

The therapeutic effects of oncolytic herpes simplex virus type 1 G47Δ in patient-derived glioblastoma stem-like cells (GSCs) under normoxia (21% oxygen) and hypoxia (1% oxygen) were analyzed and compared. Findings suggest that G47Δ represents a promising therapeutic strategy to target and kill GSCs, not only in normoxic areas of glioblastoma but also within the hypoxic niche.
Keywords: Gene transfer, Glioma, Herpes simplex virus, Cancer stem cells, Hypoxia
Abstract
Glioblastoma (GBM), a fatal malignant brain tumor, contains abundant hypoxic regions that provide a “niche” to promote both the maintenance and enrichment of glioblastoma stem-like cells (GSCs) and confer resistance to chemo- and radiotherapy. Since GSCs, with an ability to resist conventional therapies, may be responsible for tumor recurrence, targeting GSCs located in such a hypoxic environment may be critical to improving the therapeutic outcome for GBM patients. Oncolytic viral therapies have been tested in the clinic as a promising therapeutic approach for GBM. In this study, we analyzed and compared the therapeutic effects of oncolytic herpes simplex virus (oHSV) type 1 G47Δ (γ34.5−ICP6−LacZ+α47−) in patient-derived GSCs under normoxia (21% oxygen) and hypoxia (1% oxygen). GSCs cultured in hypoxia showed an increased ability to form neurospheres and expressed higher levels of the putative stem cell marker CD133 compared with GSCs cultured in normoxia. G47Δ exhibited a comparable ability to infect, replicate, and kill GSCs in normoxia and hypoxia in vitro. Importantly, G47Δ could counteract hypoxia-mediated enhancement of the stem-like properties of GSCs, inhibiting their self-renewal and stem cell marker expression. Using orthotopic human GSC xenografts in mice, we demonstrated that intratumoral injection of G47ΔUs11fluc, a newly developed G47Δ derivative that expresses firefly luciferase driven by a true late viral promoter, led to an equivalent frequency of viral infection and replication in hypoxic and nonhypoxic tumor areas. These findings suggest that oHSV G47Δ represents a promising therapeutic strategy to target and kill GSCs, not only in normoxic areas of GBM but also within the hypoxic niche.
Introduction
Glioblastoma (GBM; World Health Organization grade IV astrocytoma) is the most frequent and malignant type of primary brain tumor in adults, and despite the combined use of all currently available treatments (maximal surgical resection, radiotherapy, and chemotherapy), the median survival remains about 15 months after diagnosis [1].
Histologically, GBM is characterized by cellular pleomorphism, nuclear atypia, mitotic activity, microvascular proliferation, and/or necrosis. The aberrant vasculature and rapidly dividing tumor mass induce the establishment of a hypoxic microenvironment with poor oxygen diffusion across the growing lesion, leading to necrosis [2–4]. The majority of examined GBMs exhibit low oxygen concentrations ranging from 2.5% to 0.1% (pO2 from 2 to 0.75 mmHg) [5]. The presence of severe hypoxia in gliomas is associated with an early, mostly local recurrence of the tumor and decreased patient survival [6, 7]. Hypoxia has been thought, at least in part, to contribute to increased tumor invasion, loss of apoptosis, gene expression alteration, oncogene activation, angiogenesis induction, and chemoradiation resistance [8]. A reduction in radiation-induced free radicals in hypoxic regions diminishes the effectiveness of radiotherapy, whereas hypoxic cells distally located from blood vessels are less exposed to systemically administrated chemotherapy. In addition, hypoxia indirectly modifies GBM phenotypes and mediates chemoradiation resistance through the regulation of transcription factors such as the hypoxia inducible factors (HIFs) [6]. There is a significant decrease in survival in glioma patients with high HIF2α expression compared with patients expressing moderate or low levels of HIF2α mRNA [9].
The identification of a subpopulation of tumor cells with efficient tumorigenicity and stem cell-like properties was firstly described by Bonnet and Dick in human acute myeloid leukemia [10] and then for various solid tumors, including GBM [11, 12]. These cells have been called glioblastoma stem-like cells (GSCs) [12, 13]. GSCs share some, although not necessarily all, of the characteristics of adult neural stem cells (NSCs) including the expression of neural stem cell markers (i.e., CD133, Nestin, etc.), the ability to self-renew, the capacity to differentiate into different nervous system cell lineages, and responses to hypoxia [14–17]. Physiologically, NSCs are located in the brain in relatively hypoxic niches where neural precursors maintain their undifferentiated state and self-renewal capacity [3, 18]. Similarly, a hypoxic microenvironment has been demonstrated to maintain the stem-like status of GSCs, blocking their differentiation and modulating tumorigenicity mostly via HIF transcriptional pathways [3, 5, 7, 9, 19–21]. HIFs are involved in promoting clonogenicity, increasing the expression of stem cell markers such as CD133, SOX2, Nestin, and Oct4, and are required for GSC growth and survival [9, 20–22]. Accordingly, knockdown of HIFs impairs neurosphere formation, reduces VEGF promoter activity, decreases GSC tumor formation, and increases survival in mice bearing intracranial GSC-derived tumors [9].
CD133 has been considered a putative GSCs marker. CD133+ glioma cells survived radiation treatment better than CD133− cells, both in vitro and in vivo, and had increased expression of antiapoptotic genes [23, 24]. There is a correlation between CD133 expression and patient outcomes [25–27]. However, controversy exists regarding the role and specificity of CD133 since GSC populations with no CD133 expression have been described [28, 29]. The observation that GSCs contribute to GBM resistance to chemotherapy (temozolomide, etoposide, and paclitaxel) and radiation supports the concept that GSCs may be responsible for tumor recurrence and poor clinical outcome [24, 26]. Since hypoxia is strongly related to GSC maintenance and enhances the potential to escape conventional therapies, developing ways to target GSCs in hypoxic regions is critical to improving GBM therapy.
Oncolytic virus (OV) therapy represents a promising strategy for treating GBM, including the GSC component. Among OVs engineered to selectively infect and kill tumor cells, oncolytic herpes simplex virus (oHSV) type 1 vectors have been tested in phase I and II clinical trials, with demonstrated safety; however, improved efficacy is still needed [30–32]. In previous studies, we demonstrated that the replication of G207, a multimutated oHSV, can be enhanced by hypoxia in the U87MG glioma cell line [33]. We also showed that oHSV, such as G47Δ, exhibited significant antitumor activity against GSCs, depending upon the specific viral mutations [15]. However, it was not known whether GSCs are susceptible to oHSV when exposed to low oxygen levels (1% O2). Furthermore, G207 does not replicate well in human GSCs, whereas G47Δ does [15]. Thus, we sought to characterize the efficacy of G47Δ on human GSCs under hypoxia versus normoxia. We show for the first time that G47Δ can replicate and kill human GSCs cultured in hypoxia and that G47Δ infection counteracts hypoxia-related GSC enrichment in vitro. Direct intratumoral injection of newly developed G47ΔUs11fluc in orthotopic human GSC xenografts in mice showed that oHSV infection and replication occur in hypoxic areas of tumors in vivo.
Materials and Methods
Cell Culture
Human GSC cultures GBM4, GBM6, and GBM8, derived from primary GBM surgical specimens, have been described [15]. The cells were cultured as spheres in EF20 medium: neurobasal medium (Invitrogen, Carlsbad, CA, http://www.invitrogen.com) supplemented with 3 mmol/l l-glutamine (Mediatech, Manassas, VA, http://www.cellgro.com), 1× B27 supplement (Life Technologies, Rockville, MD, http://www.lifetech.com), 0.5× N2 supplement (Life Technologies), 2 μg/ml heparin (Sigma-Aldrich, St. Louis, http://www.sigmaaldrich.com), 20 ng/ml recombinant human EGF (R&D Systems Inc., Minneapolis, http://www.rndsystems.com), 20 ng/ml recombinant human FGF2 (Peprotech, Rocky Hill, NJ, http://www.peprotech.com), and 0.5× penicillin G-streptomycin sulfate-amphotericin B complex (Mediatech). The cultures were fed every 3 days with one-third volume of fresh medium. Cell passaging was performed by dissociation of spheres using NeuroCult chemical dissociation kit (StemCell Technologies, Vancouver, BC, Canada, http://www.stemcell.com). Primary adherent cultures from GBM4 and GBM8 were obtained by growing cells in Dulbecco's modified Eagle's medium (Mediatech) supplemented with 10% fetal calf serum (FCS) as previously described [15]. The medium was changed every 4 days, and passaging was performed by trypsinization. Hereafter we refer to these cells as 4FCS and 8FCS. The cells were cultured under either normoxic (5% CO2, 21% O2, 74% N2) or hypoxic (5% CO2, 1% O2, 94% N2) conditions at 37°C in humidified Heraeus incubators (Hera Cell; Thermo Scientific, Asheville, NC, http://www.thermoscientific.com).
Viruses
G47Δ, derived from G207, contains deletions of both copies of γ34.5 and α47, and a LacZ insertion inactivating ICP6 [34]. G47ΔBAC was derived by homologous recombination between G47Δ DNA and pBAC-ICP6EF and contains a cytomegalovirus promoter driving expression of the enhanced green fluorescent protein (EGFP) in place of LacZ [34, 35].
G47ΔUs11fluc was derived from G47Δ with the addition of the firefly luciferase gene driven by the true late Us11 gene promoter of herpes simplex virus 1 (HSV-1). The Us11 promoter was polymerase chain reaction (PCR)-cloned from the genome of wild-type HSV-1 strain F using the following primers: forward primer, CCGGATCCTGAGATCAATAAAAGGGGGCGTGAG, and reverse primer, CCGCCATGGTCCGCCCAGAGACTCGGGTGATG. This promoter sequence contains the following cis-acting regulatory elements of HSV-1 late promoters: the TATA box and the cap/leader sequences (initiator element), which together confer true late regulation, as well as the downstream activation sequence that allows transcriptional activation [36, 37]. The Us11 promoter and the luciferase gene codon-optimized for mammalian expression (from pGL4.10[luc2]; Promega, Madison, WI, http://www.promega.com) were subcloned into shuttle plasmid pFLS-IE4-Express, removing the HSV IE4 promoter. This pFLS-Us11luc shuttle plasmid was recombined with pG47Δ-BAC using Cre recombinase as described [38]. The resulting bacterial artificial chromosome (BAC) plasmid and an FLPe-expressing plasmid were cotransfected to Vero cells to remove BAC-derived sequences and the EGFP gene, which enables efficient HSV packaging [38]. The resulting virus progeny was plaque-purified, and clones selected based on luciferase expression were expanded.
Limiting Dilution Sphere Formation Assay
GSC single-cell suspensions were seeded in uncoated 96-well plates at three dilutions (100, 10, and 1 cells per well) in 100 μl of EF20 medium. The plates were fed weekly with 100 μl of fresh medium, and spheres were counted when an adequate size (∼80–100 μm) was achieved (12 days for GBM4 and 16 days for GBM6 and GBM8). Limiting dilution assays were also performed with infected GSCs. GBM4 and GBM8 single-cell suspensions were infected with G47Δ at a multiplicity of infection (MOI) of 0.2 or mock (vehicle only, 10% glycerol in phosphate-buffered saline [PBS]) for 1 hour at 37°C. The inoculum was removed, and the cells were seeded at 10 cells per well in 100 μl and incubated in normoxia or hypoxia for 12 days for GBM4 and 16 days for GBM8. Sphere numbers were counted for each condition, and the results were normalized to the normoxic mock condition.
Cell Growth Curves
After sphere dissociation, 2 × 104 GBM4, GBM6, and GBM8 cells were seeded in a 24-well plate and cultured in normoxia or hypoxia. Viable cells (trypan blue-excluding) were counted on days 3 and 6 on a hemocytometer. For cell proliferation from a clonogenic density, dissociated GBM4 cells were seeded at one cell per μl in 4 ml (T25 flask), kept in culture for 12 days in normoxia and hypoxia, and then counted on a hemocytometer after trypan blue.
Reverse Transcription-PCR
GBM8 cells were grown in hypoxia or normoxia for 24 hours and then harvested. Total RNA was extracted using TRIzol (Invitrogen). Reverse transcription was performed using the SuperScript III first-strand synthesis system for reverse transcription (RT)-PCR (Invitrogen). PCR was performed on a Peltier Thermal Cycler (PTC 200; MJ Research, Watertown, MA, http://www.mjr.com) using human CD133 (forward: CAGGTAAGAACCCGGATCAA; reverse: TCAGATCTGTGAACGCCTTG) and human glyceraldehyde-3-phosphate dehydrogenase (forward: CAATGACCCCTTCATTGACC; reverse: GACAAGCTTCCCGTTCTCAG) primers combined with the HotStarTaq Plus kit (Qiagen, Hilden, Germany, http://www1.qiagen.com). All of the reactions began with 5 minutes at 95°C for the polymerase activation; followed by 35 cycles at 95°C for 15 seconds (denaturation), 55°C for 10 seconds (annealing), and 72°C for 15 seconds (extension); and then a 10-minute final extension at 72°C.
Virus Yield Assay
GBM4 and GBM8 cells were dissociated, seeded in EF20 medium at 2 × 104 cells per well in 24-well plates, infected with G47Δ at an MOI of 1.5 and incubated in normoxia or hypoxia. The cells were harvested with supernatant at 12, 24, and 48 hours after infection and processed with three freeze/thaw cycles and sonication. The virus titer was then determined by plaque assay on Vero cells (American Type Culture Collection, Manassas, VA, http://www.atcc.org).
For GBM4, additional experiments were performed. A total of 2 × 104 cells seeded per well in 24-well plates were infected with G47Δ (MOI = 1.5), incubated for 1 hour in normoxia to allow the virus to enter cells, and then incubated in normoxia or hypoxia. To test whether hypoxic pretreatment changes virus yield, an equal number of cells were incubated 24 hours in normoxia and hypoxia before infection with G47Δ (MOI = 1.5) and immediately incubated under the same oxygen concentrations after infection. The cells were harvested with supernatant at 12, 24, and 48 hours after infection and processed for plaque assay as described.
To characterize G47ΔUs11fluc, Vero cells were seeded in 96-well plates, infected at an MOI of 0.5 in the presence or absence of acyclovir (10 μM; Sigma-Aldrich), and cultured in normoxia. After adding d-luciferin (Gold Bio, St. Louis, http://goldbio.com) to cells to 2 mM, bioluminescence was immediately detected using a microplate reader (Synergy-HT; Bio-Tek, Winooski, VT, http://www.biotek.com) at different time points. The virus titer from G47ΔUs11fluc-infected Vero cells (MOIs of 0.01, 0.1, and 1) was determined by plaque assay and correlated with bioluminescence emission.
To assess the impact of hypoxia on late viral gene expression, dissociated GSCs were seeded at a density of 8,000 cells per well in 96-well plates, incubated overnight in normoxia and hypoxia, infected with G47ΔUs11fluc at MOI of 1 or mock, and incubated in the respective oxygen concentrations. At 24 and 48 hours postinfection, bioluminescence was measured as described above. The experiments were done in triplicate.
Cell Killing Assay
Single-cell suspensions of GBM4, GBM6, and GBM8 were seeded in 24-well plates at a density of 2 × 104 cells per well and incubated in normoxia or hypoxia overnight. They were then infected with G47Δ at an MOI of 0.2 and incubated again in normoxia or hypoxia, respectively. The cells were harvested at days 3 and 6 and dissociated with trypsin/EDTA, and viable trypan blue-excluding cells were counted on a hemocytometer.
Flow Cytometric Analysis
GSCs grown in normoxia or hypoxia for 48 hours were dissociated, stained with allophycocyanin (APC)- or phycoerythrin-conjugated anti-CD133/2 (Miltenyi Biotec, Bergisch Gladbach, Germany, http://www.miltenyibiotec.com) following the manufacturer's protocol, and then analyzed using a FACSCalibur (BD Biosciences, San Diego, http://www.bdbiosciences.com). 4FCS and 8FCS cells grown in normoxia or hypoxia for 48 hours were treated with Versene (Gibco, Grand Island, NY, http://www.invitrogen.com) and subjected to the same procedures.
CD133 expression, oHSV-1 infection, and cell death were simultaneously analyzed using a three-color flow cytometric analysis. GSCs were cultured in hypoxia for 48 hours, infected with G47ΔBAC (EGFP+, MOI 0.2) or mock (vehicle only, 10% glycerol in PBS), and incubated in hypoxia again. The cells were collected 72 hours after infection and stained with APC-conjugated anti-CD133 antibody and 7-aminoactinomycin D (7AAD; BD Biosciences) before fluorescence-activated cell sorting (FACS) analysis. The data were analyzed using FlowJo software (Tree Star, Ashland, OR, http://www.treestar.com).
Western Blots
Following treatment, the cells were washed once with PBS, resuspended in RIPA buffer (Boston BioProducts, Ashland, MA, http://www.bostonbioproducts.com) with protease inhibitors (Complete Mini; Roche Diagnostics, Mannheim, Germany, http://www.roche-applied-science.com), lysed on ice for 30 minutes, and centrifuged for 10 minutes to separate cell debris. Protein concentrations of each sample were determined by modified Bradford assay (Bio-Rad, Hercules, CA, http://www.bio-rad.com). Twenty micrograms of proteins were separated on a 7.5% or 12% SDS-polyacrylamide gel electrophoresis gel, transferred to polyvinylidene difluoride membrane (Bio-Rad), and incubated with primary antibodies overnight at 4°C. The next day membranes were incubated with secondary peroxidase-conjugated antibodies for 1 hour at room temperature. The proteins were visualized using the ECL Plus Western blotting detection system (GE Healthcare, Little Chalfont, U.K., http://www.gehealthcare.com) on Kodak films.
The primary antibodies used were as follows: 1:500 rabbit anti-CD133 monoclonal (Cell Signaling Technology, Beverly, MA, http://www.cellsignal.com); 1:500 mouse anti-HIF1α monoclonal (BD Biosciences); 1:2,000 mouse anti-vinculin monoclonal (Thermo Scientific); 1:2,000 rabbit anti-actin polyclonal (Sigma-Aldrich); and 1:2,000 mouse anti-ICP4 monoclonal (United States Biological, Swampscott, MA, http://www.usbio.net). Secondary antibodies were 1:2,000 horseradish peroxidase-conjugated anti-rabbit IgG and anti-mouse IgG (Promega).
In Vivo Studies: Immunohistochemistry
Fifty thousand GBM4 cells resuspended in 3 μl were stereotactically implanted in the right striatum (2.5 mm lateral from bregma and 2.5 mm deep) of SCID mice (National Cancer Institute) under anesthesia with pentobarbital as previously described [15]. Forty days later, 2 × 106 plaque-forming units of G47ΔUs11fluc (in 4 μl) (three mice) or 4 μl of PBS (three mice) were stereotactically injected using the same coordinates as for tumor cell implantation. Twenty-four hours after viral/PBS injections, the animals received 60 mg/kg Hypoxyprobe (pimonidazole; Hypoxyprobe, Burlington, MA, http://www.hypoxyprobe.com) i.p. and were sacrificed 1 hour later. Brains were embedded in Tissue-Tek OCT Compound (Sakura Finetek, Torrance, CA, http://www.sakura.com), and frozen sections were obtained (7 μm). The sections were fixed with 4% paraformaldehyde (PFA) and stained with hematoxylin and eosin and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) to visualize the lesion and the viral infection, respectively.
For immunohistochemistry, the sections were fixed with 4% PFA and cold acetone, washed with PBS, blocked with 10% goat serum for 30 minutes at room temperature, and incubated overnight at 4°C with anti-HP (mouse, 1:50) and/or fluorescein isothiocyanate-conjugated anti-luciferase (goat, 1:500; Lifespan Biosciences, Seattle, WA, http://www.lsbio.com) antibodies. Cy3-conjugated anti-mouse IgG antibody (1:150; Jackson Immunoresearch Laboratories, West Grove, PA, http://www.jacksonimmuno.com) was applied at room temperature for 90 minutes. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Fluorescence microscopic pictures were captured from four to six randomly chosen slides, and approximately 1,000–2,000 cells were counted from each tumor.
Statistics
Differences between normoxia and hypoxia in cell growth, limiting dilution, sphere size, viral yield, and virus killing assays were compared using a two-tailed unpaired t test. p values <.05 were considered significant. Prism (GraphPad Software, Inc., San Diego, http://www.graphpad.com) and Excel (Microsoft, Redmond, WA, http://www.microsoft.com) were used for analysis.
Results
Hypoxia Enhances GSC Self-Renewal, Proliferation, and CD133 Expression
We first studied the effects of hypoxia on the stem-like characteristics of our GSCs in vitro. Neurospheres of GBM4, GBM6, and GBM8 GSC were dissociated and cultured in normoxia or hypoxia, and we noted that the average size of the resulting spheres was significantly larger when grown in hypoxia than in normoxia (Fig. 1A, 1B), suggesting that hypoxia promoted GSC proliferation [9, 16]. To assess the impact of oxygen concentrations on self-renewal of GSCs, a limiting dilution clonogenicity assay was performed. When compared with normoxia, hypoxia significantly increased the fraction of cells capable of forming spheres in the three different GSC cultures at three clonogenic cell densities (Fig. 1C). These data suggest that hypoxia promotes GSC self-renewal, which is consistent with the observations published previously [21, 22].
Figure 1.

Effect of hypoxia on in vitro growth of glioblastoma stem-like cells. (A): Phase contrast microscopic images of GBM4 spheres cultured in EF20 medium in normoxia (left panel) and in hypoxia (right panel). Both are ×10 magnification. Scale bar = 100 μm. (B): Sphere diameter of GBM4 and GBM8 cells cultured at clonogenic density in normoxia (white bars) or hypoxia (black bars). (C): Sphere forming assay with GBM4, GBM6, and GBM8 cells plated at 10, 100, or 1,000 cells per milliliter and cultured in normoxia (white bars) or hypoxia (black bars). Spheres were measured 12 days after plating GBM4 and 16 days after plating GBM6 and GBM8 for both normoxic and hypoxic cells. (D): Dissociated GBM4 cells were plated at clonal density and cultured in normoxia and hypoxia, and viable cells were counted 12 days later. (E): Dissociated GBM4 (left panel) and GBM8 (right panel) cells were plated at nonclonal density, and viable cells were counted on days 3 and 6 of culture. The error bars represent standard deviations. ∗, p < .05; ∗∗, p < .01. Abbreviation: GBM, glioblastoma.
Cell counting assay confirmed that hypoxic culture led to a higher number of viable cells than normoxic culture when GBM4 GSCs were seeded at a clonogenic density (Fig. 1D). However, there was no significant difference in cell number between hypoxic and normoxic conditions when cells were seeded at a nonclonogenic density (Fig. 1E). Thus, hypoxia-enhanced GSC proliferation may be cell density-dependent.
To further characterize a possible hypoxia-induced enrichment of GSCs, we analyzed the expression of the putative GSC marker CD133. RT-PCR analysis showed an increase of CD133 mRNA levels when GBM8 cells were incubated in hypoxic conditions (1% oxygen for 24 hours) (Fig. 2A). In GBM4 and GBM8 cells, CD133 protein expression progressively increased over a 24-hour period in hypoxia, whereas the expression of HIF1α increased and peaked at 12 hours (Fig. 2B). Western blot analysis of GBM4 cells grown under normoxia and hypoxia and collected at the same time points showed significant upregulation of CD133 expression only in hypoxic conditions (Fig. 2C). Flow cytometric analysis demonstrated different levels of CD133 expression in GBM4 (64.6%), GBM6 (0.5%), and GBM8 (73.7%) in normoxia. When cells were cultured in hypoxia for 48 hours, the fractions of CD133-positive cells rose in all of the three GSC cultures to 79.7% (GBM4), 3.35% (GBM6), and 90.4% (GBM8) (Fig. 2D and supplemental online Fig. 1A). In contrast, flow cytometric analysis of established 4FCS and 8FCS cells (the non-GSC tumor cell counterparts of GBM4 and GBM8 GSCs) cultured for 48 hours at 21% or 1% of oxygen did not reveal any change in CD133 expression (supplemental online Fig. 1B). Taken together, these results demonstrate the increased stem-like properties of GSCs cultured at 1% as opposed to 21% oxygen, as illustrated by enhanced self-renewal and stem cell marker expression and described by others [9, 16, 19, 22].
Figure 2.

Effect of hypoxia on CD133 in glioblastoma stem-like cells. (A): Reverse transcription-polymerase chain reaction showing hypoxia-induced CD133 mRNA expression in GBM8 (24-hour incubation in hypoxia). (B): Western blot showing the expression of CD133 and HIF1α following the initiation of hypoxic culture of GBM4 and GBM8. Note the difference in upregulation kinetics between the two proteins. Vinculin was used as a loading control. (C): Western blot showing time course of CD133 protein expression in GBM4 cells cultured in normoxia and hypoxia. β-Actin was used as loading control. (D): Hypoxia-related CD133 upregulation quantified by fluorescence-activated cell sorting analysis in GBM4, GBM6, and GBM8 incubated in hypoxia for 48 hours (right panel). In the left panel is the normoxic control. Abbreviations: APC, allophycocyanin; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GBM, glioblastoma; H, hypoxia; HIF, hypoxia inducible factor; N, normoxia; PE, phycoerythrin.
G47Δ Infects, Replicates in, and Kills GSCs in Hypoxia
We previously showed that oHSV-1 G47Δ (ICP6−, γ34.5−, and ICP47−) was able to infect, replicate, and spread in human GSCs [15]. Herein, we characterized the effect of hypoxia on the efficacy of G47Δ in GSCs. We first tested whether normoxic and hypoxic conditions influence G47Δ replication in GSCs. Virus yields were comparable in normoxic and hypoxic GBM8 cells at 12–48 hours after infection, whereas there was a significant decrease in viral yield in hypoxic versus normoxic GBM4 cells (1.9–2.1-fold) and at 24 hours (2.1-fold) with GBM6 cells (Fig. 3A). To understand how low oxygen levels reduced virus yield in GBM4, additional experiments were performed. To test whether hypoxia during infection inhibited virus entry or early infection events, the cells were infected and incubated for 1 hour in normoxia followed by incubation in hypoxia (Fig. 3B, left panel). This 1-hour normoxic incubation following infection did not change the profile of virus replication in hypoxic GBM4, suggesting that the reduced virus yield in hypoxia is due to slightly suppressed viral replication and not early events in infection. We next examined whether prior exposure to hypoxia, as would occur in a tumor, would further reduce virus yield. The cells were pretreated for 24 hours in hypoxia before infection and then cultured in hypoxia postinfection (Fig. 3B, right panel). This also did not further reduce virus yields in GBM4 cells, indicating that hypoxic preincubation of GBM4 cells, with an increase in the GSC population (Fig. 1), does not affect virus replication (Fig. 3B, right panel). In 4FCS cells, virus yield was not significantly reduced in hypoxia but overall was similar to GBM4 (Fig. 3C left). In contrast, the virus yields in 8FCS were significantly reduced in hypoxia (2.3–4.3-fold) compared with GBM8 (Fig. 3C, right panel).
Figure 3.

Effect of hypoxia on G47Δ replication and late herpes simplex virus gene expression in glioblastoma stem-like cells. (A): G47Δ (MOI = 1.5) replicates in GBM4 (left panel), GBM6 (middle panel), and GBM8 (right panel) in the hypoxic condition (1% O2; black bars), as compared with normoxia (white bars). (B): Virus yield in GBM4 cells did not vary with timing of hypoxia: incubated in normoxia for 1 hour after infection and then cultured in hypoxia for 48 hours (left panel) or preincubated in hypoxia for 24 hours before infection and then incubated for 48 hours after infection (right panel). (C): G47Δ virus yield in 4FCS (left panel) and 8FCS (right panel) cells, in hypoxia (black bars) compared with normoxia (white bars). (D): Luciferase activity in GBM4 (left panel), GBM6 (middle panel), and GBM8 (right panel) cells infected with G47ΔUs11fluc (MOI = 1) in hypoxia (black bars) and normoxia (white bars). The error bars represent standard deviations. ∗, p < .05; ∗∗, p < .01. Abbreviations: FCS, fetal calf serum; GBM, glioblastoma; MOI, multiplicity of infection.
Productive infection of HSV involves a regulated cascade of expression of α (immediate early), β (early), and γ (late) genes, with the expression of true late genes (γ2) strictly requiring replication of viral DNA. To follow oHSV replication in vivo and measure the impact of hypoxia on late gene expression, we constructed recombinant oHSV G47ΔUs11fluc that carries the firefly luciferase gene driven by the true late Us11 promoter. Measurement of bioluminescence after G47ΔUs11fluc infection of Vero cells was detectable only after 16 hours postinfection, and bioluminescence intensity correlated with virus yield (supplemental online Fig. 2). GBM4, GBM6, and GBM8 GSCs were infected with G47ΔUs11fluc and cultured in normoxia and hypoxia, and bioluminescence was measured at 24 and 48 hours after infection. In the GSCs, bioluminescence level profiles recapitulated those of the G47Δ virus yield determined by plaque assay (Fig. 3D); bioluminescence, representing true late promoter activity, was significantly decreased in hypoxic GBM4 cells compared with normoxic cells (2.1-fold), whereas there was no difference between normoxic and hypoxic GBM8 cells, and overall levels were lower in GBM6, with a 49% decrease in hypoxia at 24 hours after infection.
These data demonstrate that hypoxia does not interfere to a great extent with G47Δ replication in GSCs. Interestingly, GSCs and FCS cells derived from the same tumors can display different G47Δ replication profiles in normoxia versus hypoxia.
G47Δ Kills GSCs and Reduces Their Sphere-Forming Ability in Hypoxia
We next sought to determine whether G47Δ is capable of killing GSCs cultured in hypoxia as well as in normoxia. G47Δ killing of normoxic or hypoxic GSCs was evaluated at 3 and 6 days after infection (MOI of 0.2) (Fig. 4A). Although cytotoxicity was comparable on day 3 between the two oxygen concentrations, the fraction of GBM4 GSCs killed 6 days after infection was somewhat less in hypoxia, likely because of reduced viral replication in GBM4 (as shown in Fig. 3). GBM8 was more susceptible to G47Δ killing on day 3 than GBM4, possibly because of increased virus spread in GBM8, and nearly 95% killing was observed in both normoxia and hypoxia.
Figure 4.
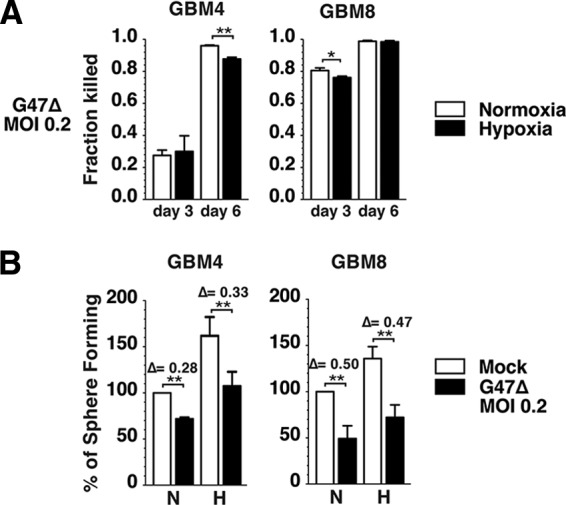
G47Δ kills glioblastoma stem-like cells (GSCs) and suppresses GSC sphere formation in hypoxia. (A): G47Δ mediates killing of normoxic (white bars) and hypoxic (black bars) GBM4 (left panel) and GBM8 (right panel) cells. The data from each infected condition were normalized to those of the corresponding mock control. (B): Sphere forming assay (100 cells per milliliter) performed in normoxia and hypoxia with GBM4 (left panel) and GBM8 (right panel) cells uninfected (mock; white bars) or infected with G47Δ (MOI = 0.2; black bars). Sphere numbers were counted 12 days after plating GBM4 and 16 days after plating GBM8 in both normoxic and hypoxic samples. The data of normoxic-infected, hypoxic-mock, and hypoxic-infected conditions were normalized to the normoxic-mock condition, which was considered 100%. There was no significant difference in virus-mediated reductions between normoxia and hypoxia. The error bars represent standard deviations. Δ, reduction, indicating the effect of G47Δ normalized to mock for both normoxia and hypoxia. ∗, p < .05; ∗∗, p < .01. Abbreviations: GBM, glioblastoma; H, hypoxia; MOI, multiplicity of infection; N, normoxia.
Because self-renewal represents one of the paramount characteristics of GSCs, we asked whether oHSV infection has an impact on self-renewal enhanced by hypoxia. To test this, a limiting dilution assay of GBM4 and GBM8 was performed using 100 GSCs per milliliter in normoxia and hypoxia after either mock or G47Δ infection. G47Δ infection at a relatively low MOI (0.2) significantly suppressed the ability of GSCs to form spheres in normoxia (Fig. 4B). Importantly, G47Δ infection resulted in a similar degree of clonogenicity suppression in both normoxia and hypoxia in GBM4 (28% reduction in normoxia vs. 33% hypoxia) and GBM8 (50% reduction in normoxia vs. 47% hypoxia) (Fig. 4B). Thus, G47Δ efficiently kills GSCs and inhibits their sphere-forming ability in both normoxic and hypoxic conditions.
G47Δ Infects and Kills CD133+ Enriched Population and Mediates CD133 Downregulation
We next determined whether oHSV infection could counteract the hypoxia-induced upregulation of stem cell marker CD133 and alter the proportion of CD133+ cells. GBM4 and GBM8 cells were dissociated, incubated for 48 hours in hypoxia, and then infected with green fluorescent protein (GFP)-expressing G47ΔBAC (MOI of 0.2) or mock. Hypoxic culture was continued for 72 hours, and flow cytometric analysis was performed to measure GFP+ (infected cells) and CD133+ and 7AAD+ (cell death) cells (Fig. 5A). G47ΔBAC infection of GBM4 and GBM8 cells yielded 64% and 59% GFP positivity, respectively, and reduced the fraction of CD133-positive cells (alive and dead) by 39% (from 83% to 44%) in GBM4 and 52% (from 77% to 25%) in GBM8. Virus infection also resulted in cell death as demonstrated by 7ADD positivity in 39% of GBM4 cells (compared with 5% in mock) and 72% of GBM8 cells (compared with 15% in mock) (Fig. 5A), confirming the difference in G47Δ sensitivity between the two GSCs demonstrated in Figure 4A (day 3). As a result, the percentage of viable (7AAD-negative) CD133+ cells remaining in infected samples was markedly reduced to 28% and 12%, as opposed to 78% and 68% in mock-infected samples, in GBM4 and GBM8 cells, respectively.
Figure 5.

G47Δ infection counteracts hypoxia-induced enrichment of CD133+ glioblastoma stem-like cells. (A): After 48-hour hypoxic culture, GBM4 (left panels) and GBM8 (right panels) cells were mock-infected (upper panels) or oncolytic herpes simplex virus (oHSV)-infected (enhanced green fluorescent protein-expressing G47ΔBAC; MOI = 0.2; lower panels) and cultured for 72 hours in hypoxia. CD133, GFP (infection), and 7AAD (cell death) were quantified by flow cytometry, and the values in each quadrant are the percentages of cells. (B): Western blot showing G47Δ-mediated abrogation of hypoxia-induced upregulation of CD133 protein in GBM4 (left panel) and GBM8 (right panel) cells. ICP4 (HSV immediate-early protein) bands demonstrate oHSV infection, and vinculin served as loading control. −, mock cell culture conditions (24 hours); +, G47Δ cell culture conditions (MOI = 1.5 for 24 hours). Abbreviations: 7AAD, 7-aminoactinomycin D; GBM, glioblastoma; GFP, green fluorescent protein; H, hypoxia; MOI, multiplicity of infection; N, normoxia.
We also examined CD133 protein expression under different assay conditions, with dissociated GBM4 and GBM8 cells cultured in normoxia or hypoxia for 24 hours, then infected with G47Δ at MOI 1.5, and then incubated in the same oxygen conditions for an additional 24 hours. Western blot analysis showed that G47Δ infection, evidenced by viral ICP4 expression, abrogated the CD133 overexpression mediated by hypoxia in the GSCs (Fig. 5B). Taken together, these data indicate that CD133+ cells expanded via hypoxia are susceptible to infection and killing by G47Δ, which is associated with a concomitant reduction in CD133 levels and the proportion of CD133+ cells.
G47ΔUs11fluc Infects and Replicates in Hypoxic Cells In Vivo
Lastly, we sought to determine whether the in vitro ability of oHSV to infect and replicate in hypoxic GSCs is translatable to an in vivo setting. Orthotopic engraftment of GBM4 GSCs in mice produced a discrete nodular tumor (Fig. 6A, left) [15]. A single intratumoral injection of G47ΔUs11fluc resulted in widespread viral infection within the tumor, as visualized by X-gal histochemistry (LacZ-expressing cells; Fig. 6A, right). In mock-treated tumors, hypoxic areas, as visualized by Hypoxyprobe immunofluorescence, were scattered and frequently situated in the central core of tumors, whereas no immunoreactivity was detected in the contralateral, non-tumor-bearing hemisphere (Fig. 6B, CTRL and PBS). G47ΔUs11fluc infection seemed to change the distribution pattern of hypoxic lesions, possibly through alterations in vascularity and/or immune responses, leading to less circumscribed Hypoxyprobe-positive areas compared with the PBS-treated tumors (Fig. 6B). There was no significant difference in the proportion of Hypoxyprobe-positive cells between mock- and G47ΔUs11fluc-treated tumors (Fig. 6C). Immunofluorescent staining of firefly luciferase demonstrated that a significant fraction of the tumor cells were immunopositive in virus-injected tumors, with no signal in PBS-treated tumors (Fig. 6B, Luciferase). Double immunofluorescent staining for Hypoxyprobe and luciferase revealed the presence of tumor cells that were positive for both Hypoxyprobe and luciferase (Fig. 6D, white arrows). The percentage of luciferase+ cells in nonhypoxic (Hypoxyprobe-negative) versus hypoxic (Hypoxyprobe-positive) areas was comparable in different animals (Fig. 6E, left panel) and not statistically different (Fig. 6E, right panel). Since luciferase expression is driven by the late Us11 promoter, the results indicate that oHSV replication takes place in vivo in hypoxic areas of GBM and that G47Δ does not discriminate between normoxic and hypoxic cells within the tumor.
Figure 6.

G47ΔUs11fluc infects and replicates in hypoxic GBM cells in vivo. (A): Brain coronal sections of GBM4 xenografts stained with hematoxylin and eosin (left) and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (blue; right), showing the nodular tumor (left) and viral spread in the tumor (right). (B): Immunofluorescent staining of contralateral non-tumor-bearing brain hemisphere (left panel) and GBM4 xenografts treated with PBS (middle panel) or G47ΔUs11fluc (right panel). Hypoxic areas (Hypoxyprobe, red; upper row), luciferase immunopositivity (green; middle row), and cellular nuclei (DAPI, blue; lower row) (magnification, ×10). (C): Percentages of hypoxic cells within tumors treated with PBS (white bar) and G47ΔUs11fluc (black bar). The percentages represent the number of Hypoxyprobe+ cells among total number of nucleated (DAPI-positive) cells. (D): Double immunofluorescence (merged images) using anti-Hypoxyprobe (red) and anti-luciferase (green) antibodies, with double positive cells (yellow, white arrows) (magnification, ×10 in the left panel and ×20 in the right panel). Scale = 100 μm (B, D). (E): Percentages of luciferase+ cells in normoxic (white bar) and hypoxic (black bar) tumor regions. Results from individual mice are shown in the left panel, and the averages of the three mice are shown in the right panel. The error bars represent standard deviations. Abbreviations: CTRL, contralateral; DAPI, 4′,6-diamidino-2-phenylindole; GBM, glioblastoma; n.s., not significant; PBS, phosphate-buffered saline.
Discussion
Hypoxia is a well-characterized feature of many solid tumors, including GBM [4, 8]. The role of hypoxia in promoting the expansion of the GSC compartment and maintaining its undifferentiated state has recently been described, and it may be one of the factors contributing to chemotherapy and radiotherapy resistance in hypoxic tumor cells [9, 16, 18, 22]. Therefore, the effect of hypoxia on GSC populations poses an unsolved challenge, and developing new therapeutic strategies to counteract the hypoxia-enhanced pathological potency of GSCs is important.
Following on our previous demonstration of the ability of oHSV-1 to kill GSCs in normoxia, we evaluated the efficacy of G47Δ in hypoxic GSCs in vitro and in vivo [15, 39]. First we tested whether hypoxia induces enrichment in the GSC compartment in our patient-derived GBM neurosphere cultures in vitro. Next we investigated the replication and cell killing properties of oHSV G47Δ in hypoxic GSCs in vitro and the effects of G47Δ on GSC self-renewal and CD133 expression, which are enhanced by hypoxia. Lastly we examined G47Δ replication in hypoxic tumor cells in vivo, using a newly developed oHSV vector, G47ΔUs11fluc. We found the following: (a) established GBM neurosphere cultures respond to hypoxia by increasing the GSC compartment in terms of self-renewal and CD133 expression; (b) G47Δ can infect, replicate, and kill GSCs in hypoxia and in normoxia; (c) G47Δ infection reduces sphere-forming ability and CD133 expression in GSCs cultured in hypoxia; and (d) G47ΔUs11fluc effectively infects and replicates in vivo in hypoxic tumor areas.
Our established cultures of human GSCs were tested for their response to hypoxia. Hypoxic culture of GSCs resulted in larger sphere size and enhanced self-renewal, which is reported to be an independent outcome predictor in GBM [40]. Although CD133 is controversial as a GSC identification marker and was shown to be a marker for bioenergetic stress in U251MG human glioma cell lines cultured at 1% oxygen, the link between its expression and clinical outcome in glioma patients suggests a role for CD133+ cells in GBM progression and therapeutic resistance [22, 27, 28, 41–44]. Studies have shown that hypoxia promotes the expansion of CD133+ populations through the activation of HIFs [21, 22]. We show that GSCs cultured in hypoxia have increased expression of CD133 at both the mRNA and protein levels in a time-dependent manner. In accord with what we have reported, we found no clear correlation between the proportion of CD133 positivity and sphere-forming ability in the GSCs cultures we tested, because CD133-low GBM6 had higher efficiency of sphere formation than CD133-high GBM8 [15]. We speculate that a fraction of CD133-negative cells have a clonogenic potential, as described by other investigators [14, 28, 29]. CD133 may not be a marker of GSCs but may respond to in vitro conditions that foster GSCs. In vivo, CD133-high GBM4 and GBM8 GSCs are more efficient in generating aggressive orthotopic tumors than CD133-low GBM6 GSCs, suggesting a possible correlation of CD133 and tumorigenicity [15]. Hypoxic culture resulted in a concomitant increase in CD133 levels and clonogenicity in the tested GSCs, irrespective of their basal CD133 levels, but a possibility remains that hypoxia-induced stem-like cells contain both CD133+ and CD133− fractions. Our findings demonstrate that hypoxia enhances the GSC compartment in GBM neurosphere cultures and provide the basis for investigating the effect of hypoxia on oHSV replication and the ability to target GSCs in the hypoxic niche.
G47Δ is able to infect hypoxia-induced GSC-enriched cultures and to replicate to a relatively similar extent at different oxygen levels (1% vs. 21%). In GBM4, there was a reduction of virus yield in hypoxia compared with normoxia, which was not due to impaired viral entry in hypoxia. Upon virus infection, the cells activate double-stranded RNA-dependent protein kinase (PKR) and endoplasmic reticulum-resident kinase (PERK) that phosphorylate eukaryotic translation initiation factor (eIF) 2α, leading to inhibition of protein synthesis, as part of a natural host response [45]. HSV encodes two proteins to overcome this response: γ34.5, which dephosphorylates phospho-eIF2α, and Us11, which inhibits PKR activation [46]. During hypoxia, both PERK and the amino acid starvation-dependent general control of amino acid biosynthesis kinase GCN2 are activated to phosphorylate eIF2α, which would not be blocked by G47Δ, possibly contributing to decreased replication in hypoxia [47, 48]. Further investigation is needed to clarify the role of phospho-eIF2α in viral infection of hypoxic GSCs. Aghi et al. reported that hypoxia enhanced the replication of the oHSV G207 in U87MG glioma cell lines because of increased GADD34 expression, which complements the lack of γ34.5 [33]. Fasullo et al. similarly found that hypoxia enhanced HSV-1 R3616 (γ34.5-deleted) replication in MDA-MB-231 breast cancer cells [33, 49]. In contrast to G207, we found that G47Δ replication was somewhat reduced in U87MG cells in hypoxia compared with normoxia (data not shown). In comparing GSC versus non-GSC-cultured tumor cells, it is interesting that viral replication in 4FCS cells, in contrast to GBM4, was not decreased in hypoxia, whereas viral replication in 8FCS cells, in contrast to GBM8, was decreased, revealing a different profile of oHSV replication in these two related cultures derived from the same GBM patient specimens. Cell differentiation status may affect cellular responses to hypoxia and susceptibility to virus in a way that has still to be determined. In accord with comparable replication, oHSV G47Δ killing of GSCs was similar in normoxia and hypoxia in vitro. Because hypoxia causes resistance to common therapies such as chemotherapy and radiotherapy, our results highlight a unique and promising property of oHSV G47Δ [8, 33].
G47Δ infection at low MOI (0.2) prior to seeding cells in wells reduced the proportion of cells capable of forming spheres. This occurred with the same efficiency in normoxia and hypoxia (Fig. 4B). G47Δ also counteracts the increase of CD133+ cells induced by hypoxia (Fig. 5). Thus, G47Δ infection inhibits the “stemness” of the hypoxia-enriched GSCs. This is in contrast to radiation therapy, from which CD133+ cells were better able to survive, generating tumors with nearly the same efficiency as nonirradiated cells [23]. CD133 expression was higher in recurrent GBM tumors, following radiation and chemotherapy, than in primary pretreatment tumors from the same patients [24, 26]. Because hypoxia induces the proliferation of GSCs and GSCs are considered as possibly responsible for tumor recurrence, the G47Δ efficacy in reducing the GSCs population in hypoxia may be useful to impede the potential of GSCs to cause recurrence [8, 9, 16, 21, 22, 50]. Further investigations are needed to clarify mechanisms involved, such as the role of HIFs in the “stemness” properties of GSCs and HIF responses to oHSV infection.
In this article, we introduce a new vector, G47Δ Us11fluc, derived from G47Δ that expresses firefly luciferase only during the late phase of virus infection (i.e., after viral DNA replication). With a linear correlation between luciferase activity and virus yield, this virus provides a fast and quantitative reagent to measure virus replication, which was used to compare infected GSCs in normoxia and hypoxia. Importantly, this also allowed us to test whether G47Δ replicates in tumor hypoxic areas in vivo. Visualization of luciferase expression with an anti-luciferase antibody revealed its presence in hypoxic cells inside the tumor with the same frequency as in nonhypoxic tumor cells. Our finding that normoxic and hypoxic tumor cells are similarly permissive to G47Δ in this GSC orthotopic xenograft model suggests that hypoxia does not influence viral activity in vivo.
Conclusion
G47Δ can infect and replicate in GSCs in hypoxic conditions in vitro and in vivo and counteract the hypoxia-enhanced stem cell-like properties of GSCs. Treatment with G47Δ should be an effective strategy to eliminate hypoxic GSCs resistant to conventional therapies and to improve therapeutic outcomes for GBM patients. G47Δ is currently in a clinical trial for recurrent GBM [51]. Further studies to better understand and enhance this effect are indicated for GBM and other cancers.
Acknowledgments
We thank Prof. Pierluigi Longatti (University of Padova) for grant support (to D.S.). This work was supported in part by NIH Grant NS032677 (to R.L.M.).
Author Contributions
D.S. and H.W.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; R.K.: collection and/or assembly of data, data analysis and interpretation, final approval of manuscript; S.D.R.: conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript; R.L.M.: conception and design, financial support, administrative support, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Wen P, Kesari S. Malignant gliomas in adults. New Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD, et al. Lyon, France: IARC Press; 2007. WHO Classification of Tumours of the Central Nervous System. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A. Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell. 2010;7:150–161. doi: 10.1016/j.stem.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 4.Heddleston JM, Li Z, Lathia JD, et al. Hypoxia inducible factors in cancer stem cells. Br J Cancer. 2010;102:789–795. doi: 10.1038/sj.bjc.6605551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bar EE. Glioblastoma, cancer stem cells and hypoxia. Brain Pathol. 2011;21:119–129. doi: 10.1111/j.1750-3639.2010.00460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amberger-Murphy V. Hypoxia helps glioma to fight therapy. Curr Cancer Drug Targets. 2009;9:381–390. doi: 10.2174/156800909788166637. [DOI] [PubMed] [Google Scholar]
- 7.Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–472. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen R. Brain tumor hypoxia: Tumorigenesis, angiogenesis, imaging, pseudoprogression, and as a therapeutic target. J Neurooncol. 2009;92:317–335. doi: 10.1007/s11060-009-9827-2. [DOI] [PubMed] [Google Scholar]
- 9.Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 11.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 12.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 13.Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 14.Kelly JJ, Stechishin O, Chojnacki A, et al. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells. 2009;27:1722–1733. doi: 10.1002/stem.98. [DOI] [PubMed] [Google Scholar]
- 15.Wakimoto H, Kesari S, Farrell CJ, et al. Human glioblastoma-derived cancer stem cells: Establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69:3472–3481. doi: 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heddleston JM, Li Z, McLendon RE, et al. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle. 2009;8:3274–3284. doi: 10.4161/cc.8.20.9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hadjipanayis CG. Tumor initiating cells in malignant gliomas: Biology and implications for therapy. J Mol Med. 2009;87:363–374. doi: 10.1007/s00109-009-0440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panchision DM. The role of oxygen in regulating neural stem cells in development and disease. J Cell Physiol. 2009;220:562–568. doi: 10.1002/jcp.21812. [DOI] [PubMed] [Google Scholar]
- 19.Bar EE, Lin A, Mahairaki V, et al. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol. 2010;177:1491–1502. doi: 10.2353/ajpath.2010.091021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ezashi T, Das P, Roberts RM. Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci USA. 2005;102:4783–4788. doi: 10.1073/pnas.0501283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seidel S, Garvalov BK, Wirta V, et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2α. Brain. 2010;133:983–995. doi: 10.1093/brain/awq042. [DOI] [PubMed] [Google Scholar]
- 22.Soeda A, Park M, Lee D, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1α. Oncogene. 2009;28:3949–3959. doi: 10.1038/onc.2009.252. [DOI] [PubMed] [Google Scholar]
- 23.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 24.Dey M, Ulasov IV, Tyler MA, et al. Cancer stem cells: The final frontier for glioma virotherapy. Stem Cell Rev. 2011;7:119–129. doi: 10.1007/s12015-010-9132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beier D, Wischhusen J, Dietmaier W, et al. CD133 expression and cancer stem cells predict prognosis in high-grade oligodendroglial tumors. Brain Pathol. 2008;18:370–377. doi: 10.1111/j.1750-3639.2008.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeppernick F, Ahmadi R, Campos B, et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res. 2008;14:123–129. doi: 10.1158/1078-0432.CCR-07-0932. [DOI] [PubMed] [Google Scholar]
- 28.Beier D, Hau P, Proescholdt M, et al. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 29.Chen R, Nishimura MC, Bumbaca SM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17:362–375. doi: 10.1016/j.ccr.2009.12.049. [DOI] [PubMed] [Google Scholar]
- 30.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: Results of a phase I trial. Gene Ther. 2000;7:867–874. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 31.Aghi M, Martuza RL. Oncolytic viral therapies: The clinical experience. Oncogene. 2005;24:7802–7816. doi: 10.1038/sj.onc.1209037. [DOI] [PubMed] [Google Scholar]
- 32.Markert JM, Liechty PG, Wang W, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre- and post-tumor resection for recurrent GBM. Mol Ther. 2009;17:199–207. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aghi MK, Liu TC, Rabkin S, et al. Hypoxia enhances the replication of oncolytic herpes simplex virus. Mol Ther. 2009;17:51–56. doi: 10.1038/mt.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Todo T, Martuza RL, Rabkin SD, et al. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci USA. 2001;98:6396–6401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fukuhara H, Ino Y, Kuroda T, et al. Triple gene-deleted oncolytic herpes simplex virus vector double-armed with interleukin 18 and soluble B7–1 constructed by bacterial artificial chromosome-mediated system. Cancer Res. 2005;65:10663–10668. doi: 10.1158/0008-5472.CAN-05-2534. [DOI] [PubMed] [Google Scholar]
- 36.Kibler PK, Duncan J, Keith BD, et al. Regulation of herpes simplex virus true late gene expression: Sequences downstream from the US11 TATA box inhibit expression from an unreplicated template. J Virol. 1991;65:6749–6760. doi: 10.1128/jvi.65.12.6749-6760.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guzowski JF, Singh J, Wagner EK. Transcriptional activation of the herpes simplex virus type 1 UL38 promoter conferred by the cis-acting downstream activation sequence is mediated by a cellular transcription factor. J Virol. 1994;68:7774–7789. doi: 10.1128/jvi.68.12.7774-7789.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuroda T, Martuza RL, Todo T, et al. Flip-Flop HSV-BAC: Bacterial artificial chromosome based system for rapid generation of recombinant herpes simplex virus vectors using two independent site-specific recombinases. BMC Biotechnol. 2006;6:40. doi: 10.1186/1472-6750-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanai R, Wakimoto H, Martuza RL, et al. A novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cells. Clin Cancer Res. 2011;17:3686–3696. doi: 10.1158/1078-0432.CCR-10-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laks DR, Masterman-Smith M, Visnyei K, et al. Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells. 2009;27:980–987. doi: 10.1002/stem.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griguer CE, Oliva CR, Gobin E, et al. CD133 is a marker of bioenergetic stress in human glioma. PLoS One. 2008;3:e3655. doi: 10.1371/journal.pone.0003655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pallini R, Ricci-Vitiani L, Montano N, et al. Expression of the stem cell marker CD133 in recurrent glioblastoma and its value for prognosis. Cancer. 2011;117:162–174. doi: 10.1002/cncr.25581. [DOI] [PubMed] [Google Scholar]
- 43.Nishide K, Nakatani Y, Kiyonari H, et al. Glioblastoma formation from cell population depleted of Prominin1-expressing cells. PLoS One. 2009;4:e6869. doi: 10.1371/journal.pone.0006869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Campos B, Herold-Mende CC. Insight into the complex regulation of CD133 in glioma. Int J Cancer. 2011;128:501–510. doi: 10.1002/ijc.25687. [DOI] [PubMed] [Google Scholar]
- 45.Cheng G, Feng Z, He B. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2α dephosphorylation by the γ134.5 protein. J Virol. 2005;79:1379–1388. doi: 10.1128/JVI.79.3.1379-1388.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mulvey M, Arias C, Mohr I. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J Virol. 2007;81:3377–3390. doi: 10.1128/JVI.02191-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koumenis C, Naczki C, Koritzinsky M, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol Cell Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, Laszlo C, Liu Y, et al. Regulation of G1 arrest and apoptosis in hypoxia by PERK and GCN2-mediated eIF2α phosphorylation. Neoplasia. 2010;12:61–68. doi: 10.1593/neo.91354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fasullo M, Burch AD, Britton A. Hypoxia enhances the replication of oncolytic herpes simplex virus in p53− breast cancer cells. Cell Cycle. 2009;8:2194–2197. doi: 10.4161/cc.8.14.8934. [DOI] [PubMed] [Google Scholar]
- 50.Sermeus A, Cosse JP, Crespin M, et al. Hypoxia induces protection against etoposide-induced apoptosis: Molecular profiling of changes in gene expression and transcription factor activity. Mol Cancer. 2008;7:27. doi: 10.1186/1476-4598-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ino Y. A clinical study of a replication-competent, recombinant herpes simplex virus type 1 (G47Δ) in patients with progressive glioblastoma. [Accessed January 16, 2012];WHO International Clinical Trials Registry. 2009 Available at http://apps.who.int/trialsearch/trial.aspx?trialid=JPRN-UMIN000002661. [Google Scholar]