

This research (Part I) draws attention to the importance of understanding the complexity of a global regulatory environment applicable to biologic drugs, including stem cells or their progeny. It demonstrates considerable differences in the regulatory approach to biologic drugs' development and application, particularly with respect to terminology and definitions used in regional regulatory frameworks.
Keywords: Cellular therapy, Clinical translation, Clinical trials, Ethics
Abstract
Recent development of a wide range of regulatory standards applicable to production and use of tissues, cells, and other biologics (or biologicals), as advanced therapies, indicates considerable interest in the regulation of these products. The objective of this study was to analyze and compare high-tier documents within the Australian, European, and U.S. biologic drug regulatory environments using qualitative methodology. Cohort 1 of the selected 18 high-tier regulatory documents from the European Medicines Agency (EMA), the U.S. Food and Drug Administration (FDA), and the Therapeutic Goods Administration (TGA) regulatory frameworks were subject to a manual documentary analysis. These documents were consistent with the legal requirements for manufacturing and use of biologic drugs in humans and fall into six different categories. Manual analysis included a terminology search. The occurrence, frequency, and interchangeable use of different terms and phrases were recorded in the manual documentary analysis. Despite obvious differences, manual documentary analysis revealed certain consistency in use of terminology across analyzed frameworks. Phrase search frequencies have shown less uniformity than the search of terms. Overall, the EMA framework's documents referred to “medicinal products” and “marketing authorization(s),” the FDA documents discussed “drug(s)” or “biologic(s),” and the TGA documents referred to “biological(s).” Although high-tier documents often use different terminology they share concepts and themes. Documents originating from the same source have more conjunction in their terminology although they belong to different frameworks (i.e., Good Clinical Practice requirements based on the Declaration of Helsinki, 1964). Automated (software-based) documentary analysis should be obtained for the conceptual and relational analysis.
Introduction
Recent development of a wide range of regulatory standards applicable to production and use of tissues, cells, and other biologics (or biologicals)—as advanced therapies—has stimulated considerable interest in the regulation of biologic drugs (including but not limited to human cell and tissue therapies) as part of the vast array of biopharmaceuticals. Biologic drug regulations have been widely developed by mature regulatory agencies; for example, the Australian framework is administered by the Therapeutic Goods Administration (TGA), the European Union's by the European Medicines Agency (EMA), and that of the U.S. by the Food and Drug Administration (FDA) [1].
A significant number of documents are currently available in the analyzed regulatory frameworks (supplemental online data 1) in the form of standards, guidelines, recommendations, or regulations [2]. Conditions of product manufacturing and release have been established and outlined in a spectrum of documents from the high-tier regulatory documents down to procedural documents. However, one of the major issues facing manufacturers, clinical practitioners, patients, patient advocacy groups, investors, and the general public is confusion over terminology at various levels and in types of documents applicable to specific products (i.e., stem cells vs. somatic cells vs. biologics vs. biologicals) [2–6]. Additional misperceptions can be attributed to the unclear level of regulatory oversight required, different levels of scrutiny (i.e., control of an entire manufacturing facility vs. control of a manufacturing process), differences in availability of specific formal written procedures (i.e., some regulatory documents are deemed overly prescriptive or overly generic), and different product testing and release requirements [2–6]. Nonetheless, complex regulations are meant to be adhered to at all times and in every aspect of the manufacturing of biologic drugs (including but not limited to human cell and tissue therapies).
For the purpose of this paper all the products mentioned above will be referred to as biologic drugs, although other terminology may be used in the literature [3]; additionally, a consensus has not been reached entirely on the use of the term “drug” [4], the regulatory oversight required [5], and established industry models [6]. An extensive overview of regulatory documents (available on websites) and the list of abbreviations and additional definitions used in this study are presented in supplemental online data 1 and supplemental online data 2, respectively.
The intention of the research is to compare the content and terminology in high-tier regulatory documents related to biologic drugs in Australia, Europe, and the U.S., where these products are also referred to as biologicals, advanced therapy medicinal products, and biologics, respectively. Since stem cells are “biologics,” these regulations are of major importance to all those attempting to translate stem cells into clinical trials and clinical practice.
Definitions
The TGA regulatory documents refer to biological as follows:
Essentially a biological comprises, contains or is derived from human cells or human tissues … and is used to: treat or prevent disease, ailment, defect or injury; or diagnose the condition of a person; or influence, inhibit or modify a physiological process in persons; or test the susceptibility of persons to a disease or ailment; or replace or modify parts of the anatomy in persons [7].
The official FDA definition of biological products or biologics can be summarized as “any virus, therapeutic serum, toxin, antitoxin or analogous product applicable to the prevention, treatment or cure of diseases or injuries of man,” whereas the FDA Consumer Information page states:
Biological products include a wide range of products such as vaccines, blood and blood components, allergenics, somatic cells, gene therapy, tissues, and recombinant therapeutic proteins. Biologics can be composed of sugars, proteins, or nucleic acids or complex combinations of these substances, or may be living entities such as cells and tissues. Biologics are isolated from a variety of natural sources—human, animal, or microorganism—and may be produced by biotechnology methods and other cutting-edge technologies. Gene-based and cellular biologics, for example, often are at the forefront of biomedical research, and may be used to treat a variety of medical conditions for which no other treatments are available [8].
The lengthy official definition (codified in 21 CFR 600.3) vaguely defines biologics on the basis of analogies (i.e., products similar to viruses, serums, toxins, and antitoxins) [9]. This definition avoids terms and concepts in use for generations (i.e., proteins, antibodies, genes, microbes, cells, viruses, and DNA/RNA). In practice, biologics include “a wide range of products such as vaccines, blood and blood components, allergenics, somatic cells, gene therapy, tissues and recombinant therapeutic proteins” [9].
The European Parliament Regulation 1394/2007 (amending directive 2001/83/EC and Regulation 726/2004) identifies an advanced therapy medicinal product (ATMP) as
a medicinal product as defined in Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use (the Directive), as amended to reflect new innovative therapeutic products. Specifically, an ATMP is a biological medicinal product which is either:
a gene therapy medicinal product as defined in Part IV of Annex I to Directive 2001/83/EC;
a somatic cell therapy medicinal product as defined in Part IV of Annex I to Directive 2001/83/EC; or
a tissue engineered product as defined in Article 21(b) of the ATMP Regulation (1394/2007/EC) [10].
Aim, Purpose, and Scope
The purpose of this study is to provide a comparison of regulatory documents pertinent to biologic drugs in different regulatory frameworks. To accomplish this, an assessment of particular documents was obtained. The scope of the study included regulations of biologic drugs in Australia, Europe, and the U.S.; the objective was to derive data shared between the regulatory frameworks in order to make them usable in a harmonization process. Exploiting the shared terminology and mutually recognized scope or aim of the regulations was easiest to define via documentary analysis of high-tier documents in each regulatory scheme. It was hypothesized that there are similarities between high-tier regulatory documents related to biologic drugs in Australia, Europe, and the U.S., although the terminology and focus of these regulatory frameworks differ to some extent. It was also anticipated that high-tier regulatory documents could be taken as a central representation of the regulatory frameworks from which they were drawn. The overall aim of the study was to establish a correlation between a manual analysis (Part I) and a software analysis (Part II) [11], to assess usability of the software analysis, and to ascertain the use of a qualitative methodology in the highly specialized area of regulatory science.
Complexity
Taking newly created biomedical discoveries from the bench to the bedside has proven to be a highly challenging and costly exercise [12]. It has been defined as translational research and recognized by researchers and funding agencies and more recently by regulatory agencies as an imminent need [12]. Success in translational research requires not only highly trained experts and complex skill sets within a team but also an understanding of the application of data in different regulatory sets of principles [13]. Independent development of regulatory authorities in Australia, Europe, and the U.S. has led to differences in the regional approach to pharmaceutical manufacturing. Some of these were favorable for the conduct of early clinical trials with minimal supporting data. Others have been affected by external factors (e.g., cultural) or have expanded their scope through the years. Recent efforts have been made to harmonize the three frameworks, mostly in the area of active pharmaceutical ingredient inspections [14], and are expected to be followed by other joint programs.
Although therapeutics derived from biological sources have been subject to regulatory oversight for some time (i.e., monoclonal antibodies), the biologic products used in transplantation procedures (i.e., cells and tissues) have historically been exempted from such oversight [15]. The unique source of the “active ingredients” renders cell and tissue therapies difficult for assessment by the traditional regulatory approaches; these have been geared to support traditional pharmaceutical manufacturing practice [16]. It has been recognized that therapeutic claims for biologics need to be supported by strong scientific evidence [17] but also by comprehensive regulatory principles. New considerations have led the existing regulatory agencies of the developed world to propound new regulatory approaches for the sector of biologic drugs or biopharmaceuticals, which “has become one of the most research-intensive sectors with a great potential for delivering innovative human medicines in the future” [18].
The biopharmaceutical industry is defined through a definition of biopharmaceutical products or as business activities related to the development of new biopharmaceutical drugs and medicines [19]. Defining the term “biopharmaceutical” for the industry's stakeholders, including the regulatory agencies, has proven to be a difficult task. Regulatory definitions of biopharmaceuticals are generally based on a broad biotechnology definition but often use other terms with their own intricate definitions (i.e., “biologicals” in Australia, “advanced therapy medicinal products” in Europe, and “biologics” in the U.S.) [9]. Documentary analysis is a useful methodology to clarify the complexity of biologic drug regulations, as part of the complex regulatory arena of biopharmaceuticals [20].
Regulation of cell and tissue products is closely linked not only to sensitive areas of public health policy and funding but also to the development of novel disciplines such as regulatory science [19, 21]. This positions regulators in a challenging position as they attempt to reconcile their roles as independent assessors with public health needs and perceptions [19, 21]. The historical background—in the development of regulatory agencies of interest—has been explored previously [22], and the regional differences were illustrated by observations pertinent to their clinical trial requirements [23–29]. These differences inevitably contribute to the complexity of the regulations applicable to the development of biologic drugs.
Materials and Methods
Materials
Documents and written material used in this research provided a way of gaining access to sets of processes that cannot be observed or documented otherwise and are not easily described in verbal or other reconstructions [20]. Documents used in this study were readily available on websites because of their purpose. They were complete, well constructed, and clearly presented.
The main tasks in the analysis were (a) the selection process of documents to be analyzed, due to the vastness of the material available, and (b) the thorough reading and analysis of selected documents. The type of sampling used in this research was theoretical or purposive sampling for high-tier regulatory documents that are, for purposes of this research, taken as the central representation of the regulatory frameworks from which they were drawn. The sampling approach is outlined in Table 1, indicating the inclusion and exclusion criteria. An extensive list of analyzed documents is presented in Table 2.
Table 1.
Inclusion and exclusion criteria for selection of documents

aOnly one document was exempt, as it was originally published in 1996 (see Table 4).
Abbreviations: ATMP, advanced therapy medicinal products; E.U., European Union; GCP, Good Clinical Practice; GMP, Good Manufacturing Practice.
Table 2.
List of analyzed documents
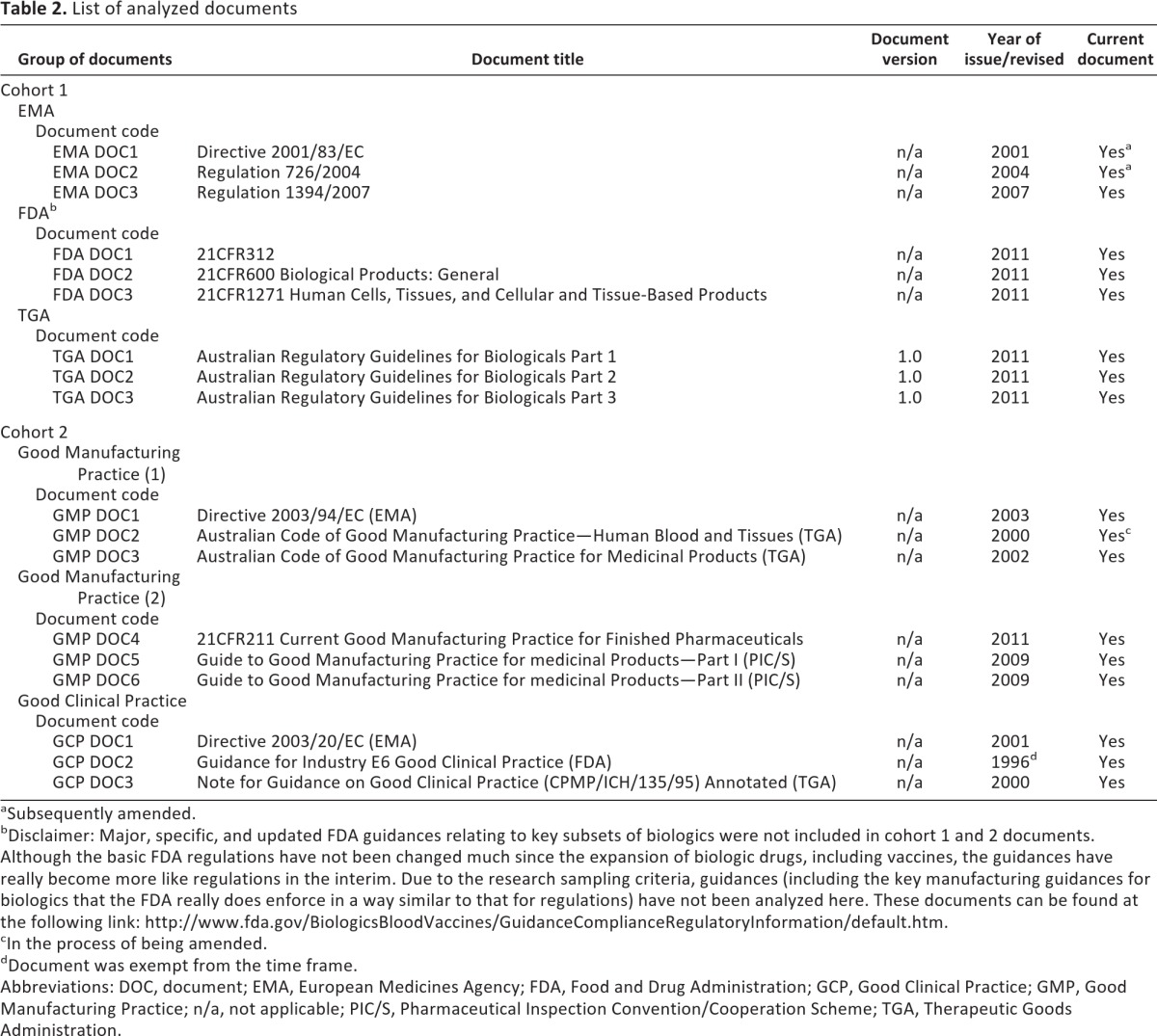
aSubsequently amended.
bDisclaimer: Major, specific, and updated FDA guidances relating to key subsets of biologics were not included in cohort 1 and 2 documents. Although the basic FDA regulations have not been changed much since the expansion of biologic drugs, including vaccines, the guidances have really become more like regulations in the interim. Due to the research sampling criteria, guidances (including the key manufacturing guidances for biologics that the FDA really does enforce in a way similar to that for regulations) have not been analyzed here. These documents can be found at the following link: http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/default.htm.
cIn the process of being amended.
dDocument was exempt from the time frame.
Abbreviations: DOC, document; EMA, European Medicines Agency; FDA, Food and Drug Administration; GCP, Good Clinical Practice; GMP, Good Manufacturing Practice; n/a, not applicable; PIC/S, Pharmaceutical Inspection Convention/Cooperation Scheme; TGA, Therapeutic Goods Administration.
Methods
Manual analysis of selected documents identified main terms and phrases in individual documents. While undertaking data analysis, important consideration was given to selectivity and the perspective of the research, that is, how to maintain focus in the process of documentary analysis. This was related to the question of what the documents were expected to provide in an ontological and epistemological sense. The process of analysis involved several steps. First, the context was established and the complexity and historical background were explored for each of the analyzed sets of regulatory principles. Second, extensive preparation for analysis and selection of documents was obtained. Finally, selected documents were copied from the official regulatory agencies' websites and examined. Since the interest of the research was predominantly in the literal wording and terminology, as a demonstration of the documents' focus and intent, a manual method (Table 3) was devised to record these.
Table 3.
Literal wording, form, and sequence manual recording in the documents

An overview of the new TGA website structure and content is presented in supplemental online data 1a. It includes only the new Biologicals Framework established in July 2011 [26–29]. Relevant documents were selected from the newly established TGA regulations [26–29]. In terms of the European Union (E.U.) regulatory requirements, selection of the documents was not straightforward due to the complexity of national and EMA regulations (presented in supplemental online data 1b). It is acknowledged by the EMA that new technologies, therapies, and medicines are emerging in the form of regenerative medicine and more personalized treatments [23, 24]. The lack of an E.U.-wide regulatory framework in the past led to divergent national approaches that hindered patient access to products, and as a result the central procedure was established [23, 24]. As a further illustration of the complexity and vastness of written material available in the area of biologic drug regulations, an overview of the relevant FDA regulations since 2004 [30–34] is briefly presented in supplemental online data 1c. Some of these documents were selected for the analysis, based on the inclusion and exclusion criteria, and a provision was made in relation to so called “nonbinding” documents in the form of guidances (see the Disclaimer in Table 2). In terms of sampling, it was essential to decide what was significant in the context of the research, its theoretical and empirical referents, and how it related to the broader universe [35–37].
Results
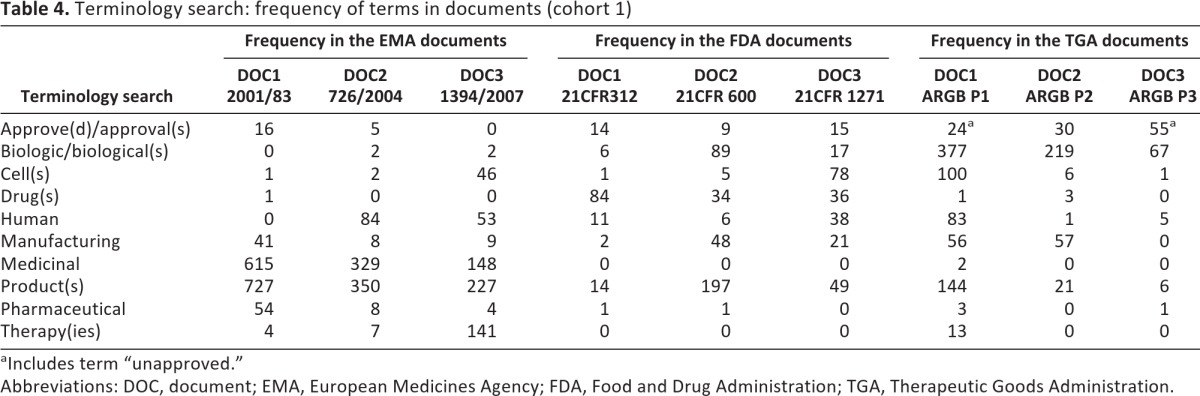
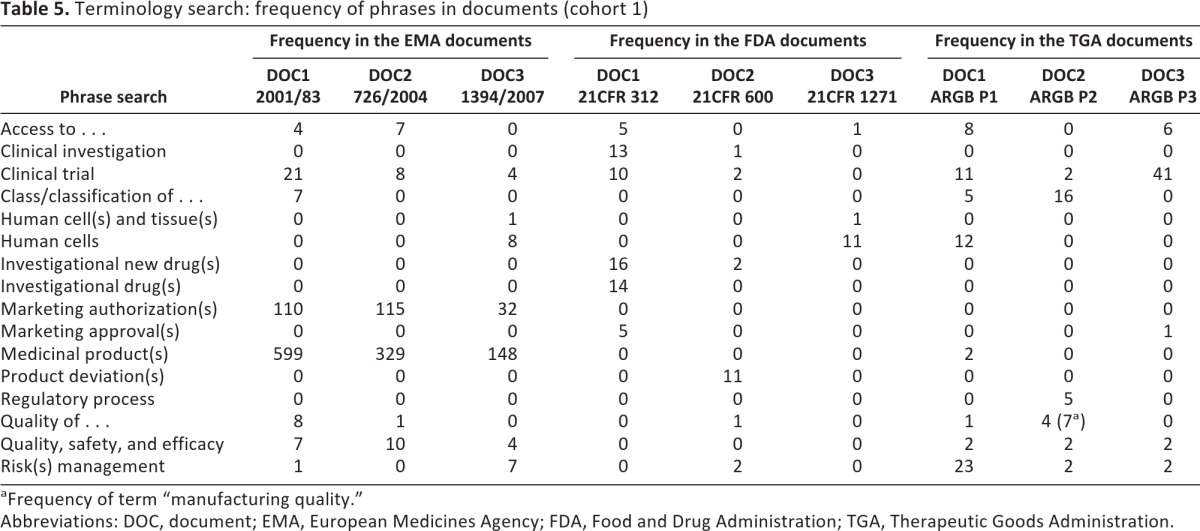
Data presented in Tables 4 and 5 were derived from the manual analysis, using the PDF search feature to assess the documents in cohort 1 (see Table 2). Rather than treating the data as an end product (i.e., variable), it was classified and subsequently parts or “slices” were treated as unfinished resources for further use [20]. Manual analysis included a terminology search with a list of 10 specific terms (Table 4) and 16 phrases (Table 5). The frequencies of terms and phrases in the documents are also illustrated in Figures 1 and 2.
Table 4.
Terminology search: frequency of terms in documents (cohort 1)
aIncludes term “unapproved.”
Abbreviations: DOC, document; EMA, European Medicines Agency; FDA, Food and Drug Administration; TGA, Therapeutic Goods Administration.
Table 5.
Terminology search: frequency of phrases in documents (cohort 1)
aFrequency of term “manufacturing quality.”
Abbreviations: DOC, document; EMA, European Medicines Agency; FDA, Food and Drug Administration; TGA, Therapeutic Goods Administration.
Figure 1.

Terminology search graph: frequency of terms in documents (cohort 1). Abbreviations: DOC, document; EMA, European Medicines Agency; FDA, Food and Drug Administration; TGA, Therapeutic Goods Administration.
Figure 2.

Terminology search graph: frequency of phrases in documents (cohort 1). Abbreviations: DOC, document; EMA, European Medicines Agency; FDA, Food and Drug Administration; TGA, Therapeutic Goods Administration.
Frequency of terms presented in Table 4 reveals certain consistency across regions in the use of “manufacturing” (58, 71, and 113 occurrences in EMA, FDA, and TGA documents, respectively) and “human” (137, 55, and 89 rates, respectively). The terms “drug(s),” “cell(s),” and “biologic(s)/biological(s)” were to some extent used across the entire spectrum of analyzed documents.
More frequent and specific use of “medicinal” was observed in the EMA documents (a total of 1,092 occurrences in three documents), whereas “pharmaceutical” was very sporadically used in any documents other than the EMA DOC1 (54 occurrences). The term “therapy” or “therapies” was not mentioned in any of the FDA documents and was seldom used in the TGA documents (13 times in total), whereas it was mentioned 141 times in the EMA DOC3, likely because of the definition by the EMA of cell therapies as advanced therapeutic medicinal products.
Whereas the most frequent word used in the EMA documents was “product(s)” along with “medicinal,” the term “drug(s)” was consistently mentioned within all FDA documents (84, 34, and 36 times) but seldom mentioned in the other two frameworks (a total of five occasions). The TGA documents featured “biologic(s)/biological(s)” most consistently, whereas “cell(s)” was most used in a document from the FDA framework (78 occurrences in FDA DOC3) and the TGA framework (100 occurrences in TGA DOC1). Considering the relevant definitions, these search terms were expected to be dominant [23–32].
The definition of cell therapies by each of the regulatory agencies was as follows.
- Somatic cell therapy and gene therapy-related definitions and considerations by the FDA:Somatic cell therapy is the administration to humans of autologous, allogeneic, or xenogeneic living cells which have been manipulated or processed ex vivo. Manufacture of products for somatic cell therapy involves the ex vivo propagation, expansion, selection (see: “A Proposed Approach to the Regulation of Cellular and Tissue-based Products,” Feb. 28, 1997, (62 FR 9721)), or pharmacologic treatment of cells, or other alteration of their biological characteristics. Such cellular products might also be used for diagnostic or preventive purposes. Recently, various innovative therapies involving the introduction of somatic cells into humans have been used or proposed. For the purpose of this Guidance, the term somatic cell therapy refers to the administration to humans of autologous, allogeneic, or xenogeneic living non-germline cells, other than transfusable blood products, for therapeutic, diagnostic, or preventive purposes…. Examples of somatic cell therapies include implantation of cells as an in vivo source of a molecular species such as an enzyme, cytokine or coagulation factor; infusion of activated lymphoid cells such as lymphokine activated killer cells and tumor-infiltrating lymphocytes (addressed in a separate Points to Consider document: see below); and implantation of manipulated cell populations, such as hepatocytes, myoblasts, or pancreatic islet cells, intended to perform a complex biological function. Because of the complexities of potential interactions with the cells and other constituents, additional components should be considered as part of the final biological product for purposes of preclinical evaluation [25, 32].
- Human cell and tissue-based therapeutic goods-related definitions and considerations by the TGA:The Australian Regulatory Guidelines for Biologicals (ARGB) provide information for manufacturers, sponsors, healthcare professionals and the general public on the legal arrangements in Australia for the supply and use of human cell and tissue-based therapeutic goods. These products are collectively defined as ‘biologicals’…. The Biologicals Regulatory Framework is the term for legislation that came into force in 2011 to regulate human cell and tissue-based products as a distinct group of therapeutic goods called ‘biologicals’. The framework is administered by the Therapeutic Goods Administration (TGA), who has produced this document—the Australian Regulatory Guidelines for Biologicals (ARGB)—to inform manufacturers, sponsors, healthcare professionals and all other interested parties about the framework…. The Biologicals Regulatory Framework provides a comprehensive system of assessment and controls that must be completed before products are allowed to be marketed in Australia (premarket), and follow-up and further controls after they are marketed (post-market)… or included on the Australian Register of Therapeutic Goods (ARTG) otherwise exempted, approved or authorised [26].
- ATMP-related definitions and considerations by the EMA:Advanced-therapy medicinal products (ATMPs) are medicines for human use that are based on gene therapy, somatic-cell therapy or tissue engineering. They offer groundbreaking new opportunities for the treatment of disease and injury. The regulatory framework for ATMPs is established in Regulation (EC) No 1394/2007 [23].For reasons of clarity, complex therapeutic products require precise legal definitions. Gene therapy medicinal products and somatic cell therapy medicinal products have been defined in Annex one to Directive 2001/83/EC, but a legal definition of tissue engineered products remains to be laid down. When products are based on viable cells or tissues, the pharmacological, immunological or metabolic action should be considered as the principle mode of action. It should also be clarified that products which do not meet the definition of a medicinal product, such as products made exclusively of non-viable materials which act primarily by physical means, cannot by definition be advanced therapy medicinal products [10].
Phrase search results are shown in Table 5. The phrase “access to” was the one most uniformly present in all documents across the frameworks although not frequently used altogether (a total of 31 occurrences across all frameworks' documents).
The phrases “clinical investigation” and “clinical trial” were interchangeably used by FDA documents, whereas EMA and TGA documents referred only to “clinical trial.” “Human cell(s) and tissue(s)” and “human cells” were only referred to by a single document in each framework (9 occasions in the EMA DOC3, 12 in the FDA DOC3, and 12 in the TGA DOC1), whereas “investigational new drug” was mentioned only in two FDA documents (a total of 32 occasions in FDA DOC1 and DOC2).
Finally, the TGA documents referred often to “manufacturing” (along with other two frameworks' documents to a lesser extent) and “approved/unapproved,” while mentioning “cell(s)” more than 100 times in only one of the TGA documents (TGA DOC1). The TGA framework relied heavily on the terms “product(s)” and “risk management” on 171 and 27 occasions, respectively. “Risk management” was mentioned less frequently in the EMA documents and only twice in all FDA documents.
The term “therapy” or “therapies” was not mentioned in any of the FDA documents and was seldom used in the TGA documents (13 times in total), whereas it was mentioned 141 times in the EMA DOC3.
Overall, the EMA framework's documents referred to “medicinal products” and “marketing authorisation(s),” the FDA documents discussed “drug(s)” or “biologic(s),” and the TGA documents mentioned “biological(s).” Hence, all the documents signified “product” as such—although they referred to it using different terminology (“medicinal product” vs. “drug/biologic” vs. “biological”).
Furthermore, the EMA documents were mostly concerned with “marketing authorisation(s)” while mentioning “quality, safety and efficacy.” In the same manner the FDA documents discussed “drug(s)” or “investigational drug(s)” that were not cited in the other two frameworks at all. They also used “product deviation” exclusively and used “clinical investigation” and “clinical trial” interchangeably.
Discussion
Biologic drugs must be developed and manufactured through disciplined and specific mechanisms [3]. This is the case even when full compliance with traditional concepts of pharmaceutical manufacturing practice is sometimes not possible (although strived for), particularly for life-saving therapies and new modalities in investigational drug development [13]. These systems incorporate considerations of risk-benefit ratios and include mechanisms for the transparent and accountable release of products [17].
Conditions of product requirements and release are established and outlined in a spectrum of documents from the high-tier regulatory documents down to the procedural documents. Hence, the documents outlining the highest level of regulatory requirements in three mature regulatory frameworks were analyzed by a documentary analysis method in order to establish concomitance between different regional regulations applicable to biologic drugs as a source of a specialized knowledge in regulatory science.
The commissioner of the FDA stated recently “that a failure to invest ‘adequately’ in regulatory science has been one of the reasons why fewer new drugs have been making it to the market” [38]. In October 2010 the FDA promised to bring its “regulatory science” into the 21st century [39] and in August 2011 issued its strategic plan for regulatory science [40], called Advancing Regulatory Science in FDA [19]. The Common Fund's Regulatory Science program was initiated in the second half of 2010 as a partnership between the U.S. National Institutes of Health (NIH) and the FDA [41].
At the same time, the EMA stated on its website that “the European Medicines Agency is a key leader in the development and application of regulatory science” [42] and referred to the conference hosted by the Agency in December 2010, which resulted in the report “Regulatory science: Are regulators leaders or followers?” [43].
The origin of the term “regulatory science” is unknown; it is believed that it was created in the 1970s when the newly formed U.S. Environmental Protection Agency was required to make decisions based on incomplete and sometimes nonexistent scientific information [44]. Although there is still some debate as to whether it is necessary to use this new terminology “as science is science regardless how it is applied” [44], the FDA currently defines regulatory science as “the science of developing new tools, standards, and approaches to assess safety, efficacy, quality, and performance of all (FDA) regulated products” [45]. There is an inevitable need to capture regulatory knowledge and expertise required for the development of future regulatory policies to enable safe application of new biologic drugs [46].
This study revealed numerous differences between the regulatory frameworks high-tier documents and the terminology they employ. For instance, “marketing authorisation” was exclusively used by the EMA regulatory framework, as was the phrase “medicinal products.” Neither of these was mentioned at all in the FDA and the TGA frameworks' documents. “Class” and “classification of” were not mentioned at all in the FDA documents, whereas the same phrases were used minimally in the EMA documents (7 occurrences) but more frequently in the TGA documents (54 occurrences).
Whereas the most frequent word used in the EMA documents was “product(s),” along with “medicinal,” the term “drug(s)” was consistently mentioned within all FDA documents (84, 34, and 36 times) but seldom mentioned in the other two frameworks (a total of 5 occasions). The TGA documents featured “biologic(s)/biological(s)” most consistently, whereas “cell(s)” was most used in a document from the FDA framework (78 occurrences in FDA DOC3) and the TGA framework (100 occurrences in TGA DOC1).
“Quality, safety and efficacy” was a phrase barely used in the EMA and the TGA documents, and it was not present in any of the FDA documents. This also applied to the phrases “quality of” and “manufacturing quality.”
This paper has described the processes that were used in the research while developing a method of manual documentary analysis for high-tier regulatory documents. Although the data were studied out of context and the selectiveness of the data or representativeness of the sample can be questioned, this approach to documentary analysis is particularly useful when the researcher is faced with the vast amount of complex documents with no common format, scope, or terminology.
Similar studies, aimed at analyzing or comparing regulatory documents, have been performed in the pharmaceutical and other industries (i.e., aviation and education) [47–50]. Irrespective of the area (industry) and a specific methodology (manual or automated documentary analysis) used in each study, they have added benefit to their respective areas. The purpose of these studies was to provide a procedure for assessing connections between the current regulations and incorporation of new requirements [47], to increase understanding and knowledge about the trends in success of regulatory submissions [48], or to compare the regulatory versus the educational value of documents (i.e., guidances for undergraduate education for medical practitioners) [49]. Some of the research looked into establishing a clear definition of requirements or identifying inconsistencies between those definitions [50].
In the process of choosing the documents for the study, an ontological position was adopted that suggested that written words, texts, and documents are meaningful constituents of the process in the way in which they are changed and used [18]. In addition, an epistemological position was adopted that suggested that texts, documents, and written material can provide or count toward evidence of these ontological properties. The benefits of studies aimed at analyzing or comparing regulatory documents have been already recognized in the health care industry [47], the pharmaceutical industry [48, 50], and aviation and education fields [49], and qualitative documentary review and analysis demonstrated its benefits to a number of other sectors, such as political science [51]; environmental health research [52]; ethics [53]; and the World Health Organization's health, economics, and policy-making research [54]. We have observed that the main tasks in documentary analysis are a thorough understanding of the analyzed documents, familiarity with relevant terminological differences, and robust quality assurance of the methodology [55–57].
The main advantages for the use of existing documents in a research study are that the data are usually easy to access, it takes a relatively short period of time to collect the data, and to some extent it provides a relatively inexpensive form of data collection [35]. In addition, the documents are unbiased by the data collection process (i.e., nonreactivity) and may be useful for hypothesis and problem formulation. Nonreactivity is one of the main documentary analysis methodological strengths—“the data selection method itself generally does not change the data being collected” [36]. The stability of a measure refers to the extent to which the same results are obtained on repeated administrations of the instrument and it focuses on the instrument's susceptibility to “extraneous factors from one administration to the next” [57]. We considered it appropriate to rely on the “stability” of the documents since a retest measurement facility was possible due to the nature of high-tier documents available to the public and researchers. Reliability refers to the accuracy of the research methods and techniques [58, 59]. The process of reading, understanding, selecting, and comparing documents added a further dimension of construction as well as reflexivity—“textual analysis involves the mediation between the frames of reference of the researcher and those who produced the text … the researcher's own frame of reference becomes the springboard from which the circle is entered, and so the circles reaches back to encompass the dialogue between the researcher and the text” [60]. Careful consideration of the validity of data generation requires a precise application of the logic used previously in determining methodological approaches—the chosen methodology has to be more valid than other methodological approaches [20]. To some extent there is a blurring of the distinction between validity and reliability. The main question posed here was how well the analyzed documents represented the concepts of the research or the hypothesis in an accurate, authentic, and relevant manner [37]. Since the issue of similarities and differences between the regulatory approaches was examined, it appears appropriate to use high-tier regulatory documents that establish the hallmark of these frameworks. It would be difficult to justify the use of any other documentary sources for this type of the research since the other documents (i.e., lower-tier procedural texts or guidelines) are either overly specialized in their aim or scope or not considered as mandatory for every product.
Generally, qualitative research cannot entirely rely on standardized procedures to deal with concepts such as bias and reproducibility [58]. In this process, reproducibility is often replaced with transparency or an audit trail (a record of the researcher's design decision) about gaining access, collecting and analyzing the data, or the actual research design [59]. Manual analysis with its quantified data (i.e., frequencies of terms and phrases) along with the software analysis in Part II of this study [11] (i.e., identifying concepts and themes) can be repeated accurately on many occasions and with no operator dependence. The only variable potentially influencing the reproducibility of the study relies upon selection of the documents to be analyzed. Construct validity concerns the degree to which results of documentary analysis documents reflect the underlying theoretical concepts or exposure pointers. This would be “the extent to which the theoretical concepts have been successfully operationalised” and “ the process of translating research concepts into measurable phenomena” [35]. Whereas the manual documentary analysis obtained in Part I (this paper) provides semantics analysis, the software-based analysis in Part II [11] is expected to assist in conveying contexts and relations between the concepts supported by visual representation [60]. Validity of the construct was established by the assumption that differences in the regulatory frameworks were dictated by different historical and cultural backgrounds, regional dynamics, and various complexities of the biologic drugs' development. Nevertheless, some shared concerns were considered an underpinning factor for similarities among the regulatory frameworks, such as public and individual safety and a uniform scientific knowledge. Validity of interpretation is another way to think about the validity of the research. It is concerned with the data analysis validity and the interpretation on which it is based; it is contingent upon the end result, including how that interpretation was reached [20]. The validity of interpretation was established in two ways: first, by making the process of reaching our interpretations as transparent as possible, and second, by following the same logic in making methodological choices and identifying the sources to be used (i.e., analyzed documents). Furthermore, the intent of the research was to provide some level of generalization, since it examined a broad set of regulations in the developed world (i.e., Europe, the U.S., and Australia). Generalization is sometimes perceived in two distinct ways: empirical (i.e., one empirical population is representative of a wider population) and theoretical [20]. Theoretical generalization is often seen as more productive as it encompasses a range of strategies based on different logics, some of which are more “theoretical” than others (i.e., attaining a wider perspective) [20]. The aim in the theoretical generalization of this research was to demonstrate that high-tier regulatory documents are a central representation of the regulatory frameworks from which they are drawn. The comparison aimed to contribute more to the generalization than a simple statement of similarity or discrepancy. It was also designed to pose questions for future regulatory studies. What questions do all regulatory agencies want answered? What are the specific areas they emphasize? In what specific areas do they converge/diverge, and how can the lessons learned from a conventional pharmaceutical model(s) be applied to biopharmaceutical(s) development? It is well known that the biggest risk in drug development is failure to convince a regulator that a therapeutic product meets the requirements for the next stage of development or marketing approval. It is also becoming clear that regulatory education and precedence information analysis [61], along with thorough knowledge of regulatory framework(s) and their focus, are the most acceptable development strategies for a smarter product development pathway. Not only do they require fewer clinical and nonclinical trials while reducing time and related cost [61], but they also potentially bring development of a biologic drug closer to market approval stage in different regions [62].
Conclusion
This research draws attention to the importance of understanding the complexity of a global regulatory environment applicable to biologic drugs, including stem cells or their progeny. It demonstrates considerable differences in the regulatory approach to biologic drugs' development and application, particularly with respect to terminology and definitions used in regional regulatory frameworks. The EMA documents were mostly concerned with “marketing authorisation(s)” but occasionally mentioned “quality, safety and efficacy”; the FDA documents discussed “drug(s)” or “investigational drug(s)” that were not cited in the other two frameworks at all, used “product deviation” exclusively, and used “clinical investigation” and “clinical trial” interchangeably. Whereas the most frequent word used in the EMA documents was “product(s),” along with “medicinal,” the term “drug(s)” was consistently mentioned within all FDA documents but seldom mentioned in the other two frameworks. The TGA documents featured “biologic/biological(s)” most consistently, whereas “cell(s)” was most used in a document from the FDA framework and the TGA framework.
Nevertheless, this study identifies some conjoint aspects (i.e., the focus on the “product” whether it is referred to as “medicinal product,” “drug/biologic,” or “biological,” so the terms “drug(s),” “cell(s),” and “biologic/biological(s)” were to some extent used across the entire spectrum of analyzed documents) and common principles (i.e., the TGA documents refer often to “manufacturing” whereas the documents of the other two frameworks referred to it to a lesser extent, and all documents use the term “human” in a comparable manner).
Although the data were studied out of context and the selectiveness of the data or representativeness of the sample can be questioned, this approach to documentary analysis is particularly useful when the researcher is faced with the vast amount of complex documents with no common format, scope and terminology. Hence, further documentary analysis has been generated in Part II of this study [11].
See the accompanying article on pages 909–920 of this issue.
Author Contributions
N.I.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; S.S., E.S., and K.A.: final approval of manuscript; L.T.: conception and design, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
E.S. holds a compensated employment/leadership position in Ground Zero Pharmaceuticals, Inc.
References
- 1.Burger SR. Current regulatory issues in cell and tissue therapy. Cytotherapy. 2003;5:289–298. doi: 10.1080/14653240310002324. [DOI] [PubMed] [Google Scholar]
- 2.Dutton RL, Scharer JM. Advanced Technologies in Biopharmaceutical Processing. Ames, IA: Blackwell Publishing Ltd.; 2007. [Google Scholar]
- 3.Prince HM, Wall DP, Stokes KH, et al. Cell processing for clinical trials and commercial manufacture. Cell Gene Ther. 2004;5:15–21. [Google Scholar]
- 4.Kaiser J. U.S Federal Court says stem cell treatments are drugs. [Accessed August 6, 2012]. Available at http://news.sciencemag.org/scienceinsider/2012/07/us-federal-court-says-stem-cell-.html.
- 5.Freeman M, Fuerst M. Does the FDA have regulatory authority over adult autologous stem cell therapies? 21 CFR 1271 and the emperor's new clothes. J Transl Med. 2012;10:60–65. doi: 10.1186/1479-5876-10-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenblatt B. Meeting regulatory challenges for cell-based therapies. Bioprocess Int. 2012;10(suppl 3):8–11. [Google Scholar]
- 7.TGA Biologicals Framework Newsletter. Issue 4. Australia: Therapeutic Goods Administration; 2010. Nov, [Accessed May 20, 2012]. Available at http://www.tga.gov.au/pdf/biologicals-newsletter-2010-04.pdf. [Google Scholar]
- 8.Drugs@FDA Glossary of Terms. Information on Drugs. [Accessed May 20, 2012]. Available at http://www.fda.gov/Drugs/InformationOnDrugs/ucm079436.htm.
- 9.Rader RA. (Re)defining biopharmaceutical. Nat Biotechnol. 2008;26:743–751. doi: 10.1038/nbt0708-743. [DOI] [PubMed] [Google Scholar]
- 10.Regulation (EC) No. 1394/2007 of the European Parliament and of the Council, Official Journal of the European Union (10.12.2007) [Accessed May 20, 2012]. Available at http://eur-lex.europa.eu/LexUriServ/site/en/oj/2007/l_324/l_32420071210en01210137.pdf.
- 11.Ilic N, Savic S, Siegel E, et al. Examination of the regulatory frameworks applicable to biologic drugs (including stem cells and their progeny) in Europe, the U.S., and Australia: Part II—A method of software documentary analysis. Stem Cells Translational Medicine. 2012;1:909–920. doi: 10.5966/sctm.2012-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.To thwart disease, apply now (Editorial). Nature, 453 7197 (2008) [Accessed August 12, 2010]. Available at http://www.nature.com/nature/journal/v453/n7197/full/453823a.html. [DOI] [PubMed]
- 13.Kirouac DC, Zandstra PW. The systematic production of cells for cell therapies. Cell Stem Cell. 2008;3:369–381. doi: 10.1016/j.stem.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Final report on the International API inspection Pilot Programme, European Medicines Agency. 16 June 2011 EMA/453741/2011. [Accessed October 17, 2011]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Report/2011/07/WC500108655.pdf.
- 15.Halme DG, Kessler DA. FDA regulation of stem-cell-based therapies. N Engl J Med. 2006;355:1730–1735. doi: 10.1056/NEJMhpr063086. [DOI] [PubMed] [Google Scholar]
- 16.FDA Guidance for Industry: PAT—A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance. 2004. [Accessed January 13, 2011]. Available at http://www.fda.gov/downloads/Drugs/…/Guidances/ucm070305.pdf.
- 17.Burger SR. CTP/GMP cell engineering for cell and gene therapies. BioProcessing J. 2003;2:66–69. [Google Scholar]
- 18.The financing of biopharmaceutical product development in Europe. Final report. European Commission, Enterprise and Industry. 2009. Oct, [Accessed December 5, 2011]. Available at http://ec.europa.eu/enterprise/sectors/biotechnology/files/docs/financing_biopharma_product_dev_en.pdf.
- 19.Advancing Regulatory Science at FDA: A Strategic Plan (August 2011). Food and Drug Administration, U.S. Department of Health and Human Services. [Accessed December 5, 2011]. Available at http://www.fda.gov/ScienceResearch/SpecialTopics/RegulatoryScience/ucm267719.htm.
- 20.Mason J. Qualitative Researching. 2nd ed. London, U.K.: Sage Publications; 2002. [Google Scholar]
- 21.Implementing the European Medicines Agency's Road Map to 2015: The Agency's Contribution to Science, Medicines, Health—“From Vision to Reality.” EMA/MB/550544/2011. 2011. Oct 6, [Accessed November 22, 2011]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Other/2011/10/WC500115960.pdf.
- 22.Ilic N, Khalil D, Hancock S, et al. Regulatory considerations applicable to manufacturing of human placenta-derived mesenchymal stromal cells (MSC) used in clinical trials in Australia and comparison to USA and European regulatory frameworks. In: Chase LG, Vemuri MC, editors. Mesenchymal Stem Cell Therapy. Springer/Humana Press; (in press) [Google Scholar]
- 23.The European Medicines Agency. [Accessed October 15, 2011]. Available at http://www.ema.europa.eu/ema/
- 24.About us. The European Medicines Agency (EMA) [Accessed October 15, 2011]. Available at http://www.ema.europa.eu/ema/index.jsp?curl=pages/about_us/general/general_content_000235.jsp&murl=menus/about_us/about_us.jsp|.
- 25.The Food and Drug Administration (FDA) [Accessed October 27, 2011]. Available at http://www.fda.gov/default.htm.
- 26.TGA basics. Therapeutic Goods Administration. [Accessed September 1, 2011]. Available at http://www.tga.gov.au/about/tga.htm.
- 27.Clinical trials. Therapeutic Goods Administration. [Accessed September 1, 2011]. Available at http://www.tga.gov.au/industry/clinical-trials.htm.
- 28.Clinical trials at a glance. Therapeutic Goods Administration. [Accessed September 1, 2011]. Available at http://www.tga.gov.au/industry/clinical-trials-glance.htm.
- 29.Note for guidance on good clinical practice. Therapeutic Goods Administration. [Accessed August 6, 2011]. Available at http://www.tga.gov.au/industry/clinical-trials-note-ich13595.htm.
- 30.Development and Approval Process (CBER) Food and Drug Administration. [Accessed July 20, 2011]. Available at http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/default.htm.
- 31.About the Center for Drug Evaluation and Research. The Center for Drug Evaluation and Research (CDER), Food and Drug Administration. [Accessed July 20, 2011]. Available at http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/default.htm.
- 32.New Drug Application (NDA) Process (CBER) Food and Drug Administration. [Accessed July 21, 2011]. Available at http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/NewDrugApplicationNDAProcess/default.htm.
- 33.Anderson M. FDA abandons Declaration of Helsinki for international clinical trials. Social Medicine Portal. 2008. Jun 1, [Accessed April 3, 2011]. Available at http://www.socialmedicine.org/2008/06/01/ethics.
- 34.The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH); [Accessed October 29, 2011]. Available at http://www.ich.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas J, Harden A. Methods for the thematic synthesis of qualitative research in systematic reviews. BMC Med Res Methodol. 2008;8:45–55. doi: 10.1186/1471-2288-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bailey KD. Methods of Social Research. 2nd ed. New York, NY: Free Press; 1982. [Google Scholar]
- 37.Robson C. Real World Research. 2nd ed. Oxford, U.K.: Blackwell; 2002. [Google Scholar]
- 38.Sukkar E. Lack of investment in regulatory science is part of “innovation gap,” believes FDA commissioner. [Accessed January 5, 2012];Br Med J. 2011 343:d2017. doi: 10.1136/bmj.d8017. Available at http://www.bmj.com/content/343/bmj.d8017. [DOI] [PubMed] [Google Scholar]
- 39.Lowes RR. FDA vows to bring its “regulatory science” into 21st century. Medscape Medical News. Oct 2010, [Accessed January 22, 2011]. Available at http://www.medscape.com.
- 40.FDA: Regulatory science plan positions agency to foster innovation through better science (news release) 2011. Aug 17, [Accessed September 4, 2011]. Available at http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm268293.
- 41.The Common Fund's Regulatory Science program between NIH and FDA. [Accessed January 5, 2012]. Available at http://commonfund.nih.gov/regulatoryscience/overview.aspx.
- 42.European Medicines Agency, Special Topics: Regulatory science. [Accessed January 5, 2012]. Available at http://www.ema.europa.eu/ema.
- 43.EMA Report. Regulatory science: Are regulators leaders or followers? [Accessed September 4, 2011]. Available at http://www.ema.europa.eu/ema.
- 44.What Is Regulatory Science? Institute for Regulatory Science, USA. [Accessed January 5, 2012]. Available at http://www.nars.org/whatis.html.
- 45.Advancing Regulatory Science: Moving Regulatory Science into the 21st Century. [Accessed January 5, 2012]. Available at http://www.fda.gov/scienceresearch/specialtopics/regulatoryscience/default.htm.
- 46.Ilic N, Siegel TL. Conference Proceedings: E: Regulation of Medicines, Access to New Investigational Drugs: Cell Therapies; November 14–18, 2010; New Orleans, LA. 2010. FIP Pharmaceutical Sciences World Congress. [Google Scholar]
- 47.Munns TE, Beard RE, Culp AM, et al. Analysis of Regulatory Guidance for Health Monitoring. NASA/CR-2000-210643. [Accessed May 20, 2012]. Available at http://ntrs.nasa.gov/archive/nasa/casi.ntrs.nasa.gov/20010017836_2001019668.pdf.
- 48.Putzeist M, Mantel-Teeuwisse AK, Gispen-De Wied CC, et al. Regulatory Scientific advice in drug development: Does company size make a difference? Eur J Clin Pharmacol. 2011;67:157–164. doi: 10.1007/s00228-010-0919-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gray T, Grant J. Good Medical Practice or CanMEDS for education? J Clin Pathol. 2012;65:565–567. doi: 10.1136/jclinpath-2011-200583. [DOI] [PubMed] [Google Scholar]
- 50.Rozet E, Ceccato A, Hubert C, et al. Analysis of recent pharmaceutical regulatory documents on analytical method validation. J Chromatogr A. 2007;1158:111–125. doi: 10.1016/j.chroma.2007.03.111. [DOI] [PubMed] [Google Scholar]
- 51.Wesley JJ. Qualitative document analysis in political science. Paper presented at: T2PP Workshop, Vrije Universiteit Amstardam; April 9–10, 2010; Amsterdam, Netherlands. [Google Scholar]
- 52.Scammell MK. Qualitative environmental health research: An analysis of literature, 1991–2008. Cien Saude Colet. 2011;16:4239–4255. doi: 10.1590/s1413-81232011001100030. [DOI] [PubMed] [Google Scholar]
- 53.Sixsmith J, Murray CD. Ethical issues in the documentary data analysis of Internet posts and archives. Qual Health Res. 2001;11:423–432. doi: 10.1177/104973201129119109. [DOI] [PubMed] [Google Scholar]
- 54.Hanney SR, Gonzalez-Block MA, Buxton MJ, et al. The Utilisation of Health Research in Policy-Making: Concepts, Examples, and Methods of Assessment. A report to the Research Policy and Co-operation Department, World Health Organization; Geneva. 2002. Oct, [Accessed August 6, 2012]. HERG Research Report No. 28. Available at http://www.brunel.ac.uk/depts/herg/pubs/internal.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miles MB, Huberman AM. Qualitative Data Analysis. 2nd ed. Thousand Oaks, CA: Sage Publications; 1994. [Google Scholar]
- 56.Patton MQ. Qualitative Research & Evaluation Methods. 3rd ed. Thousand Oaks, CA: Sage Publications; 2002. [Google Scholar]
- 57.Weber RP. Basic Content Analysis. 2nd ed. Thousand Oaks, CA: Sage Publications; 1990. [Google Scholar]
- 58.Creswell JW. Qualitative Inquiry & Research. 2nd ed. Thousand Oaks, CA: Sage Publications; 2007. [Google Scholar]
- 59.Maxwell JA. Qualitative Research Design. 2nd ed. Thousand Oaks, CA: Sage Publications; 2005. [Google Scholar]
- 60.Scott J. A Matter of Record. Cambridge, U.K.: Polity Press; 1990. [Google Scholar]
- 61.Kumar M. Taming the regulatory beast: Role of regulatory trend analysis in successful FDA approval (Editorial) [Accessed May 20, 2012]. Available at http://www.mastercontrol.com/newsletter/pharmaceutical/regulatory_trend_analysis_fda_approval_1009.html?source=n3w5.
- 62.EMA News Release: European Medicines Agency sees benefits of interaction with Japanese regulators. 2012. Jul 30th, [Accessed August 6, 2012]. Available at http://www.emea.europa.eu/ema.
- 63.Advanced Therapy Medicinal Products FAQs. Human Tissue Authority (U.K.) [Accessed August 6, 2012]. Available at http://www.hta.gov.uk/licensingandinspections/sectorspecificinformation/tissueandcellsforpatienttreatment/advancedtherapymedicinalproductsfaqs.cfm.