paecilomyces Bainier sp. 229 [18], Thermus caldophilus [19], and Microbacterium esteraromaticum [20] had optima at pH 3.5, 5.0, and 7.0, respectively. The optimal temperature for BglF3 activity was 37℃ and the enzyme was stable at lower than 37℃. The enzyme lost 35% of its activity at 45℃, while no thermostability was determined at 55℃ (Fig. 2B). However, the purified β-glucosidase from Paecilomys Bainier sp. 229 [18], T. caldophilus [19], and Sulfolobus solfataricus [21] had the optimal temperatures of 55℃, 75℃, and 90℃, respectively.
Fig. 2. Effects of pH (A) and temperature (B) on the stability and activity of BglF3.
The effects of metal ions, EDTA, β-mercaptoethanol, and sodium dodecyl-sulfate ( SDS) on BglF3 activity were investigated (Table 1). BglF3 activity was not affected by dithiothreitol or β-mercaptoethanol, which is a well-known thiol group inhibitor. Na+, K+, or Mg2+ had positive effects on the activity of the enzyme while the enzyme activity was inhibited in the presence of both 1 mM and 10 mM Mn2+, Co2+, Zn2+, Ca2+, Cu2+, or Hg2+. The chelating agents EDTA and SDS also inhibited BglF3 activity.
Table 1.
Effects of metal ions and chemical agents on the activity of purified BglF3
| Metal ions or reagents | Relative activity±SD (%) | |
|---|---|---|
|
| ||
| 1 mM | 10 mM | |
|
| ||
| NaCl | 143.9±1.6 | 108.2±1.5 |
| KCl | 128.2±3.5 | 110.2±1.6 |
| MgCl2 | 152.6±3.3 | 112.5±3.4 |
| MnCl2 | 53.3±1.0 | 39.7±2.4 |
| CoCl2 | 58.8±3.5 | 44.3±2.7 |
| ZnCl2 | 4.7±1.7 | 2.9±2.5 |
| CaCl2 | 92.5±2.6 | 81.0±1.9 |
| CuCl2 | 6.3±1.4 | 6.0±2.0 |
| HgCl2 | 0 | 0 |
| SDS | 39.9±3.2 | 1.98±1.7 |
| EDTA | 76.4±2.7 | 68.9±3.7 |
| β-mercaptoethanol | 93.8±1.7 | 91.9±1.8 |
| DTT | 96.0±4.1 | 97.9±3.4 |
| Control | 100.0±2.6 | 100.0±1.5 |
SDS, sodium dodecyl-sulfate; EDTA, ethylenediaminetetraacetic acid; DTT, dithiothreitol.
The substrate specificity of BglF3 was tested using 2.0 mM of PNP and ONP-glycosides with α and β configurations (total 17 kinds), as reported previously [7,17]. BglF3 was maximally active against PNP-β-Dglucopyranoside, followed by ONP-β-D-glucopyranoside (53.2% compared to PNP-β-D-glucopyranoside), and other substrates were not hydrolyzed. This explained why BglF3 did not have catalytic activity against ginsenoside Rb2 and Rc, which had an outer arabinose moiety at the C-20 position of aglycon. The kinetic parameters for recombinant BglF3 had apparent Km values of 0.91±0.02 and 2.84±0.05 mM and Vmax values of 5.75±0.12 and 0.71±0.01 μmol·min-1·mg of protein-1 against p-nitrophenyl-β-D-glucopyranoside and Rb1, respectively.
Biotransformation of ginsenosides
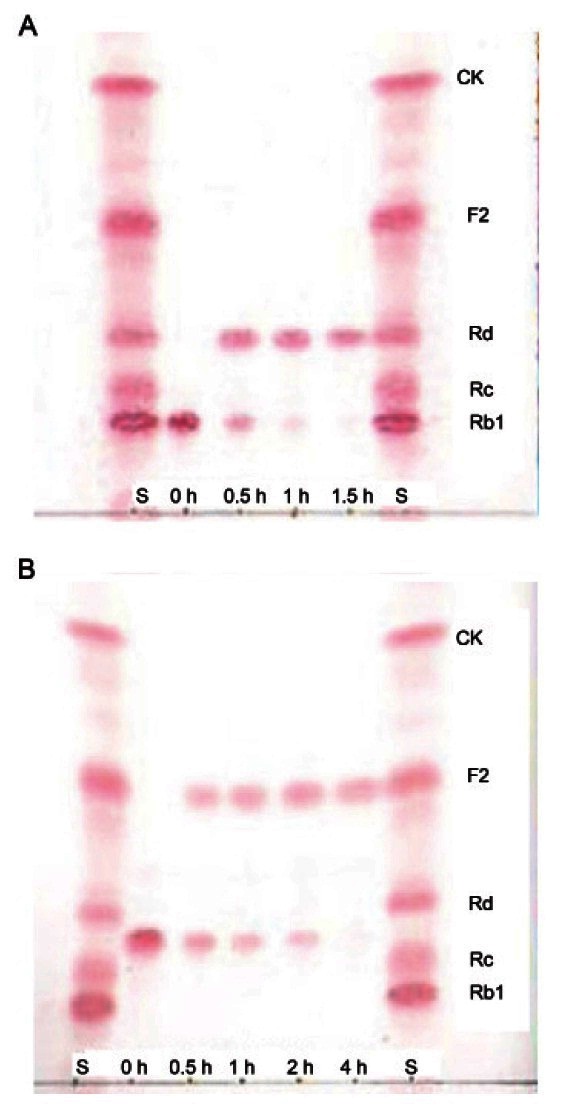
For a thorough investigation of ginsenoside transformation by recombinant BglF3, 9 ginsenosides (Rb1, Rb2, Rc, Rd, gypenoside X VII, Rg3(S), Re, Rg1, and F2) were used as substrates and recombinant BglF3 could hydrolyze only ginsenoside Rb1 and gypenoside XVII. The hydrolysates of ginsenoside Rb1 and gypenoside XVII were examined at regular intervals by TLC. Ginsenoside Rb1 (1 mg/mL) and gypenoside XVII (1 mg/mL) were completely converted into ginsenosides Rd and F2 after 1.5 h and 4 h, respectively, and no further production occurred during longer reaction time (Fig. 3). This conversion speed was faster than that obtained with the usage of crude enzymes extracts from Paecilomyces sp. 229-7 [22] and Bifidobacterium sp. SH5 [23], which converted 1 mg/mL of ginsenoside Rb1 to Rd within 24 h.
Fig. 3. TLC analyses of biotransformation of ginsenoside Rb1 (A) and gypenoside XVII (B) by recombinant BglF3. S, saponin standards; CK, compound K.
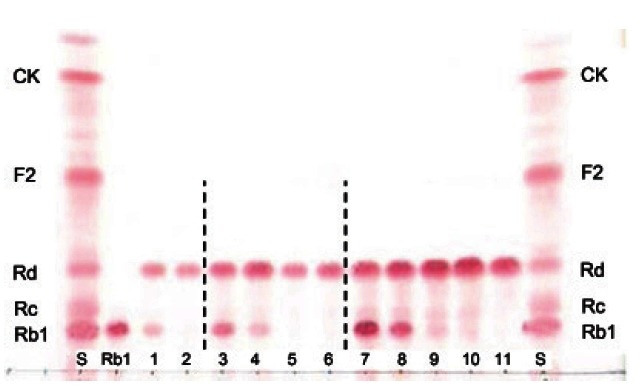
These results indicated that BglF3 showed substrate specificity for ginsenoside Rb1 and gypenoside XVII, which had glucose moieties at the C-3 and C-20 positions, and showed specific affinity to only the outer C-20 glucose moiety (Fig. 4). To evaluate the usage of BglF3 at an industrial scale, a high concentration of ginsenosides Rb1 was used as the substrate. BglF3 could wholly hydrolyze 2.5 and 5.0 mg/mL of the ginsenosides Rb1 into Rd within 20 and 72 h, respectively (Fig. 5).
Fig. 4. Biotransformation pathways from ginsenoside Rb1 and gypenoside XVII to ginsenosides Rd and F2 by recombinant BglF3, respectively.
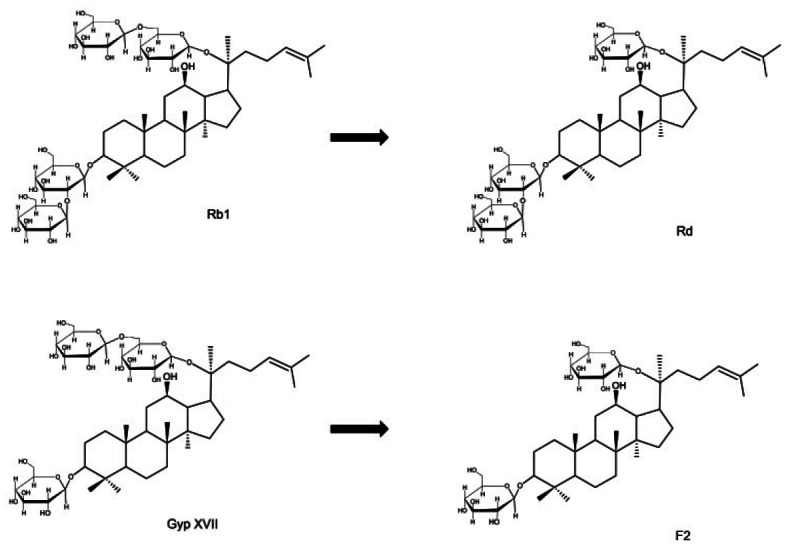
Fig. 5. TLC analyses of ginsenoside Rd production from 1.0, 2.5, and 5.0 mg/mL ginsenoside Rb1 by recombinant BglF3. Reaction mixture of 1.0 mg/mL Rb1: 1, 2; reaction mixture of 2.5 mg/mL Rb1: 3, 4, 5, 6; reaction mixture of 5.0 mg/mL Rb1: 7, 8, 9, 10, 11; reaction time: 1, 0.5 h; 2, 1.5 h; 3, 1 h; 4, 3 h; 5, 12 h; 6, 20 h; 7, 1 h; 8, 3 h; 9, 20 h; 10, 48 h; 11, 72 h. S, saponin standards; CK, compound K.
Thus, it would be a very practical tool to prepare a large amount of ginsenoside Rd with high-yield if a purified ginsenoside mixture (Rb1, Rc, and Rd) is used as the substrate.
Theoretically, β-glucosidase can hydrolyze the outer and inner glucoses attached to ginsenosides at the C-3 and C-20 positions in various transformation pathways. With regard to glycoside hydrolase family 3, where BglF3 is included, there have been several reports on ginsenoside conversion. For example, Bgp A derived from Terrabacter ginsenosidimutans Gsoil 3082T preferentially hydrolyzed the outer glucose moiety at the C-3 position followed by the inner glucose moiety to make gypenoside XVII and gypenoside LXXV and further into compound K by hydrolyzing the C-20 glucose moiety [17]. Bgp1 derived from M. esteraromaticum preferentially hydrolyzed both the outer and inner glucose moieties at the C-20 position until ginsenoside Rg3 was produced finally [24]. The other enzyme, β-glucosidase from Cladosporium cladosporioides, converted gypenoside XVII into ginsenoside F2 by hydrolyzing the outer glucose at the C-20 position and further into compound K by hydrolyzing the C-3 glucose moiety [25]. However, in this study, BglF3 could selectively hydrolyze only one outer glucose at the C-20 position for the major ginsenoside Rb1 and gypenoside XVII.
In conclusion, we have constructed recombinant β-glucosidase (BglF3) from F. johnsoniae KACC 11414T for biotransformation of ginsenoside Rb1 and gypenoside XVII to more pharmacologically active ginsenosides Rd and F2, respectively, by selectively hydrolyzing their outer glucose moiety at the C-20 position. Optimal reaction conditions for the enzyme are 37℃ and pH 6.0. The recombinant BglF3 provides effective ginsenoside Rb1 transformation with high productivity. Therefore, BglF3 would offer a shortcut to prepare ginsenosides Rd and F2 in large-scale industrial production.
Acknowledgments
This work was supported by a grant from the Next- Generation BioGreen 21 Program (no. PJ008193), Rural Development Administration, and by the Intelligent Synthetic Biology Center of Global Frontier Project funded by the Ministry of Education, Science and Technology (2011-0031967), Republic of Korea.
References
- 1.Ernst E. Panax ginseng: An overview of the clinical evidence. J Ginseng Res. 2010;34:259–263. doi: 10.5142/jgr.2010.34.4.259. [DOI] [Google Scholar]
- 2.Kim MH, Lee YC, Choi SY, Cho CW, Rho J, Lee KW. The changes of ginsenoside patterns in red ginseng processed by organic acid impregnation pretreatment. J Ginseng Res. 2011;35:497–503. doi: 10.5142/jgr.2011.35.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang L, Liu QM, Sung BH, An DS, Lee HG, Kim SG, Kim SC, Lee ST, Im WT. Bioconversion of ginsenosides Rb1, Rb2, Rc and Rd by novel β-glucosidase hydrolyzing outer 3-O glycoside from Sphingomonas sp. 2F2: cloning, expression, and enzyme characterization. J Biotechnol. 2011;156:125–133. doi: 10.1016/j.jbiotec.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 4.Wang DM, Yu HS, Song JG, Xu YF, Jin FX. Enzyme kinetics of ginsenosidase type IV hydrolyzing 6-O-multiglycosides of protopanaxatriol type ginsenosides. Process Biochem. 2012;47:133–138. [Google Scholar]
- 5.Tawab MA, Bahr U, Karas M, Wurglics M, Schubert-Zsilavecz M. Degradation of ginsenosides in humans after oral administration. Drug Metab Dispos. 2003;31:1065–1071. doi: 10.1124/dmd.31.8.1065. [DOI] [PubMed] [Google Scholar]
- 6.Akao T, Kida H, Kanaoka M, Hattori M, Kobashi K. Intestinal bacterial hydrolysis is required for the appearance of compound K in rat plasma after oral administration of ginsenoside Rb1 from Panax ginseng. J Pharm Pharmacol. 1998;50:1155–1160. doi: 10.1111/j.2042-7158.1998.tb03327.x. [DOI] [PubMed] [Google Scholar]
- 7.Kim HS, Lee EH, Ko SR, Choi KJ, Park JH, Im DS. Effects of ginsenosides Rg3 and Rh2 on the proliferation of prostate cancer cells. Arch Pharm Res. 2004;27:429–435. doi: 10.1007/BF02980085. [DOI] [PubMed] [Google Scholar]
- 8.Keum YS, Han SS, Chun KS, Park KK, Park JH, Lee SK, Surh YJ. Inhibitory effects of the ginsenoside Rg3 on phorbol ester-induced cyclooxygenase-2 expression, NF-kappaB activation and tumor promotion. Mutat Res. 2003;523-524:75–85. doi: 10.1016/s0027-5107(02)00323-8. [DOI] [PubMed] [Google Scholar]
- 9.Kim S, Nah SY, Rhim H. Neuroprotective effects of ginseng saponins against L-type Ca2+ channel-mediated cell death in rat cortical neurons. Biochem Biophys Res Commun. 2008;365:399–405. doi: 10.1016/j.bbrc.2007.10.048. [DOI] [PubMed] [Google Scholar]
- 10.Shin JY, Lee JM, Shin HS, Park SY, Yang JE, Cho SK, Yi TH. Anti-cancer effect of ginsenoside F2 against glioblastoma multiforme in xenograft model in SD rats. J Ginseng Res. 2012;36:86–92. doi: 10.5142/jgr.2012.36.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guan YY, Zhou JG, Zhang Z, Wang GL, Cai BX, Hong L, Qiu QY, He H. Ginsenoside-Rd from Panax notoginseng blocks Ca2+ influx through receptor- and store-operated Ca2+ channels in vascular smooth muscle cells. Eur J Pharmacol. 2006;548:129–136. doi: 10.1016/j.ejphar.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Lee JK, Choi SS, Lee HK, Han KJ, Han EJ, Suh HW. Effects of ginsenoside Rd and decursinol on the neurotoxic responses induced by kainic acid in mice. Planta Med. 2003;69:230–234. doi: 10.1055/s-2003-38475. [DOI] [PubMed] [Google Scholar]
- 13.Yang ZG, Sun HX, Ye YP. Ginsenoside Rd from Panax notoginseng is cytotoxic towards HeLa cancer cells and induces apoptosis. Chem Biodivers. 2006;3:187–197. doi: 10.1002/cbdv.200690022. [DOI] [PubMed] [Google Scholar]
- 14.Cheng LQ, Na JR, Kim MK, Bang MH, Yang DC. Microbial conversion of ginsenoside Rb1 to minor ginsenoside F2 and gypenoside XVII by Intrasporangium sp. GS603 isolated from soil. J Microbiol Biotechnol. 2007;17:1937–1943. [PubMed] [Google Scholar]
- 15.Park CS, Yoo MH, Noh KH, Oh DK. Biotransformation of ginsenosides by hydrolyzing the sugar moieties of ginsenosides using microbial glycosidases. Appl Microbiol Biotechnol. 2010;87:9–19. doi: 10.1007/s00253-010-2567-6. [DOI] [PubMed] [Google Scholar]
- 16.Kim BN, Yeom SJ, Kim YS, Oh DK. Characterization of a β-glucosidase from Sulfolobus solfataricus for isoflavone glycosides. Biotechnol Lett. 2012;34:125–129. doi: 10.1007/s10529-011-0739-9. [DOI] [PubMed] [Google Scholar]
- 17.An DS, Cui CH, Lee HG, Wang L, Kim SC, Lee ST, Jin F, Yu H, Chin YW, Lee HK, et al. Identification and characterization of a novel Terrabacter ginsenosidimutans sp. nov. beta-glucosidase that transforms ginsenoside Rb1 into the rare gypenosides XVII and LXXV. Appl Environ Microbiol. 2010;76:5827–5836. doi: 10.1128/AEM.00106-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan Q, Zhou W, Li X, Feng M, Zhou P. Purification method improvement and characterization of a novel ginsenoside-hydrolyzing beta-glucosidase from Paecilomyces Bainier sp. 229. Biosci Biotechnol Biochem. 2008;72:352–359. doi: 10.1271/bbb.70425. [DOI] [PubMed] [Google Scholar]
- 19.Son JW, Kim HJ, Oh DK. Ginsenoside Rd production from the major ginsenoside Rb1 by beta-glucosidase from Thermus caldophilus. Biotechnol Lett. 2008;30:713–716. doi: 10.1007/s10529-007-9590-4. [DOI] [PubMed] [Google Scholar]
- 20.Quan LH, Min JW, Sathiyamoorthy S, Yang DU, Kim YJ, Yang DC. Biotransformation of ginsenosides Re and Rg1 into ginsenosides Rg2 and Rh1 by recombinant β-glucosidase. Biotechnol Lett. 2012;34:913–917. doi: 10.1007/s10529-012-0849-z. [DOI] [PubMed] [Google Scholar]
- 21.Noh KH, Son JW, Kim HJ, Oh DK. Ginsenoside compound K production from ginseng root extract by a thermostable beta-glycosidase from Sulfolobus solfataricus. Biosci Biotechnol Biochem. 2009;73:316–321. doi: 10.1271/bbb.80525. [DOI] [PubMed] [Google Scholar]
- 22.Ye L, Zhou CQ, Zhou W, Zhou P, Chen DF, Liu XH, Shi XL, Feng MQ. Biotransformation of ginsenoside Rb1 to ginsenoside Rd by highly substrate-tolerant Paecilomyces Bainier 229-7. Bioresour Technol. 2010;101:7872–7876. doi: 10.1016/j.biortech.2010.04.102. [DOI] [PubMed] [Google Scholar]
- 23.Chi H, Ji GE. Transformation of ginsenosides Rb1 and Re from Panax ginseng by food microorganisms. Biotechnol Lett. 2005;27:765–771. doi: 10.1007/s10529-005-5632-y. [DOI] [PubMed] [Google Scholar]
- 24.Quan LH, Min JW, Yang DU, Kim YJ, Yang DC. Enzymatic biotransformation of ginsenoside Rb1 to 20(S)-Rg3 by recombinant β-glucosidase from Microbacterium esteraromaticum. Appl Microbiol Biotechnol. 2012;94:377–384. doi: 10.1007/s00253-011-3861-7. [DOI] [PubMed] [Google Scholar]
- 25.Wu L, Jin Y, Yin C, Bai L. Co-transformation of Panax major ginsenosides Rb1 and Rg1 to minor ginsenosides C-K and F1 by Cladosporium cladosporioides. J Ind Microbiol Biotechnol. 2012;39:521–527. doi: 10.1007/s10295-011-1058-9. [DOI] [PubMed] [Google Scholar]