

The aim of this study was to test the effect of a range of medium supplements on mesenchymal stem cell (MSC) self-renewal and differentiation potential. Overall, medium supplementation with fibroblast growth factor-2, ascorbic acid, epidermal growth factor, and platelet-derived growth factor-BB greatly enhanced the total in vitro expansion capacity of MSC cultures, although differentiation potentials were lost prior to reaching senescence despite the persistence of expression of MSC surface markers throughout.
Keywords: Adult stem cells, Differentiation, Cell culture, Self-renewal
Abstract
Mesenchymal stem cells (MSCs) possess great potential for use in regenerative medicine. However, their clinical application may be limited by the ability to expand their cell numbers in vitro while maintaining their differential potentials and stem cell properties. Thus the aim of this study was to test the effect of a range of medium supplements on MSC self-renewal and differentiation potential. Cells were cultured until confluent and subcultured continuously until reaching senescence. Medium supplementation with fibroblast growth factor (FGF)-2, platelet-derived growth factor (PDGF)-BB, ascorbic acid (AA), and epidermal growth factor (EGF) both increased proliferation rate and markedly increased number of cell doublings before reaching senescence, with a greater than 1,000-fold increase in total cell numbers for AA, FGF-2, and PDGF-BB compared with control cultures. Long-term culture was associated with loss of osteogenic/adipocytic differentiation potential, particularly with FGF-2 supplementation but also with AA, EGF, and PDGF-BB. In addition FGF-2 resulted in reduction in expression of CD146 and alkaline phosphatase, but this was partially reversible on removal of the supplement. Cells expressed surface markers including CD146, CD105, CD44, CD90, and CD71 by flow cytometry throughout, and expression of these putative stem cell markers persisted even after loss of differentiation potentials. Overall, medium supplementation with FGF-2, AA, EGF, and PDGF-BB greatly enhanced the total in vitro expansion capacity of MSC cultures, although differentiation potentials were lost prior to reaching senescence. Loss of differentiation potential was not reflected by changes in stem cell surface marker expression.
Introduction
Mesenchymal stem cells (MSCs) are multipotent, self-renewing cells with the capacity to differentiate into cells of mesenchymal origin, including osteoblasts, adipocytes, and chondrocytes, in vitro and give rise to bone, fat, cartilage, and muscle tissues in vivo [1]. Their differentiation potential, together with their ease of isolation and cultivation, has made them a potentially important source of stem cells for use in tissue engineering and regenerative medicine. Indeed, MSCs have been used for treatment of bone disorders such as in children with osteogenesis imperfecta, in whom allogeneic MSCs were engrafted and shown to enhance growth [2], repair of large defects in long bones when incorporated with biomaterial scaffolds [3], treatment of an atrophic tibial diaphyseal nonunion [4], reconstruction of a major maxillary defect, and reconstruction of jaw defects prior to dental implant placement [5, 6]. Other promising clinical applications for MSCs come from their indirect immune-regulatory and paracrine action in a variety of clinical situations, such as reducing graft-versus-host disease after hemopoietic stem cell (HSC) transplantation [7]; improving engraftment of HSCs [8]; and enhancing cardiac [9, 10], liver [11], and neural repairs [12]. MSCs have been also used as vehicles for delivery of therapeutic gene products and agents to tumor and cancer sites [13].
Despite the vast therapeutic potentials for application of MSCs, these cells are a rare population that comprises only approximately 0.001%–0.01% of the total bone marrow mononuclear cells [1]. Therefore, MSCs isolated for clinical application typically require extensive expansion in vitro prior to their therapeutic use. However, MSCs have a finite life span, undergo senescence on long-term culture in vitro, and may lose some of their differentiation potentials with increased time in culture. For successful expansion in vitro, MSCs need to maintain their stem cell phenotype and show repeated self-renewal; that is, they need to undergo extensive proliferation while maintaining their multipotent differentiation potentials. Consequently a considerable effort has been made to optimize the culture conditions for in vitro expansion of MSCs. These include the assessment of culture media, serum content, cell-plating density, culture surfaces, and the addition of growth factor supplements [14–16]. Of these variables, the use of growth factor and cytokine supplements has been most widely investigated and has been shown to have marked effects on MSC proliferation and self-renewal. The use of fibroblast growth factor (FGF)-2 supplement has been described and used extensively to increase the expansion of MSCs [15, 17–20]. The use of other cytokines, including platelet-derived growth factor (PDGF)-BB, epidermal growth factor (EGF), and interleukin (IL)-6 has also been described for increasing growth and expansion of MSCs [21–23]. In general, most of these experiments have only been short-term studies and have not thoroughly investigated the maintenance of stem cell phenotype and differentiation potential of MSCs following long-term culture with these factors. It is therefore essential to examine directly the effect of factors known to enhance the proliferation of MSCs to determine the most suitable agent to enhance the in vitro expansion of MSCs while maintaining their differentiation potential and stem cell phenotype.
For this reason the aim of the study described here was to carry out a comprehensive investigation of the effect of growth factor and other supplements on MSC self-renewal and differentiation potential. Specifically we have investigated the effect of ascorbic acid (AA), EGF, FGF-2, IL-6, PDGF-BB, transferrin, or Wnt3a on in vitro expansion of MSCs in long-term continuous cultures through to cellular senescence.
Materials and Methods
Experimental Overview
MSC cultures were grown in culture medium with test supplements to confluence, at which time the increase in cell number was calculated by cell counting. An aliquot of these cells was replated into test medium again and grown to confluence as before. This was repeated until cells reached senescence. At the end of every second passage, a further aliquot of the cells was taken for flow cytometric analysis of expression of MSC-related cell surface markers, together with two aliquots which were transferred to osteogenic or adipogenic media and cultured for up 14 days to test for differentiation potentials. Osteogenic and adipogenic differentiation of MSCs was assessed by quantitative reverse transcription-polymerase chain reaction (qRT-PCR) for expression of phenotypic marker genes and for production of mineralization or lipid-containing adipocytes in cultures.
Cell Culture
Primary human MSCs from young male and female adults were purchased from Lonza (Slough, U.K., http://www.lonza.com) (a total of three different cell lines). Cells already at passage 2 when purchased were expanded in culture (this was considered passage 3) in normal growth medium consisting of α-Minimal Essential Medium, penicillin (50 U/ml), streptomycin (50 μg/ml) (all from Sigma-Aldrich, Poole, Dorset, U.K., http://www.sigmaaldrich.com), Glutamax (2 mM) (Invitrogen, Paisley, U.K., http://www.invitrogen.com), and 10% fetal bovine serum (Sigma-Aldrich) and maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Upon reaching confluence cells were seeded at a density of 2,000 cells per cm2 in T-75 flasks and grown in normal medium alone or medium supplemented with 0.2 mM AA (Sigma-Aldrich), 10 ng/ml EGF (Invitrogen), 10 ng/ml FGF-2 (Peprotech, London, U.K., http://www.peprotech.com), 10 ng/ml IL-6 (Invitrogen and Peprotech), 10 ng/ml PDGF-BB (Peprotech), 10 μg/ml transferrin (Sigma-Aldrich), or 50 ng/ml Wnt3a (50 ng/ml) (R&D Systems Inc., Minneapolis, MN, http://www.rndsystems.com) and kept in continuous culture until cells reached senescence. Cell number was counted at each passage using a Z2 Coulter counter (Beckman Coulter, High Wycombe, U.K., http://www.beckmancoulter.com) and subcultured for further expansion at a density of 2,000 cells per cm2. After every second passage cells were also analyzed for proliferation (CellTiter 96 AQueous One Solution Cell Proliferation assay; Promega, Southampton, U.K., http://www.promega.com), cell surface marker and gene expression, and differentiation potential.
Differentiation Potential
MSCs were seeded at a density of 5,000 cells per cm2 in 12-well plates (Nunc, Fisher Scientific, Loughborough, U.K., http://www.fisherscientific.com). After 24 hours of incubation osteogenic differentiation was induced by replacing the medium with the osteogenic medium (growth medium supplemented with 0.1 μM dexamethasone, 0.05 mM AA, and 10 mM glycerophosphate, all from Sigma-Aldrich). To assess osteogenic differentiation, RNA was isolated after 4 and 14 days of incubation, mRNA expression of markers of differentiation (i.e., Runx-2, alkaline phosphatase [ALP], collagen 1 [Col-1], and osteocalcin [OSC]) was determined by qRT-PCR, and accumulation of calcium deposits was visualized by staining with alizarin red dye. Briefly, cells were fixed (15 minutes with 4% formaldehyde in phosphate-buffered saline [PBS]), stained for 10 minutes with alizarin red S (1:100 dilution in H2O), and washed (five times) in 50% ethanol and air-dried.
To induce adipogenic differentiation cells were seeded at a density of 40,000 cells per cm2 in 12-well plates (Nunc) and incubated for 24 hours before switching to adipogenic medium (growth medium supplemented with 1 μM dexamethasone, 0.25 mM isobutylmethylxanthine, 50 μM indomethacin, and 10 μg/ml insulin, all from Sigma-Aldrich). Adipogenesis was assessed by qRT-PCR analysis of markers of differentiation, that is, peroxisome proliferator-activated receptor (PPAR)-γ, Ccaat enhancer binding protein-α (CEBP-α), fatty acid binding protein 4 (FAB-4), and lipoprotein lipase (LPL), and visualized by light microscopy alone or following staining with Oil Red O dye as described previously [24]. Briefly, cells were fixed (15 minutes with 4% formaldehyde in PBS), stained for 15 minutes, and washed with 60% isopropanol and with PBS.
qRT-PCR Analysis
Total RNA was extracted using TRI reagent (Ambion, Warrington, U.K., http://www.ambion.com) and Phase Lock Gel Heavy tubes (five prime; VWR, Lutterworth, U.K., https://us.vwr.com) according to the manufacturers' instructions. RNA purity and quantity were assessed by Nanodrop (Fisher Scientific) (A260/A280 1.8–2 was considered suitable for further analysis), possible contaminating DNA was removed, and cDNA was prepared from 1 μg of RNA using QuantiTect Reverse Transcription Kit (Qiagen, Crawley, U.K., http://www.qiagen.com) according to the manufacturer's instructions. qRT-PCR was performed on a Rotor-Gene 6000 thermal cycler (Qiagen) using Brilliant III Ultra-Fast SYBR Green qPCR Master Mix (Stratagene, Agilent Technologies, Stockport, U.K., http://www.stratagene.com) and primer pairs as listed in supplemental online Figure 1. PCR conditions consisted of 1 cycle of 95°C for 3 minutes and 40 cycles of 95°C for 10 seconds and 60°C for 10 seconds followed by melting analysis of 1 cycle with gradual increase from 65°C to 95°C. RPL13a was used as an invariant housekeeping gene. The quantitative expression of gene of interest relative to the housekeeping gene was calculated, and this ratio for basal or untreated cells was assigned a value of 1. Amplification of the right product was confirmed by monitoring the dissociation/melt curve (at the end of the PCR, samples were subjected to stepwise increase in temperature from 60°C to 95°C, with fluorescence measurements taken throughout this range). Template and reverse transcription negative controls were also included in all amplification experiments.
Flow Cytometry
MSC surface marker expression was assessed using four-color flow cytometry analysis, with the FACSCanto II flow cytometer (BD Biosciences, San Jose, CA, http://www.bdbiosciences.com). Briefly, 150,000 cells were incubated with Fc block (BioLegend, Cambridge, U.K., http://www.biolegend.com) in 50 μl of fluorescence-activated cell sorting (FACS) buffer for 15 minutes at room temperature. Direct staining was performed by adding an antibody cocktail of the following: A, CD34-PerCP/Cy5.5 (clone 4H11), CD105-fluorescein isothiocyanate (FITC) (clone 43A3), CD146-phycoerythrin (PE)/Cy7 (clone SHM-57), and TNAP-PE (clone W8B2); or B, CD45-PerCP/Cy5.5 (clone HI30), CD44-FITC (clone BJ18), CD90-allophycocyanin (APC) (clone 5E10), and CD71 (clone A015). Appropriate isotype controls were also included. All antibodies and isotype controls were obtained from BioLegend U.K. (Cambridge Bioscience) and used at the recommended concentration. Cells were incubated for 15 minutes at room temperature in the dark. After being washed twice, cells were resuspended in 300 μl of FACS buffer and immediately analyzed. A total of 10,000 events were acquired for each sample, and data analysis was performed using DIVA software (BD Biosciences).
Data Analysis
Statistical comparisons between means were made by one-way analysis of variance (SPSS 17; SPSS, Chicago, IL, http://www.spss.com) and post hoc analyses using the Tukey test to evaluate the differences among the mean values between groups. If comparisons were made only between two groups Student's t test (SPSS 16; SPSS) was used. A p value of less than .05 was considered statistically significant.
The number of cell doublings at each passage was calculated using the formula Cell doublings = Log2 (Final cell number/Initial cell number), and the total number of cell doublings was calculated by the sum of doublings for each passage.
Results
MSC Expansion
To compare the stimulatory effect of various cytokines on the total proliferation capacity of MSCs in long-term culture, bone marrow-derived MSCs were grown in parallel cultures with or without AA, EGF, FGF-2, IL-6, PDGF-BB, transferrin, and Wnt3a on three separate occasions. Cells were grown until the fastest cultures reached confluence, which typically took approximately 13–14 days, and then all cultures were passaged. An increased proliferation rate was observed in cells expanded in the presence of FGF-2, AA, PDGF-BB, EGF, and Wnt3a with population doubling times of 2.5, 2.7, 2.8, 3.05, and 4.2 days, respectively, compared with 8.1, 7.8, and 7.6 days for cells expanded in normal growth medium, IL-6, or transferrin at the initial passage. Microscopic images of MSCs expanded in the presence of each factor are shown in supplemental online Figure 2. The proliferation was maintained at approximately the same rate for the first four passages but gradually slowed thereafter and eventually leveled off at the last passage when cells reached senescence. The rate of MSC population doubling for each factor is shown in Figure 1. The total cell doublings prior to senescence were estimated as up to 25.5, 21.1, 20.2, 15.6, and 16.4 in presence of FGF-2, PDGF-BB, AA, EGF, and Wnt3a, respectively, in comparison with 10.0 for MSCs cultured in normal medium alone. IL-6 or transferrin supplementation had no significant effect on cell doubling time or the total number of population doublings. Thus, supplementation with AA, FGF-2, and PDGF-BB resulted overall in a greater than 1,000-fold increase in total cell numbers compared with controls (for controls 210.05 = 895 cells; AA, 220.27 = 1,267,893 cells; FGF-2, 225.59 = 50,542,945 cells). Furthermore, an even greater proliferation rate was observed when a combination of AA, FGF-2, and PDGF-BB was added to the culture. In particular, the combination of FGF-2 with either AA or PDGF-BB or the combination of the three factors together resulted in, respectively, one and two more cell doublings compared with each factor alone after a couple of passages (supplemental online Fig. 3). Cells cultured in the presence of FGF-2, AA, and PDGF-BB also showed enhanced proliferative capacity even when subcultured in normal growth medium for 1 week, with up to 1.5-, 1.7-, and 1.4 (p < .01)-fold increase in comparison with those subcultured from normal medium (supplemental online Fig. 4).
Figure 1.

Effect of supplements on mesenchymal stem cell (MSC) cumulative population doubling. Calculated cell doubling number for MSCs cultured with or without AA, EGF, FGF-2, IL-6, PDGF-BB, transferrin, and Wnt3a from passage 3 to passage 10 (mean of three experiments). Note that error bars have been omitted for clarity of reading the figure; standard deviations for these mean values are shown in tabular form in supplemental online Figure 6. Abbreviations: AA, ascorbic acid; EGF, epidermal growth factor; FGF, fibroblast growth factor; IL, interleukin; PDGF, platelet-derived growth factor.
MSC Surface Phenotype Expression
To determine whether MSC surface marker expression alters with long-term culture or in the presence of cytokine supplements, we carried out four-color flow cytometry analysis of panels of known negative and positive markers of MSCs. As shown in Table 1, MSCs were 90%–100% positive for CD105, CD90, CD44, CD71, and CD146 but lacked the HSC marker (0%–5%) CD34 and CD45 expression. Among the positive markers CD90 and CD44 expression remained more or less constant (95%–100% positive), and both negative markers stayed negative throughout the duration of culture and with all culture conditions. Expression of ALP was elevated from 29.8% at first to 39.3% at the third passage of culture in normal medium but returned back to approximately 30% thereafter. Long-term culture was associated with slight reduction in expression of CD105, CD146, and CD71 cells (from, respectively, 95%, 92%, and 76% in initial culture to 75%, 78%, and 60% at the final passage), although this was not statistically significant. MSCs cultured with FGF-2 showed significant reduction in cells positive for CD146 at every passage (40.9%, 52%, 65%, and 61% in comparison with 92%, 91%, 91%, and 78% in control at passages 4, 6, 8, and 10) examined. AA, EGF, and PDGF-BB cultures also showed a lower level of CD146 positive cells in comparison with control. ALP expression was significantly lower in cells grown in FGF-2 (by less than half in passages 4 and 6), whereas it was enhanced in cells cultured with AA, EGF, and PDGF-BB (49%, 52%, and 40%, respectively, in comparison with 30% in control at passage 4). The reduction seen in CD146 expression following FGF-2 treatment was found to be reversible, and the expression level was restored to control levels following a week of incubation in the absence of FGF-2. ALP expression was also partly restored but still remained lower when compared with cells grown entirely in normal growth medium (supplemental online Fig. 5.1).
Table 1.
Effect of supplements on MSC surface marker expression
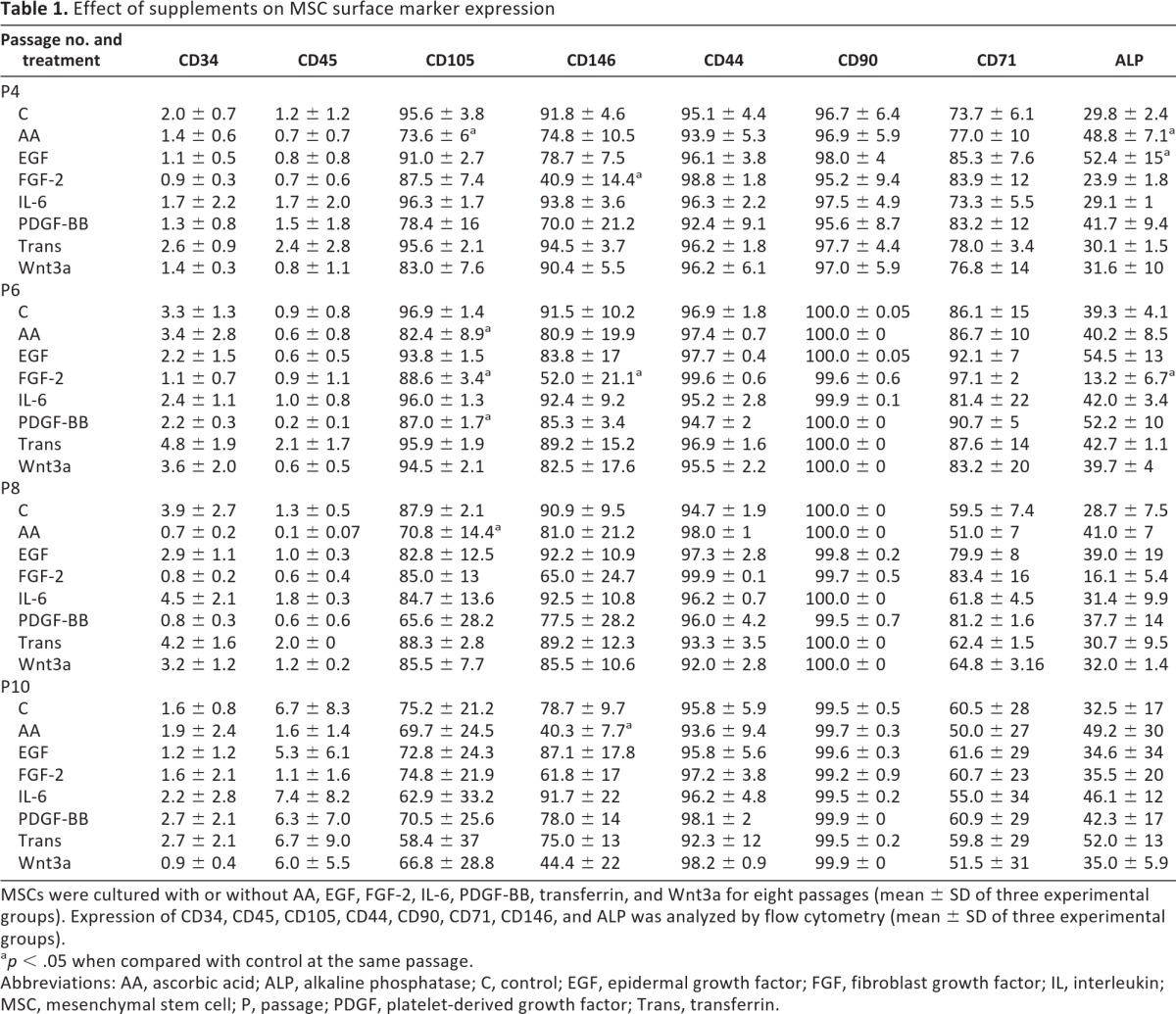
MSCs were cultured with or without AA, EGF, FGF-2, IL-6, PDGF-BB, transferrin, and Wnt3a for eight passages (mean ± SD of three experimental groups). Expression of CD34, CD45, CD105, CD44, CD90, CD71, CD146, and ALP was analyzed by flow cytometry (mean ± SD of three experimental groups).
ap < .05 when compared with control at the same passage.
Abbreviations: AA, ascorbic acid; ALP, alkaline phosphatase; C, control; EGF, epidermal growth factor; FGF, fibroblast growth factor; IL, interleukin; MSC, mesenchymal stem cell; P, passage; PDGF, platelet-derived growth factor; Trans, transferrin.
We also investigated the effect of differentiation on cell surface marker expression. Neither osteogenesis nor adipogenesis altered the expression of negative markers (CD34 and CD45) or positive markers CD44, CD71, CD90, and CD146 following a week of differentiation (supplemental online Fig. 5.2). Only CD105 was reduced significantly following both osteogenic and adipogenic differentiation. As expected the expression of ALP was significantly increased following osteogenesis, but it also increased upon adipogenic differentiation (supplemental online Fig. 5.2).
MSC Differentiation Potential
Next, we tested whether the long-term culture or incubation with cytokine and growth factors was associated with changes in MSC multipotential capacity. MSCs grown in presence of different factors or normal growth medium for more than 100 days of culture (eight passages) were induced to differentiate to osteoblasts or adipocytes at every second passage, and changes in markers of osteogenesis or adipogenesis were analyzed by qRT-PCR and phenotype changes by alizarin red and Oil Red O staining. MSCs were capable of differentiating to osteoblasts and adipocytes, as exhibited by increase in expression of marker genes and accumulation of calcium deposits and lipid droplets by the cells (Figs. 2, 3). The maximal differentiation capacity was observed at initial passage and was significantly reduced at late passages. MSCs cultured in osteogenic medium showed 5- and 10-fold increases in expression of Runx-2 and ALP mRNA at initial passage, but the level was reduced to as low as 2.5 and 1.5 (p < .01), respectively, in subsequent passages (Fig. 2A, 2B). We also observed approximately 60% and 80% (p < .01) reduction in expression of Col-1 and OSC mRNA with cultivation time (Fig. 2C, 2D). Furthermore, the deposition of calcium induced by osteogenic medium was significantly reduced by increase in culture time and at the last passage (passage 10 [P10]) was almost absent (Fig. 3A). The time taken for cells to undergo matrix mineralization also gradually increased from approximately 21 days at the initial passage to up to 40 days at late passages, and even complete loss of mineralization capacity was observed for some cultures (Fig. 3A). Similarly and even more strikingly, the adipogenic differentiation potential of MSCs was reduced by time in culture. As shown in Figure 2E and 2F the expression of PPAR-γ and CEBP-α was induced in presence of adipogenic medium to approximately 190- and 770-fold at the initial passage, but it significantly decreased (p < .01) with prolonged cultivation to 30- and 36-fold for PPAR-γ, 458- and 50-fold for CEBP-α at subsequent passages. Cultivation was also associated with reduced expression of FAB-4 and LPL by more than 75% and 85% (p < .01), respectively (Fig. 2G, 2H). Accordingly, the ability of cells to accumulate lipid following differentiation was reduced gradually and eventually completely inhibited at last passage as shown microscopically after staining with Oil Red O (Fig. 3C).
Figure 2.

Differentiation potential of mesenchymal stem cells in long-term culture. Osteogenic and adipogenic differentiation was induced for 4 and 14 days after every second passage of cultivation. Expression of osteogenic markers Runx-2, ALP, Col-1, and OSC (A–D) and adipogenic markers PPAR-γ, CEBP-α, FAB-4, and LPL (E–H) mRNA was analyzed by quantitative reverse transcription-polymerase chain reaction. (Mean ± SEM of three experiments of duplicates.) *, p < .05, **, p < .01, ***, p < .001 compared with P4. Abbreviations: ALP, alkaline phosphatase; CEBPα, Ccaat enhancer binding protein-α; Col-1, collagen 1; OSC, osteocalcin; FAB-4, fatty acid binding protein 4; LPL, lipoprotein lipase; P, passage; PPAR, peroxisome proliferator-activated receptor.
Figure 3.

Representative photograph of mesenchymal stem cells (MSCs) differentiated to osteoblasts and adipocytes following long-term culture or cultivation with various factors. MSCs expanded in presence or absence of AA, EGF, FGF-2, IL-6, PDGF-BB, transferrin, and Wnt3a. The ability for lineage-specific differentiation was assessed for osteogenesis by staining with alizarin red for calcium deposition at P6–P10 (A), and for adipogenesis by staining with Oil Red O for lipid accumulation (magnification, ×200) (B) and with Oil Red O staining in cells expanded with various factors (at P6) and with Oil Red O staining (C) with long-term cultivation (P6–P10). Abbreviations: AA, ascorbic acid; C, control; EGF, epidermal growth factor; FGF, fibroblast growth factor; IL, interleukin; P, passage; PDGF, platelet-derived growth factor; Trans, transferrin.
Parallel analysis was also carried out on the osteogenic and adipogenic potential of cells cultured with various supplements that induce proliferation of MSCs. Except with IL-6 and transferrin, which had little or no effect on either osteogenesis or adipogenesis (data not shown), the effects were vastly diverse between other cytokines, although reduced differentiation was apparent with all factors at the last passage. Cells grown in cultures supplemented with FGF-2 showed an increase in expression of Runx-2 but a reduction in ALP (Fig. 4A, 4B). These cells when induced to differentiate to osteoblasts also showed a reduction in ALP level, although no significant change was observed with other osteogenic genes. Supplementation of medium with AA resulted in a significant increase in Runx-2 and ALP expression under basal conditions, but a reduction in expression of Runx-2 was observed following osteogenic differentiation upon long incubation of cells with AA. EGF also induced the expression of ALP but had no significant effect on osteogenic potential following treatment of cell with differentiation medium. Supplementation of cultures with Wnt3a reduced the osteogenic potential of MSCs, with reduction in expression of Runx-2 and ALP. PDGF-BB had little effect on osteogenic potential at early passages (Fig. 4A, 4B). Despite the effect on mRNA expression of osteogenic genes by AA, EGF, FGF-2, PDGF-BB, and Wnt3a, supplementation of these factors over six passages completely inhibited the ability of cell to undergo mineralization, as shown by reduced alizarin staining of calcium deposits (Fig. 3A).
Figure 4.

Osteogenic marker mRNA expression in mesenchymal stem cells expanded in presence or absence of AA, EGF, FGF-2, IL-6, PDGF-BB. (A, B): Expression of Runx-2 (A) and ALP (B) in undifferentiated cells (day 0) or following 4 and 14 days of osteogenic differentiation. (C, D): Col-1 (C) and OSC (D) expression after 14 days of osteogenesis. Data (mean ± SEM) are from three experiments in duplicate. *, p < .05, **, p < .01 compared with control. Abbreviations: AA, ascorbic acid; ALP, alkaline phosphatase; C, control; EGF, epidermal growth factor; FGF, fibroblast growth factor; IL, interleukin; P, passage; PDGF, platelet-derived growth factor.
Analysis of adipogenic genes showed that supplementation of culture with FGF-2 or PDGF-BB enhanced (p < .01) the expression of “master switch ” gene PPAR-γ but not CEBP-α or FAB-4 expression under basal conditions (Fig. 5A–5D). Surprisingly, FGF-2-treated and, to a lesser extent, PDGF-BB-treated cells showed a reduction in adipogenic gene expression during the differentiation process, and phenotypic reduction in lipid containing cells was more apparent in cells grown with FGF-2 (Fig. 3B). Addition of AA in culture also significantly reduced (p < .01) the expression of genes involved in adipogenesis both under basal conditions and during the differentiation, as well as reducing the number of lipid-filled adipocytes (Figs. 3B, 5). Similar to the reduction seen with osteogenic genes and mineralization level, the potential for adipogenesis was reduced at the last passage for the cells grown in AA, EGF, FGF-2, PDGF-BB, and Wnt3a as shown by decreased gene expression and lack of lipid accumulation (Fig. 3B).
Figure 5.

Adipogenic marker mRNA expression in mesenchymal stem cells expanded in presence or absence of AA, C, control; EGF, FGF-2, IL-6, and PDGF-BB. (A, B): Expression of PPAR-γ (A) and CEBP-α (B) in undifferentiated cells (day 0) or following 4 and 14 days of adipogenic differentiation. (C, D): FAB-4 (C) and LPL (D) expression after 14 days of adipogenesis. Data (mean ± SEM) are from three experiments in duplicate. *, p < .05, **, p < .01 compared with control. Abbreviations: AA, ascorbic acid; C, control; EGF, epidermal growth factor; FAB-4, fatty acid binding protein 4; FGF, fibroblast growth factor; IL, interleukin; LPL, lipoprotein lipase; P, passage; PDGF, platelet-derived growth factor.
MSC Stemness, Cell Cycle, Senescence, and DNA Repair Gene Expression
Time in culture (number of cell doublings) or supplementation with cytokines was shown to alter the proliferative rate, self-renewal ability, and differentiation capacity of MSCs. Therefore, we investigated the expression of genes involved in maintenance of stem cell phenotype, cell cycle, senescence, and DNA repair. Human MSCs expressed “stemness” genes Nanog, Oct-4, and Klf2, but Sox-2 and TRET genes were barely detectable. Consistent with the alteration in MSC stem cell properties (i.e., differentiation potential) there was a significant reduction in expression of Nanog, Oct-4, or Klf4 with long cultivation time (Fig. 6A). Supplementation with FGF-2 and AA, however, resulted in enhanced expression (p < .01) of Oct-4 and Klf4 (Fig. 6E) only at the initial passage. Cells grown in the presence of AA also expressed more CDK2 mRNA, and CDK4 expression was higher in cultures treated with FGF-2 and EGF (p < .01). No significant change was observed in expression of cyclin D or cyclin E in the presence of growth factors. Long-term culture, however, resulted in reduction in expression of cell cycle-related genes, that is, CDK2, CDK4, cyclin D, and cyclin E (Fig. 6B) and increased expression of senescence gene P16 (p < .01) and was even further induced in the presence of FGF-2, PDGF-BB, and AA only at the last passage. Expression of other senescence-related genes, such as P21 and Rb2, remained unchanged during cultivation but was induced in the presence of AA at the last passage. Similarly, the DNA repair genes Pold3, Ercc1, and Mre11a were unaffected by culture time, although higher expression of Pold3 was observed in cells cultured with AA, EGF, FGF-2, and PDGF-BB, and Ercc1 and Mre11a expression was higher in cells treated with AA (Fig. 6G).
Figure 6.

Expression of stemness, cell cycle, senescence, and DNA repair marker mRNA in mesenchymal stem cells (MSCs) following long-term culture or cultivation with various factors. (A–D): Expression of mRNA for stemness (Nanog, Oct-4, and Klf4) (A), cell cycle (CDK2, CDK4, cyclin D, and cyclin E) (B), DNA repair (Plod3, Ercc1, and Mre11a) (C), and senescence (P16, P21, and Rb2) (D) genes in MSCs cultivated for 10 passages. Data (mean ± SEM) are from three experiments in duplicate. *, p < .05, **, p < .01 compared with P4. (E–G): mRNA expression of Nanog, Oct-4, and Klf4 (E), CDK2, CDK4, cyclin D, and cyclin E (F), and Plod3, Ercc1, and Mre11a (G) genes in MSCs cultivated in presence of AA, EGF, FGF-2, and PDGF-BB at initial passage (P4). (H): Expression of P16, P21, and Rb2 mRNA in MSCs cultivated in presence of AA, EGF, FGF-2, and PDGF-BB at the last passage (P10). Data (mean ± SEM) are from three experiments in duplicates. *, p < .05, **, p < .01 compared with control at the same time point. Abbreviations: AA, ascorbic acid; C, control; EGF, epidermal growth factor; FGF, fibroblast growth factor; P, passage; PDGF, platelet-derived growth factor.
Discussion
Growth factors and cytokines such as FGF-2, EGF, PDGF-BB, IL-6, and other supplements such as AA have all previously been shown to induce proliferation of MSCs. However, these studies have examined only the short-term effect of these factors on MSC number and stem cell properties, which represents a different set of conditions from those that may occur during continuous culture to achieve maximal in vitro expansion of stem cells. Here we have demonstrated that a number of different supplements have the capacity to enhance greatly the large-scale expansion of MSCs in vitro, although with all treatments, as the cultures approached senescence there was a marked decline in their differentiation potentials.
Under control conditions MSCs were able to undergo 10 cell doublings in approximately 100 days of culture before reaching senescence, whereas in the presence of FGF-2 the cells underwent approximately 25 cell doublings in the same time period, which is a great difference in final cell numbers/initial donor cell of approximately 900 with controls to approximately 50 million cells with FGF-2 treatment. Likewise PDGF-BB and AA supplements resulted in total cell doublings of approximately 21 and 20, respectively, in the same period. EGF and Wnt3a supplements were also able to enhance moderately the proliferative capacity of MSCs, whereas transferrin and IL-6 had little effect compared with continuous control cultures. The lack of effect of IL-6 on MSC proliferation and differentiation was rather surprising as it contrasted with recently reported data by Pricola et al. showing that IL-6 is both necessary and sufficient for enhanced MSC proliferation and inhibiting adipogenic differentiation [22]. The disparities between data could not be attributed to the differences in IL-6 or MSCs, as we used IL-6 from various companies and MSCs from different individuals.
A factor that enhances the proliferative capacity of MSCs such as FGF-2, AA, and PDGF-BB can only be considered a suitable candidate for in vitro expansion of MSCs if the expanded cells maintain their differentiation potential. We show here that long-term culture of MSCs, even under normal culture conditions, results in a gradual reduction in differentiation capability of MSCs at both phenotypic and genotypic levels. MSCs were able to undergo substantial osteogenic and adipogenic differentiation up to passage 6 and to a lesser extent at passage 8. Cultivation for 10 passages, however, resulted in considerable reduction and on some occasions lack of differentiation. The capacity of MSCs for differentiation was further reduced in cells cultivated in the presence of number of growth factors at late passages, although lineage-specific upregulation or downregulation was observed with different factors following early cultivation. However, it should be appreciated that in these supplemented cultures the actual number of cell doublings was greatly increased at any given passage number. Thus, for example, the total number of cell doublings seen with control cultures, 10, is reached in FGF-2-supplemented cultures by passage 5.
Both AA and FGF-2 were inhibitory to adipogenic differentiation and significantly reduced the expression of adipogenic markers and number of lipid-containing cells throughout the entire cultivation time. In undifferentiated cells, however, FGF-2 and also PDGF-BB significantly induced PPAR-γ expression. A previous study has also described an upregulation of PPAR-γ mRNA expression with FGF-2 treatment, but unlike our data they showed enhanced adipogenesis following induction of differentiation [25]. FGF-2 supplementation also induced the expression of the osteogenic master switch gene, Runx-2, but a lower ALP expression both in undifferentiated and differentiated cells was observed, which consequently resulted in a reduction in osteogenic mineralization. In contrast, a number of studies have reported a stimulatory role for FGF-2 in osteogenesis [17, 26], although some recently published reports support our findings [27, 28]. In addition, AA was stimulatory toward the osteogenic lineage, especially in expression of ALP and the ability of calcium deposition by cells at early passages (up to P6), but was inhibitory upon longer cultivation. Interestingly, when FGF-2 supplementation was withdrawn the cells tended to revert at least partially to their control expression levels, suggesting that these mass effects of growth factors are reversible.
Use of all the growth factors that extensively enhanced cell number (i.e., FGF-2, AA, PDGF-BB, and EGF) eventually resulted in reduction in differentiation potential at a similar time point during culture (P8) despite the number of cell doublings being different between them. In the experiments described here we investigated bipotent differentiation potentials and did not include investigation of chondrocytic potentials. Consequently, caution needs to be exercised in generalizing these results for applications that might involve such chondrocytic differentiation. However, given that loss of both osteogenic and adipocytic differentiation potentials was seen in late stage cultures, we do not have any reason to think this would not have also been the case for cartilage production as well. In considering a suitable method for therapeutic application it is therefore necessary to select a supplement that maximizes cell doubling prior to the loss of potential for differentiation. For example, at passage 6, where full differentiation potentials are largely preserved, FGF-2, AA, and PDGF-BB result in 13.8, 13, and 12.3 cell doublings respectively, whereas in controls even at passage 8 only eight cell doublings are achieved. In terms of cell numbers this is a difference of 1:28 (256) cells in controls compared with 1:213.8 (14,263) cells in FGF-2-supplemented cultures. Our data additionally suggest that although factors such as Wnt3a, AA, EGF, or FGF-2 may all regulate MSC differentiation fate, as shown previously by various groups, long-term supplementation of MSCs with these factors for enhanced expansion may have slightly distinct effects. Although these differences do not appear to be significant between AA, FGF, and PDGF for up to six passages at least, for longer term expansion an appropriate medium supplement should be included to the culture based on the downstream clinical application, for example, AA for bone reconstruction. Use of combination of two or more supplements should be also considered, as greater expansion achieved when FGF-2, PDGF, and AA were combined.
The decreased proliferation rate with increased cell doubling and in vitro cultivation has been previously studied and is known as replicative senescence [29]. In MSCs the replicative senescence is characterized by gradual reduction in proliferation, morphological alterations, and finally loss of functions, that is, differentiation potential, also known as functional senescence [30]. The mechanisms behind replicative senescence may be multifactorial and associated with telomere shortening and uncapping, DNA damage accumulation, and/or epigenetic changes. However, the mechanisms behind the functional senescence of MSCs are less well understood. Recent reports have suggested a link between DNA damage, reduction in stemness related genes, and loss of MSC self-renewal and differentiation potential [31–33]. Alves et al. have demonstrated that MSCs are susceptible to accumulate DNA damage during in vitro cultivation with an increase in expression of p16 and p21 protein, which they suggested results in the loss of their differentiation potential [31]. In line with this, we showed here an upregulation of P16 gene with culture and in the presence of growth factor supplements, although no change was observed in the P21 and RB2 genes with increased cell doubling. The expression of genes involved in DNA repair, cell cycle, and stemness was also recently investigated upon in vitro culture of rat MSCs. Analysis using semiquantitative PCR showed a significant downregulation of genes involved in stemness and DNA damage repair only after 15–30 days of culture of rat MSCs [32]. Our data partly support these results and show a reduction in expression of stemness genes, although no distinct change was observed with DNA repair genes even after 100 days of culture. The disparities in data may be related to species differences. In addition to the increase in expression of cell senescence genes during culture, we showed here a reduction in cell cycle genes CDK2, CDK4, cyclin D, and cyclin E that has not been addressed in previous studies. We suggest that in addition to upregulation of DNA damage and genes such as P16, the loss of cell cycle genes should be taken into consideration in relation to the reduction in MSC proliferation and differentiation. In addition we showed that a number of factors that induced MSC proliferation also induced cell cycle, stemness, and DNA repair genes, which may suggest that these genes may be involved in downstream mechanisms by which these factors exert their effect.
One of the phenotypic features of MSCs is expression of number of cell surface markers [34]. In fact, one of the criteria for defining bone marrow stromal cells proposed by the International Society for Cellular Therapy is the expression of CD105, CD90, and CD73 and lack of CD45, CD34, or CD14 cell surface markers [35]. More recently CD146 and CD271 were suggested as specific MSC markers [36, 37]. Interestingly, our data show little alteration in the expression of the MSC surface markers CD44 and CD90 in long-term culture, despite the reduction in stemness properties of the cells. Only a small reduction was observed with CD146, CD71, and CD105, which was predominantly apparent at the last passage when cells reached senescence. These data suggest that the use of cell surface markers as characteristic criteria for MSCs might be misleading and not indicative of MSC stemness. Use of surface markers may therefore be limited for distinguishing MSCs from HSCs during initial isolation and sorting. Similarly there was no alteration in CD44 and CD90 with culture conditions, whereas the CD146, CD105, and ALP expression profile was regulated by supplementation with FGF-2, EGF, AA, and PDGF-BB. In particular, the reduction in CD146 and ALP in FGF-2 treated cells was striking. This is particularly interesting as CD146 (a cell adhesion molecule of the immunoglobulin superfamily) has been shown by Sacchetti et al. to be expressed by a subpopulation of osteoprogenitors within bone marrow stromal cells that are able to form bone and stroma and establish a hematopoietic microenvironment following transplantation [37]. They also showed that only cells with high levels of CD146 were able to transfer the hematopoietic microenvironment in vivo, although both low and high CD146-expressing cells were able to establish differentiated osteoblasts and bone upon in vivo transplantation. Although we showed here that the reduction in CD146 expression is reversible, the use of FGF-2 for expansion of MSCs prior to clinical application should be further investigated as it may alter the potential of MSCs as a support for hematopoietic stem cell microenvironment and engraftment.
One of the limitations of the data that are presented here, particularly in relation to total cell doublings, is the fact that these experiments were all conducted on commercially obtained MSCs rather than primary isolates. Although these cells were all at passage 2, this does not equate to an exact number of previous cell doublings. However, at initial culture of these cells, more than 95% of cells expressed stem cell markers and exhibited appropriate differentiation potentials.
In practical terms, our results suggest that medium supplemented with FGF-2, PDGF, or ascorbate can all provide a suitable medium for in vitro expansion of MSCs while maintaining their differentiation potentials. Combinations of these supplements may enhance expansion efficiency further, although differences between any of these treatments are relatively minor compared with the large improvements seen compared with other media tested. As MSCs lost their differentiation potentials with repeated passages, most importantly without loss of expression of their cell surface marker profiles, our results show that using these supplements allows expansion for at least six passages (approximately 13 cell doublings) without any loss of stem cell properties, which would take approximately 84 days in culture.
Conclusion
Supplementing MSC cultures, particularly with FGF-2, AA, PDGF-BB, and EGF, is highly beneficial for achieving large-scale production of MSCs. At the same time long-term cultivation with these factors may be associated with loss of MSC differentiation potential. In cultures approaching senescence, FGF-2 in particular, which has been previously suggested as a suitable factor for expansion of MSCs and commonly used by many research groups, was associated with loss of differentiation and phenotypic changes in MSCs. Regardless of the changes in differentiation capacity and proliferation potential of MSCs, the expression of cell surface markers persisted throughout the cultivation period, calling into question their ability to be true MSC markers. Taken together, our data challenge what was previously portrayed about the role of growth factors such as FGF-B for preclinical expansion of MSCs and the use of MSC markers such as CD90, CD44, and CD105 as definitive indicators of their stemness characteristics.
See www.StemCellsTM.com for supporting information available online.
Acknowledgments
This work was supported by research funding from Guy's & St. Thomas' Charity. The authors acknowledge financial support from the Department of Health via the National Institute for Health Research Comprehensive Biomedical Research Centre award to Guy's & St. Thomas' National Health Service (NHS) Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust. We thank Dr. Susanne Heck and P.J. Chana for assistance with flow cytometry analysis.
Author Contributions
B.G.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; F.J.H.: conception and design, financial support, provision of study material or patients, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 2.Horwitz EM, Gordon PL, Koo WK, et al. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: Implications for cell therapy of bone. Proc Natl Acad Sci USA. 2002;99:8932–8937. doi: 10.1073/pnas.132252399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quarto R, Mastrogiacomo M, Cancedda R, et al. Repair of large bone defects with the use of autologous bone marrow stromal cells. N Engl J Med. 2001;344:385–386. doi: 10.1056/NEJM200102013440516. [DOI] [PubMed] [Google Scholar]
- 4.Hernigou P, Poignard A, Beaujean F, et al. Percutaneous autologous bone-marrow grafting for nonunions. Influence of the number and concentration of progenitor cells. J Bone Joint Surg Am. 2005;87:1430–1437. doi: 10.2106/JBJS.D.02215. [DOI] [PubMed] [Google Scholar]
- 5.Lee J, Sung HM, Jang JD, et al. Successful reconstruction of 15-cm segmental defects by bone marrow stem cells and resected autogenous bone graft in central hemangioma. J Oral Maxillofac Surg. 2010;68:188–194. doi: 10.1016/j.joms.2009.08.031. [DOI] [PubMed] [Google Scholar]
- 6.Mesimäki K, Lindroos B, Törnwall J, et al. Novel maxillary reconstruction with ectopic bone formation by GMP adipose stem cells. Int J Oral Maxillofac Surg. 2009;38:201–209. doi: 10.1016/j.ijom.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Le Blanc K, Frassoni F, Ball L, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: A phase II study. Lancet. 2008;371:1579–1586. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 8.Behre G, Theurich S, Weber T, et al. Reply to “The correlation between cotransplantation of mesenchymal stem cells, higher recurrence rates in hematologic malignancy patients: Outcome of a pilot clinical study” by Ning et al. Leukemia. 2009;178:21579–15793. doi: 10.1038/leu.2008.150. [DOI] [PubMed] [Google Scholar]
- 9.Timmers L, Lim SK, Hoefer IE, et al. Human mesenchymal stem cell-conditioned medium improves cardiac function following myocardial infarction. Stem Cell Res. 2011;6:206–214. doi: 10.1016/j.scr.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Amado LC, Saliaris AP, Schuleri KH, et al. Cardiac repair with intramyocardial injection of allogeneic mesenchymal stem cells after myocardial infarction. Proc Natl Acad Sci USA. 2005;102:11474–11479. doi: 10.1073/pnas.0504388102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kharaziha P, Hellström PM, Noorinayer B, et al. Improvement of liver function in liver cirrhosis patients after autologous mesenchymal stem cell injection: A phase I-II clinical trial. Eur J Gastroenterol Hepatol. 2009;21:1199–1205. doi: 10.1097/MEG.0b013e32832a1f6c. [DOI] [PubMed] [Google Scholar]
- 12.Tfilin M, Sudai E, Merenlender A, et al. Mesenchymal stem cells increase hippocampal neurogenesis and counteract depressive-like behavior. Mol Psychiatry. 2010;15:1164–1175. doi: 10.1038/mp.2009.110. [DOI] [PubMed] [Google Scholar]
- 13.Dwyer RM, Khan S, Barry FP, et al. Advances in mesenchymal stem cell-mediated gene therapy for cancer. Stem Cell Res Ther. 2010;1:25. doi: 10.1186/scrt25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernardo ME, Avanzini MA, Perotti C, et al. Optimization of in vitro expansion of human multipotent mesenchymal stromal cells for cell-therapy approaches: Further insights in the search for a fetal calf serum substitute. J Cell Physiol. 2007;211:121–130. doi: 10.1002/jcp.20911. [DOI] [PubMed] [Google Scholar]
- 15.Sotiropoulou PA, Perez SA, Salagianni M, et al. Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells. 2006;24:462–471. doi: 10.1634/stemcells.2004-0331. [DOI] [PubMed] [Google Scholar]
- 16.Both SK, van der Muijsenberg AJ, van Blitterswijk CA, et al. A rapid and efficient method for expansion of human mesenchymal stem cells. Tissue Eng. 2007;13:3–9. doi: 10.1089/ten.2005.0513. [DOI] [PubMed] [Google Scholar]
- 17.Tsutsumi S, Shimazu A, Miyazaki K, et al. Retention of multilineage differentiation potential of mesenchymal cells during proliferation in response to FGF. Biochem Biophys Res Commun. 2001;288:413–419. doi: 10.1006/bbrc.2001.5777. [DOI] [PubMed] [Google Scholar]
- 18.Solchaga LA, Penick K, Porter JD, et al. FGF-2 enhances the mitotic and chondrogenic potentials of human adult bone marrow-derived mesenchymal stem cells. J Cell Physiol. 2005;203:398–409. doi: 10.1002/jcp.20238. [DOI] [PubMed] [Google Scholar]
- 19.Martin I, Muraglia A, Campanile G, et al. Fibroblast growth factor-2 supports ex vivo expansion and maintenance of osteogenic precursors from human bone marrow. Endocrinology. 1997;138:4456–4462. doi: 10.1210/endo.138.10.5425. [DOI] [PubMed] [Google Scholar]
- 20.Bianchi G, Banfi A, Mastrogiacomo M, et al. Ex vivo enrichment of mesenchymal cell progenitors by fibroblast growth factor 2. Exp Cell Res. 2003;287:98–105. doi: 10.1016/s0014-4827(03)00138-1. [DOI] [PubMed] [Google Scholar]
- 21.Tamama K, Fan VH, Griffith LG, et al. Epidermal growth factor as a candidate for ex vivo expansion of bone marrow–derived mesenchymal stem cells. Stem Cells. 2006;24:686–695. doi: 10.1634/stemcells.2005-0176. [DOI] [PubMed] [Google Scholar]
- 22.Pricola KL, Kuhn NZ, Haleem-Smith H, et al. Interleukin-6 maintains bone marrow-derived mesenchymal stem cell stemness by an ERK1/2-dependent mechanism. J Cell Biochem. 2009;108:577–588. doi: 10.1002/jcb.22289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar A, Salimath BP, Stark GB, et al. Platelet-derived growth factor receptor signaling is not involved in osteogenic differentiation of human mesenchymal stem cells. Tissue Eng Part A. 2010;16:983–993. doi: 10.1089/ten.TEA.2009.0230. [DOI] [PubMed] [Google Scholar]
- 24.Gharibi B, Abraham AA, Ham J, et al. Adenosine receptor subtype expression and activation influence the differentiation of mesenchymal stem cells to osteoblasts and adipocytes. J Bone Miner Res. 2011;26:2112–2124. doi: 10.1002/jbmr.424. [DOI] [PubMed] [Google Scholar]
- 25.Neubauer M, Fischbach C, Bauer-Kreisel P, et al. Basic fibroblast growth factor enhances PPARgamma ligand-induced adipogenesis of mesenchymal stem cells. FEBS Lett. 2004;577:277–283. doi: 10.1016/j.febslet.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 26.Ito T, Sawada R, Fujiwara Y, et al. FGF-2 increases osteogenic and chondrogenic differentiation potentials of human mesenchymal stem cells by inactivation of TGF-β signaling. Cytotechnology. 2008;56:1–7. doi: 10.1007/s10616-007-9092-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lai W-T, Krishnappa V, Phinney DG. Fibroblast growth factor 2 (Fgf2) inhibits differentiation of mesenchymal stem cells by inducing Twist2 and Spry4, blocking extracellular regulated kinase activation, and altering Fgf receptor expression levels. Stem Cells. 2011;29:1102–1111. doi: 10.1002/stem.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Osathanon T, Nowwarote N, Pavasant P. Basic fibroblast growth factor inhibits mineralization but induces neuronal differentiation by human dental pulp stem cells through a FGFR and PLCγ signaling pathway. J Cell Biochem. 2011;112:1807–1816. doi: 10.1002/jcb.23097. [DOI] [PubMed] [Google Scholar]
- 29.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 30.Bonab MM, Alimoghaddam K, Talebian F, et al. Aging of mesenchymal stem cell in vitro. BMC Cell Biol. 2006;7:14. doi: 10.1186/1471-2121-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alves H, Munoz-Najar U, De Wit J, et al. A link between the accumulation of DNA damage and loss of multi-potency of human mesenchymal stromal cells. J Cell Mol Med. 2010;14:2729–2738. doi: 10.1111/j.1582-4934.2009.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galderisi U, Helmbold H, Squillaro T, et al. In vitro senescence of rat mesenchymal stem cells is accompanied by downregulation of stemness-related and DNA damage repair genes. Stem Cells Dev. 2009;18:1033–1042. doi: 10.1089/scd.2008.0324. [DOI] [PubMed] [Google Scholar]
- 33.Cheng H, Qiu L, Ma J, et al. Replicative senescence of human bone marrow and umbilical cord derived mesenchymal stem cells and their differentiation to adipocytes and osteoblasts. Mol Biol Rep. 2011;38:5161–5168. doi: 10.1007/s11033-010-0665-2. [DOI] [PubMed] [Google Scholar]
- 34.Chamberlain G, Fox J, Ashton B, et al. Concise review: Mesenchymal stem cells: Their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells. 2007;25:2739–2749. doi: 10.1634/stemcells.2007-0197. [DOI] [PubMed] [Google Scholar]
- 35.Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 36.Flores-Torales E, Orozco-Barocio A, Gonzalez-Ramella OR, et al. The CD271 expression could be alone for establisher phenotypic marker in bone marrow derived mesenchymal stem cells. Folia Histochem Cytobiol. 2010;48:682–686. doi: 10.2478/v10042-010-0063-6. [DOI] [PubMed] [Google Scholar]
- 37.Sacchetti B, Funari A, Michienzi S, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–336. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]