

The aims of this study were (a) to determine whether human renal cultures (human primary kidney cells [hPKC]) can be enriched in erythropoietin-positive cells (hPKC(F+)) by using magnetic-bead sorting; (b) to characterize the hPKC(F+) following cell separation; and (c) to establish that intrarenal delivery of enriched hPKC(F+) would be more beneficial in the treatment of renal injury, inflammation, and oxidative stress than unsorted hPKC cultures in a chronic kidney injury model. The results indicate that hPKC(F+) may be used as components of cell-based therapies for degenerative kidney diseases.
Keywords: Adult stem cells, Autologous stem cell transplantation, Cell biology, Cellular therapy, Tissue-specific stem cells, Renal fibrosis, Erythropoietin
Abstract
New therapeutic strategies for chronic kidney disease (CKD) are necessary to offset the rising incidence of CKD and donor shortage. Erythropoietin (EPO), a cytokine produced by fibroblast-like cells in the kidney, has recently emerged as a renoprotective factor with anti-inflammatory, antioxidant properties. This study (a) determined whether human renal cultures (human primary kidney cells [hPKC]) can be enriched in EPO-positive cells (hPKC(F+)) by using magnetic-bead sorting; (b) characterized hPKC(F+) following cell separation; and (c) established that intrarenal delivery of enriched hPKC(F+) cells would be more beneficial in treatment of renal injury, inflammation, and oxidative stress than unsorted hPKC cultures in a chronic kidney injury model. Fluorescence-activated cell sorting analysis revealed higher expression of EPO (36%) and CD73 (27%) in hPKC(F+) as compared with hPKC. After induction of renal injury, intrarenal delivery of hPKC(F+) or hPKC significantly reduced serum creatinine, interstitial fibrosis in the medulla, and abundance of CD68-positive cells in the cortex and medulla (p < .05). However, only hPKC(F+) attenuated interstitial fibrosis in the renal cortex and decreased urinary albumin (3.5-fold) and urinary tubular injury marker kidney injury molecule 1 (16-fold). hPKC(F+) also significantly reduced levels of renal cortical monocyte chemotactic protein 1 (1.8-fold) and oxidative DNA marker 8-hydroxy-deoxyguanosine (8-OHdG) (2.4-fold). After 12 weeks, we detected few injected cells, which were localized mostly to the cortical interstitium. Although cell therapy with either hPKC(F+) or hPKC improved renal function, the hPKC(F+) subpopulation provides greater renoprotection, perhaps through attenuation of inflammation and oxidative stress. We conclude that hPKC(F+) may be used as components of cell-based therapies for degenerative kidney diseases.
Introduction
Chronic kidney disease (CKD) is associated with high cardiovascular morbidity and mortality, poor quality of life, and high treatment cost [1]. Approximately 13% of the population is affected by CKD worldwide, and the estimated cost for the treatment of this disease for Medicare alone reached $42 billion in 2005 [2, 3]. Although kidney transplantation for end-stage renal disease patients has improved over recent years, the reduced supply of organs is compounded by a steady increase in CKD cases and immune-related problems, thus limiting the availability of this treatment to patients [4]. Pharmacological treatments for CKD are often directed toward blood pressure control (angiotensin-converting enzyme inhibitors and angiotensin receptor 1 antagonists), reduction in atherosclerosis (statins), and improvement of anemia (erythropoietin-like agents). Although these drugs can improve renal function and promote renal repair (particularly angiotensin-converting enzyme inhibitors and angiotensin receptor one antagonists), they require the administration of high doses, and the effect often develops only over several years [2].
The field of regenerative medicine offers new approaches for the structural and functional repair of injured organs using cells, scaffolds, and biological factors alone or in combination [2]. The major challenges in the development of successful cell therapies are to identify cell populations with therapeutic potential and to find the appropriate dose and route of administration [5]. In current practice, autologous adult cells are the preferred source for cell therapy, because they induce little or no immune response. Cell therapies targeting the native ability of renal cells for repair and regeneration either via paracrine effects or via creation of a better environment for functional tissue recovery may provide an alternative therapy that avoids immunogenicity, teratogenicity, and ethical concerns involved with some types of stem cells.
Renal repair and regeneration may involve targeting renal fibrosis and factors that may sustain renal damage, such as inflammation or oxidative stress. In this study, we sought to explore the therapeutic potential of a specific cell type, such as erythropoietin (EPO)-producing cells, in a chronic renal injury model. EPO is a cytokine produced by fibroblast-like type I interstitial cells of the renal cortex and outer medulla [6]. Its expression is significantly reduced in patients with CKD because of damage of EPO-producing cells by the disease processes. However, it has recently been shown that EPO not only stimulates erythropoiesis but also has an important role as a protective agent against ischemia-reperfusion injury in the kidney [7]. These tissue-protective effects were mostly attributed to anti-inflammatory and antioxidant properties of EPO [8–10]. Our group and others have shown that human kidney cells derived from a donor kidney can be expanded in vitro and that their function can be preserved as shown by the albumin and hydroxylase uptake assays [11, 12]. The aims of this study were (a) to determine whether human renal cultures (human primary kidney cells [hPKC]) can be enriched for EPO-expressing cells by using a magnetic-bead sorting system in which the beads are conjugated with anti-fibroblast antibodies; (b) to characterize the human renal cell cultures containing erythropoietin-positive cells (hPKC(F+)) following cell separation; and (c) to establish whether intrarenal delivery of enriched hPKC(F+) cells would be more beneficial in the treatment of renal injury, inflammation, and oxidative stress than unsorted hPKC cultures in a chronic kidney injury model.
Materials and Methods
Cell Culture
hPKC cultures were established from a discarded kidney of an adult human donor (51-year-old female) with normal renal function as previously described [11, 13]. All of the procedures were approved by the Institutional Review Board of Wake Forest School of Medicine. The kidneys were placed in Krebs-Ringer bicarbonate buffer (Sigma-Aldrich, St. Louis, http://www.sigmaaldrich.com) supplemented with 1% antibiotic (penicillin/streptomycin; Invitrogen, Carlsbad, CA, http://www.invitrogen.com). Renal capsules and adjacent connective tissues were removed. Pieces of the kidney (cortex and medulla) were minced in Krebs buffer and then digested with liberase blendzyme 3 (Roche, Indianapolis, http://www.roche.com) diluted in 10 ml on a shaker at room temperature for 40 minutes. The resulting cells were passed through a 100-μm BD Falcon strainer (BD Biosciences, San Jose, CA, http://www.bdbiosciences.com) and plated into culture dishes. Renal cells were cultured as heterogeneous populations in media containing a combination of keratinocyte growth medium (supplemented with 2.5% fetal bovine serum, 0.4% insulin transferrin selenium solution, 0.2% epidermal growth factor, and 0.2% bovine pituitary extract) and Dulbecco's modified Eagle medium (supplemented with high glucose [4,500 mg/l] and 10% fetal bovine serum) at a ratio of 1:1 [11].
Cell Separation and Characterization
Because EPO is produced by fibroblast-like cells, hPKC at passage 3 were enriched for EPO-producing fibroblast-like cells using a magnetic activated cell sorting (MACS) kit with magnetic beads attached to antifibroblast antibodies according to the manufacturer's instructions (Miltenyi Biotec, Bergisch Gladbach, Germany, http://www.miltenyibiotec.com).
Fluorescence-Activated Cell Sorting
The unfractionated hPKC and the hPKC(F+) fraction derived following MACS cell separation were characterized using a FACSCalibur flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, http://www.bd.com) as described [11]. Human renal cells were trypsinized, washed, and fixed in 2% paraformaldehyde. For EPO staining, the cells were permeabilized using 0.1% saponin in phosphate-buffered saline (PBS), blocked using serum-free protein block and goat serum, and then incubated with anti-human EPO (1:200; Santa Cruz Biotechnology Inc., Santa Cruz, CA, http://www.scbt.com) antibody. This was followed by incubation with an Alexa Fluor 488-conjugated anti-rabbit secondary antibody (Invitrogen). In addition, the following antibodies were used to characterize hPKC and hPKC(F+): CD73 (ecto-5′-nucleotidase), a surface marker of EPO-producing cells (adenomatous polyposis coli [APC] mouse anti-human; eBioscience Inc., San Diego, http://www.ebioscience.com); CD34, an endothelial cell marker (phosphatidylethanolamine [PE] mouse anti-human; Abcam, Cambridge, U.K., http://www.abcam.com); CD133, a progenitor cell marker (APC mouse anti-human; Miltenyi Biotec); and CD44 (PE mouse anti-human; eBioscience) and CD90 (APC mouse anti-human; eBioscience), markers of mesenchymal stem cells (MSCs). These antibodies were used at a concentration of 1 μg/ml and applied for 30 minutes at room temperature. Appropriate IgG isotype controls were used for each antibody (eBioscience). Cell debris, clumps, and dead cells were eliminated by the intensity of low angle forward scatter and fluorescence. Data from 10,000 events within the live cell gate were collected for each cell sample and analyzed using CellQuest software. There was no difference between the unstained cell population and the IgG control. The cells stained for each protein were compared with cells incubated with the appropriate IgG control.
To confirm the successful enrichment of hPKC populations with EPO-producing cells, we determined EPO gene expression in response to hypoxia in hPKC(F+) and hPKC populations. The cells were cultured at normoxic conditions (37°C, 5% CO2, and 95% atmospheric air) for 12 hours in serum-free medium before being moved into hypoxic conditions in a hypoxia chamber (3% oxygen; Proox model C-21; BioSpherix, Redfield, NY, http://www.biospherix.com). Cells and conditioned media were collected at the basal state (normoxia) and after 24 hours in hypoxia, and then they were frozen at −80°C before being processed for detection of EPO mRNA and protein.
RNA Isolation and Reverse-Transcription Real-Time Polymerase Chain Reaction Assay
RNA was isolated from hPKC and hPKC(F+) samples using the RNeasy kit (Qiagen, Hilden, Germany, http://www.qiagen.com) according to the manufacturer's instructions. RNA concentration was determined by measuring absorbance at 260 nm using a Nanodrop (ND-1000) spectrophotometer (Labtech International, Ringmer, U.K., http://www.labtech.co.uk). The human EPO primer-probe set was purchased from Applied Biosystems (Foster City, CA, http://www.appliedbiosystems.com). All of the reactions were performed in triplicate, and glyceraldehyde-3-phosphate dehydrogenase, amplified using the TaqMan ribosomal RNA control kit (Applied Biosystems), served as an internal control. The results were quantified as Ct values, where Ct is defined as the threshold cycle of polymerase chain reaction at which amplified product is first detected and expressed as the ratio of target to control. The release of EPO from hPKC and hPKC(F+) in response to 24 hours of hypoxia (3% oxygen) was determined in the conditioned medium by using the human EPO tissue culture kit (SECTOR Imager 6000; Meso Scale Discovery, Gaithersburg, MD, http://www.meso-scale.com) according to the manufacturer's instructions.
Animals
Athymic nude male rats (Hsd:RH-Foxn1rnu) were purchased from Harlan Laboratories (Dublin, VA, http://www.harlan.com) at the age of 10–15 weeks. The animals were housed in a temperature-controlled room with a 12-hour light/dark cycle in an Association for Assessment and Accreditation of Laboratory Animal Care-approved facility with free access to food and water. The procedures used in this study were approved by the Wake Forest institutional animal care and use committee. The rats were randomly assigned to study groups.
Ischemia-Reperfusion and Gentamicin Injury Model
Bilateral renal ischemia was achieved by clamping both renal pedicles with vascular clamps for 60 minutes. This was followed by reperfusion after the clamps were removed. Two weeks post-ischemia-reperfusion (IR) injury, gentamicin (GM) was administered in five daily doses (100 mg/kg) to establish chronic kidney disease in the animals. The time of ischemia and the dose of GM were chosen during a preliminary study in which evaluation of renal injury was based on the increases in serum creatinine and urinary albumin seen in the animals. Renal function was stably reduced at 8–10 weeks post-GM treatment.
Human Renal Cell Delivery
Eighteen weeks post-IR-GM treatment, hPKC and hPKC(F+) (passage 3) were resuspended in PBS and injected bilaterally into the lower pole of renal parenchyma at a concentration of 5 × 106 cells per 100 μl per animal. The cell concentration was chosen based on the study by Kelley et al. [14]. The vehicle-only control group received bilateral intrarenal injections of PBS. Blood was collected for the measurements of hemoglobin and serum creatinine 1 week postinjection and then every 4 weeks until the end of the study (12 weeks following cell delivery). Urine was collected at the end of the study by placing the rats in metabolic cages for 24 hours. Finally, the animals were sacrificed, and the kidneys were removed and processed for histology.
Recombinant Human Erythropoietin-α Delivery
A separate group of rats at 18 weeks post-IR-GM treatment was treated with recombinant human erythropoietin-α (rhEPO; Prospec-Tany Technogene Ltd., East Brunswick, NJ, http://www.prospecbio.com) at a dose of 100 IU/kg per day per rat for 12 weeks. rhEPO was diluted in saline and administered intraperitoneally via an osmotic minipump (28 days, 2.5 μl/hour; model 2ML4; Durect Corp., Cupertino, CA, http://www.alzet.com) [15]. Drug delivery was verified by measuring the residual volume of each pump at the end of the study. Serum creatinine and hemoglobin were determined at 1, 4, and 12 weeks following the beginning of rhEPO administration.
Masson's Trichrome Staining
Kidney sections (5 μm) were stained with Masson's trichrome stain to identify collagen fibers. Interstitial fibrosis was indicated by an increased amount of collagen between tubules; the degree of fibrosis was graded higher if more of the parenchyma was involved. The total area of collagen deposition was determined in 12 sections of the digitized images from each kidney (n = 3–4 in each group) and measured as the collagen-to-field ratio using Photoshop 7.0 (Adobe Systems Inc., San Jose, CA, http://www.adobe.com). Glomerulosclerosis was determined as increased collagen deposition within the glomerular or periglomerular area. Each lesion was graded using a 5-point scale: grade 1, minimal; grade 2, mild; grade 3, moderate; grade 4, marked; and grade 5, severe. The data are reported as injury scores that equal the product of a lesion's severity grade and the incidence (number of times lesions of that grade occurred) [16]. Using the same sections, we counted the number of tubular casts per ×200 field.
Immunostaining
Kidney sections (5 μm) were stained using the avidin-biotinylated enzyme complex method as previously described [15]. All of the stains required an antigen retrieval treatment with sodium citrate buffer, pH 6.0, at 90°C–95°C for 30 minutes. Monoclonal CD68 (1:100 dilution; Chemicon, Temecula, CA, http://www.chemicon.com), human nuclear (1:500 dilution; Thermo Scientific, Barrington, IL, http://www.thermoscientific.com), and secondary biotinylated anti-mouse antibodies (1:500 dilution; Vector Laboratories, Burlingame, CA, http://www.vectorlabs.com) were used for the staining. The stained sections were examined under a light microscope and photographed with a Leica DFC 420 camera (Leica Microsystems, Buffalo Grove, IL, http://www.leica-microsystems.com) and SimplePCI (version 6) software (Hamamatsu Corp., Bridgewater, NJ, http://www.hamamatsu.com). The CD68-positive cells were counted using automated counting software (Image-Pro 6.3; Media Cybernetics, Bethesda, MD, http://www.mediacy.com) in seven sections from each kidney (n = 4 in each group). The data are reported as the number of positive cells per ×200 field.
Injury Markers
Levels of urinary albumin and kidney injury molecule 1 (KIM-1) were measured using a rat multiplex kidney injury panel 1 kit (SECTOR Imager 6000; Meso Scale Discovery) according to the manufacturer's instructions. In order to determine the levels of monocyte chemotactic protein 1 (MCP-1) and 8-OHdG, the renal cortex was separated from the medulla, and both tissues were homogenized in a buffer containing HEPES/NaCl and protease inhibitor cocktail (Sigma-Aldrich), pH 7.4. The homogenates were then centrifuged at 20,000g for 30 minutes, and the supernatant from each was used for enzyme-linked immunosorbent assay (ELISA). In addition, urine was collected over a 24-hour period and centrifuged at 2,000g for 15 minutes to separate out debris, and the supernatant was used for the same assays. MCP-1 was measured in renal cortex and medulla and in the urine at the end of the study using a rat ELISA kit (RayBiotech, Inc. Norcross, GA, http://www.raybiotech.com), and the data are expressed as picograms per milligram of tissue, or micrograms per kilogram per day for urinary MCP-1. The levels of 8-OHdG, a marker of DNA oxidation, were determined in the renal cortex and medulla and in the urine using a DNA damage ELISA kit (Stressgen; Enzo Life Sciences, Inc., Farmingdale, NY, http://www.enzolifesciences.com). The data are expressed as nanograms per milligram of tissue weight and micrograms per kilogram per day, respectively.
Statistical Analysis
Based upon previous experience, an n value of 5–6 rats is sufficient to determine significance (80% power to detect an α of 0.05 with an SD of 10 and a difference of 15). Statistical significance of differences was evaluated using t tests or analysis of variance with p values corrected by Tukey's post hoc test. The criterion for statistical significance was p < .05.
Results
hPKC derived from a discarded kidney from an adult human donor (51 years of age, female) were grown as heterogeneous population and expanded in culture. Because EPO is produced by the fibroblast-like interstitial cells in the kidney, we attempted to enrich hPKC cultures for EPO-positive cells using a MACS technique based on fibroblast antibodies attached to magnetic beads. The phase contrast microscopy images (Fig. 1A) show different cell morphologies in hPKC cultures compared with hPKC(F+) cultures, with the majority of cells appearing as typical “fibroblast-like” cells in the hPKC(F+) cultures. In contrast, the hPKC cultures contained a mixed cell population. Growth curves were constructed from unfractionated hPKC and hPKC(F+) following MACS separation. In 3 days, both cell populations doubled [11]. The cells were cultivated for 15 days, and the population doubled approximately eight times (Fig. 1A). Figure 1B demonstrates that cells from both culture types express EPO in a typical cytoplasmic localization pattern. However, fluorescence-activated cell sorting (FACS) data showed that there were 36% more cells expressing EPO in the hPKC(F+) culture (Fig. 1C). The effect of a 24-hour incubation under hypoxic conditions on EPO mRNA levels and EPO protein release in human primary kidney cells is also shown (Fig. 1D). In response to 3% oxygen, EPO mRNA levels increased threefold in hPKC cultures and eightfold in hPKC(F+) cultures. The basal levels of EPO in the conditioned media obtained from these cultures were approximately 4–6 mU/ml in both cultures. Hypoxia (3% O2) induced the release of EPO by 1.5-fold in hPKC and by 2.5-fold in hPKC(F+) cultures (Fig. 1D).
Figure 1.

Phase contrast microscopy images and growth curves of hPKC and hPKC(F+) (A), the immunofluorescent staining (B), fluorescence-activated cell sorting (FACS) analysis for EPO (C), and the effects of 24 hours of hypoxia (3% oxygen) on EPO mRNA levels and protein release by cultured human renal cells (D). (A): The majority of cells in hPKC(F+) showed typical fibroblast-like morphology. The growth curves show that in 3 days both cell populations doubled their number. Overall, the cells were cultured for 15 days, and the population doubled approximately eight times. (B, C): Immunofluorescent staining (B) and FACS analysis (C) for EPO were performed in unfractionated hPKC (left panels) and sorted hPKC(F+) (right panels). (D): The mRNA data were obtained from three different experiments performed in triplicate and expressed as the ratio of EPO to glyceraldehyde-3-phosphate dehydrogenase (left panel). The mRNA data are means ± SE. ∗, p < .05 versus baseline at normoxic condition; n = 2–4. The release of EPO protein into conditioned media was determined using the human EPO tissue culture kit (SECTOR Imager 6000; Meso Scale Discovery). The data are expressed as mU/ml (right panel). Abbreviations: EPO, erythropoietin; hPKC, human primary kidney cell; hPKC(F+), human primary kidney cell containing erythropoietin-positive cells.
hPKC and hPKC(F+) were also characterized using FACS analysis (Fig. 2). CD73, a marker of fibroblast-like cells, was increased in hPKC(F+) cultures by 27.8%. There were minimal changes in endothelial (CD34+, 3.25%) cells. The abundance of renal progenitor (CD133+) cells was similar in both culture types following cell separation. The number of cells expressing mesenchymal stem cell markers was increased only slightly after separation (CD44+, 7.8%; CD90+, 4.75%).
Figure 2.
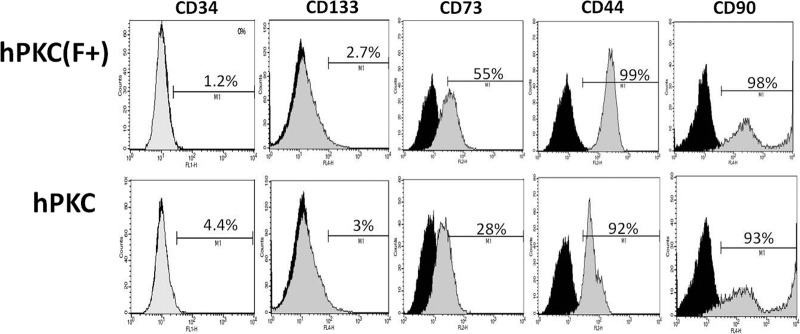
Characterization of hPKC and hPKC(F+) at passage 3 using fluorescence-activated cell sorting analysis. The positive staining for each marker is shown representing the comparison with the respective IgG control. Abbreviations: hPKC, human primary kidney cell; hPKC(F+), human primary kidney cell containing erythropoietin-positive cells.
The IR-GM injury model was found to be a reliable means of establishing a model of chronic renal injury in nude rats (Fig. 3A), and it resulted in significantly elevated serum creatinine levels (untreated: 0.43 ± 0.03 versus IR-GM: 0.76 ± 0.04 mg/dl; ∗, p < .05; n = 13–14), reduced hemoglobin (untreated: 14.45 ± 0.34 versus IR-GM: 13.35 ± 0.4 g/dl; ∗, p < .05; n = 11–12), and increased urinary albumin (untreated: 109.9 ± 26.3 versus IR-GM: 1,939 ± 562.3 μg/day; ∗, p < .05; n = 3–4) in the injured animals. This result represents pooled data from all of the groups studied. Similar levels of albumin excretion were seen as early as 8 weeks following gentamicin injections (data not shown). Thus, we used this model to test the hypothesis that cell therapy with primary renal cells improves kidney function and damage in the injured animals. To do this, cells from either hPKC or hPKC(F+) cultures were injected into both kidneys in some animals, and we looked for changes in the previously described indicators of renal injury in treated and untreated animals.
Figure 3.

Characterization of chronic kidney injury in the nude rat following IR combined with GM administration (A) and the indices of renal function and injury following intrarenal cell delivery in IR-GM rats (B–E). (A): Serum creatinine, hemoglobin, and urinary albumin are shown at 18 weeks after the final gentamicin injection as pooled data from all of the groups studied. The untreated group contains age-matched animals that did not receive the IR-GM treatment. All of the data are the means ± SE. Serum creatinine is expressed as mg/dl; ∗, p < .05 versus untreated group; n = 13–14. Hemoglobin is expressed as g/dl; ∗, p < .05 versus untreated group; n = 11–12. Urinary albumin is expressed as μg/day; ∗, p < .05 versus untreated group; n = 3–4. (B–E): Eighteen weeks post-IR-GM treatment, hPKC and hPKC(F+) at passage 3 were injected bilaterally into the lower pole of renal parenchyma at a concentration of 5 × 106 cells per 100 μl per animal. rhEPO was delivered via an osmotic minipump at the dose of 100 IU/kg per day per rat. The vehicle group consisted of IR-GM rats that received bilateral intrarenal phosphate-buffered saline injections, the hPKC group contained IR-GM rats that received unsorted primary renal cultures into renal parenchyma, and the hPKC(F+) group consisted of IR-GM rats that received primary renal cell cultures enriched in erythropoietin-producing cells. All of the data are shown as the means ± SE. (B): A time course for serum creatinine following cell delivery. The data are expressed as mg/dl, n = 4–6; ∗, p < .05 versus untreated in all groups; #, p < .05 hPKC and PKC(F+) versus vehicle; &, p < .05 versus (F+) baseline. The dotted black line shows levels of serum creatinine in the untreated nude rat. The dotted gray line shows levels of serum creatinine in the rats infused with rhEPO. (C): There were no differences in hemoglobin levels among vehicle, hPKC, and hPKC(F+) groups at 12 weeks after cell delivery. However, rhEPO significantly increased hemoglobin levels at 12 weeks from the beginning of the treatment. (D): Urinary albumin is expressed as μg/day; ∗, p < .05 versus vehicle; #, p < .05 versus hPKC; n = 4 in each group. (E): Urinary KIM-1 is expressed as ng/day; ∗, p < .05 versus vehicle; n = 4–6. Abbreviations: GM, gentamicin; IR, ischemia-reperfusion; hPKC, human primary kidney cell; hPKC(F+), human primary kidney cell containing erythropoietin-positive cells; KIM-1, kidney injury molecule 1; rhEPO, recombinant human erythropoietin-α.
Figure 3B shows the time course of changes in serum creatinine following cell delivery. There were no significant differences between study groups at baseline (preinjury) time points. IR-GM injury significantly increased serum creatinine in both hPKC (untreated: 0.41 ± 0.04 versus IR-GM: 0.77 ± 0.05 mg/dl; ∗, p < .05; n = 6) and hPKC(F+) (untreated: 0.47 ± 0.04 versus IR-GM: 0.67 ± 0.02 mg/dl; ∗, p < .05; n = 4) groups. One week following introduction of the primary renal cells into the kidneys, serum creatinine levels were significantly reduced in the animals receiving cell injections compared with the control (vehicle only) group (∗, p < .05) and remained attenuated until the end of the study (vehicle: 0.76 ± 0.02 versus hPKC: 0.53 ± 0.03 or hPKC(F+): 0.56 ± 0.04 mg/dl; ∗, p < .05; n = 4–6, 12 weeks after cell delivery). Hemoglobin levels were not different among the studied groups at 12 weeks after cell delivery (Fig. 3C). Serum creatinine levels were reduced following rhEPO administration in a manner similar to that seen when hPKC and hPKC(F+) were injected (Fig. 3B). However, hemoglobin levels were higher after rhEPO administration compared with control and with hPKC injection (Fig. 3C). Urinary albumin excretion was attenuated in the animals receiving hPKC(F+) compared with those receiving vehicle only or with those receiving hPKC (hPKC(F+): 207.9 ± 66.52 versus vehicle: 730 ± 96.7 or versus hPKC: 572.4 ± 30.5 μg/day; ∗, p < .05; n = 4 in each group); however, urinary albumin excretion was similar in animals receiving hPKC and those receiving the vehicle only (Fig. 3D). In addition, urinary KIM-1 levels were significantly attenuated in animals receiving hPKC(F+) (hPKC(F+): 1 ± 0.16 versus vehicle: 16.3 ± 6.3 ng/day; ∗, p < .05, n = 4–5). Injections of hPKC tended to decrease urinary excretion of KIM-1, but this was not significant when compared with the vehicle-only group (hPKC: 3.7 ± 1.4 ng/day, n = 5) (Fig. 3E). Body weight was significantly reduced in the vehicle group compared with the hPKC(F+) group (vehicle: 386.6 ± 11.4 versus hPKC(F+): 462.5 ± 28 g; ∗, p < .05; n = 4–5) but was not different compared with the hPKC group (450 ± 28 g; n = 5).
Masson's trichrome staining was used to visualize collagen deposition in the kidney. Collagen, which is shown as a light blue color in Figure 4, was localized to the tubulointerstitial and perivascular areas of the renal cortex and medulla, suggesting that tubulointerstitial and perivascular fibrosis was occurring in the kidney of these animals. In both areas of the kidney, collagen deposition was reduced in the hPKC(F+) group (∗, p < .05). However, in the hPKC group, collagen deposition was attenuated only in the medulla (∗, p < .05). Glomerulosclerosis was significantly reduced in the hPKC and hPKC(F+) groups (injury scores: vehicle: 185.2 ± 30.5 versus hPKC: 61.25 ± 5.2 or versus hPKC(F+): 103.4 ± 17.4; ∗, p < .05 versus vehicle; n = 3–4). The number of tubular casts was significantly reduced in the kidneys of animals receiving hPKC (vehicle: 4.9 ± 0.74 versus hPKC: 0.8 ± 0.2; ∗, p < .05; n = 8 in each group) or hPKC(F+) (vehicle: 4.9 ± 0.74 versus hPKC(F+): 1.4 ± 0.3; ∗, p < .05; n = 6–8).
Figure 4.

Collagen deposition in the kidneys of the ischemia-reperfusion-gentamicin (IR-GM) rats at 12 weeks following treatment. Total collagen deposition was determined by Masson's trichrome staining and is visible as a light blue color localized to the tubulointerstitial and perivascular areas of the renal cortex and medulla of IR-GM rats (top panel). Magnification, ×400. The lower panel shows the analysis of the total collagen deposition and glomerulosclerosis in the kidney. The data are expressed as collagen-to-field ratios; ∗, p < .05 versus vehicle group; n = 3–4. Abbreviations: hPKC, human primary kidney cell; hPKC(F+), human primary kidney cell containing erythropoietin-positive cells.
The abundance of inflammatory cells (CD68-positive macrophages and monocytes) was significantly reduced following the introduction of either hPKC or hPKC(F+) in both cortical (vehicle: 174.9 ± 11.36 versus hPKC: 33.03 ± 13.11 or versus hPKC(F+): 69.37 ± 35.8 cells; ∗, p < .05; n = 3 in each group) or medullary regions of the kidney (vehicle: 65.8 ± 19.7 versus hPKC: 4.25 ± 0.9 or versus hPKC(F+): 15.9 ± 4.8 cells; ∗, p < .05; n = 3 in each group) (Fig. 5). The levels of MCP-1, a chemoattractive factor that plays a role in the recruitment of monocytes to an injury site, were significantly reduced in the renal cortex in the hPKC(F+) group (vehicle: 112 ± 13.7 versus hPKC(F+): 60.65 ± 4.6 pg/mg tissue; ∗, p < .05; n = 4) but not in the hPKC group (Fig. 6). There were no changes in the MCP-1 levels in the renal medulla in either group 12 weeks following cell delivery. Urinary daily excretion of MCP-1 did not change in either hPKC or hPKC(F+).
Figure 5.
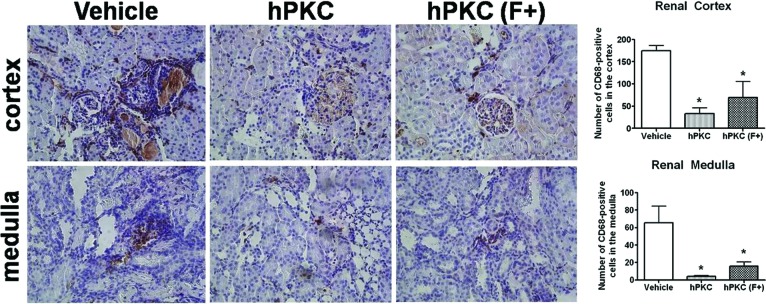
The abundance of inflammatory cells (CD68-positive) in the renal cortex and medulla of the ischemia-reperfusion-gentamicin rats. The CD68-positive cells are localized predominantly in the tubulointerstitial space of the kidney. The number of positively stained cells per ×200 area was counted using automated cell counting software (Image-Pro 6.3; Media Cybernetics). The data are the means ± SE; ∗, p < .05 versus vehicle; n = 4 per group. Magnification, ×200. Abbreviations: hPKC, human primary kidney cell; hPKC(F+), human primary kidney cell containing erythropoietin-positive cells.
Figure 6.

The levels of MCP-1 and 8-OHdG proteins in the renal cortical and medullary tissues as well as in the urine of the ischemia-reperfusion-gentamicin rats. In the left panels, MCP-1 expression was measured in renal cortical and medullary 20,000g supernatants and in the urine by enzyme-linked immunosorbent assay (ELISA). The data are the means ± SE; ∗, p < .05 versus vehicle; n = 4. In the right panel, 8-OHdG levels were measured in renal cortical and medullary 20,000g supernatants and in the urine by ELISA. The data are the means ± SE; ∗, p < .05 versus vehicle; n = 4. Abbreviations: hPKC, human primary kidney cell; hPKC(F+), human primary kidney cell containing erythropoietin-positive cells; MCP-1, monocyte chemotactic protein 1; 8-OHdG, 8-hydroxy-deoxyguanosine.
A marker of DNA oxidation, 8-OHdG (Fig. 6) was reduced only in the renal cortex in the hPKC(F+) group (vehicle: 1.96 ± 0.34 versus hPKC(F+): 0.83 ± 0.05 ng/mg tissue; ∗, p < .05; n = 4). No changes in this marker were seen in the medulla. Although there was a tendency toward a decrease in both the hPKC and hPKC(F+) groups, daily urinary excretion of 8-OHdG was not significantly different compared with the vehicle-only group.
Finally, to determine whether the injected cells survived in the kidney for 12 weeks following injection into the renal parenchyma, kidney sections were stained with human nuclear antibody to distinguish human cells from rat renal cells. There were very few human cells found in the kidneys, and these few cells were mostly located in the interstitial areas of the cortex (Fig. 7).
Figure 7.
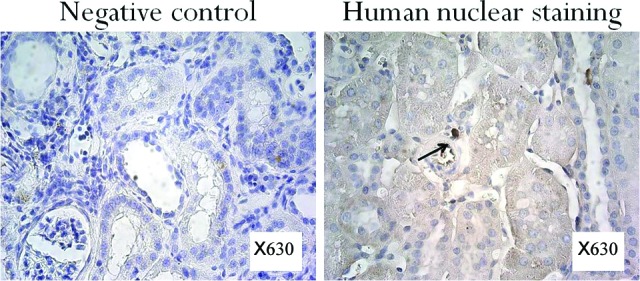
Immunostaining of the kidney tissues of ischemia-reperfusion-gentamicin rats with human nuclear antibody 12 weeks following cell transplantation. Very few stained cells were found in the kidney. The cells that were found were mostly located in the interstitial areas of the cortex. Magnification, ×630.
Discussion
This study demonstrates that cell sorting using an antifibroblast antibody may increase the proportion of EPO-producing cells in a population of primary renal cells. Moreover, this study showed that EPO-producing cells are one of the primary components of hPKC(F+) sorted cultures. We also show that IR injury combined with gentamicin treatment can be used in the nude rat to establish a chronic-after-acute model of renal injury and that this model can be used to investigate the effects of cell therapy on renal function and injury. Our data indicate that the delivery of a subpopulation of renal cells enriched in EPO-positive cells to the renal parenchyma well after the onset of disease improves renal function and reduces renal fibrosis, inflammation, and oxidative stress in this model. These data suggest that subpopulations of hPKC containing EPO-positive cells may be used as components of cell-based therapies for degenerative kidney diseases.
Cell therapy may provide an alternative treatment option for chronic kidney disease. Cell therapy uses cells to repair native tissue either by replacing the function of injured cells or by stimulating the regenerative capacities of native cells through a release of paracrine factors. Studies using stem or progenitor cells, including bone marrow mesenchymal cells, embryonic stem cells, or resident renal progenitor cells, have shown that various types of cell therapies may improve renal function and/or attenuate renal damage in models of renal injury [17–20]. However, differentiated adult renal cells may also induce renoprotective actions, and these differentiated cells may be a preferred source of cells for therapy, because they do not evoke an immune response when used autologously, and they do not form teratomas when injected. Differentiated cells with regenerative potential could be derived from the kidney, possibly through renal biopsy, and then expanded in culture before being introduced back into the tissue. Kelley et al. tested the efficacy of a differentiated, tubular cell-enriched subpopulation derived from an adult kidney in the 5/6 nephrectomy model. They found that these cells improved renal function and attenuated profibrotic signals such as transforming growth factor-β, plasminogen activator inhibitor 1, and fibronectin [14]. Their results suggested that differentiated cell populations derived from adult kidney could be used for cell therapy. In our study, we can see that our isolated cell fraction is distinctly different from that of most previously separated fractions derived from the kidney interstitial space. Previous studies have focused either on the progenitor subpopulation [18–20] with the goal of eventual differentiation of these cells into different renal cell types or on the cells of the epithelial lining (proximal and distal tubular cells) [14] with the goal of integrating these cells into the functional units of the kidney.
The renoprotective actions of EPO were previously demonstrated in an acute ischemia-reperfusion model. These renoprotective effects were not always associated with changes in hemoglobin, suggesting the involvement of other mechanisms. EPO can induce anti-inflammatory and antioxidant pathways and can act as a survival factor by reducing cell apoptosis [8–10]. By attenuating inflammation and oxidative stress in injured kidneys, EPO may reduce renal fibrosis, induce cell proliferation, and thus accelerate renal repair. The production of EPO in CKD patients is reduced. Thus, we hypothesized that the transplantation of human renal cell cultures containing EPO-positive cells to the renal parenchyma may be beneficial in renal injury and may have a better therapeutic outcome compared with unfractionated primary renal cultures.
Following cell separation, the levels of EPO expression in our primary renal cell cultures were increased. This was true for both EPO gene expression as well as EPO protein release into the media in the sorted hPKC(F+) cultures. This suggests that a higher proportion of EPO-producing cells is present in this cell population and that cell function is preserved in the cultures. Additional cell characterization demonstrated increased frequency of a surface marker of peritubular interstitial fibroblasts, CD73, in hPKC(F+) cultures (27%) [21]. Double staining for EPO and CD73 in hPKC cultures revealed that approximately 12% of the cell population was positive for both markers (data not shown), confirming the previous observation that a subset of CD73-positive peritubular fibroblasts may produce EPO [21]. The percentages of epithelial and endothelial cells in our cultures were not altered by cell sorting with anti-fibroblast antibody. CD133, a marker of a multipotent adult interstitial stem cell population, was expressed at similar levels in hPKC(F+) and hPKC cultures [22]. Bussolati et al. demonstrated that CD133+ cells that had been isolated from the interstitial compartment of the kidney homed to a site of injury and gave rise to endothelial and tubular epithelial cells in a toxic renal injury model [22]. MSCs may also induce renal repair via paracrine or endocrine actions [23]. We used CD44 and CD90 antibodies to determine whether MSCs existed in our primary renal cell cultures. Although we did find some cells that were positive for both of these MSC markers, there was only a marginal increase of these markers in hPKC(F+) compared with hPKC cultures. These data suggest that cell sorting with anti-fibroblast antibody may increase the proportion of EPO-producing cells in hPKC cultures but does not change the proportion of other expected cell types in the sorted culture. Moreover, it appears that once the cultures are sorted in this manner, EPO-producing cells are the primary components of the sorted hPKC(F+) cultures.
Chronic kidney disease often develops as a progression from an acute renal injury that does not recover over time. To test the long-term effects of cell therapy in this type of injury, we chose to combine ischemia-reperfusion and gentamicin injuries as a model of chronic-after-acute type of renal injury. Renal ischemia with low tissue oxygenation causes necrosis and apoptosis of renal cells. If ischemic insult is transitory, the cells proliferate and renal repair occurs [24]. Inefficient regeneration of renal tissue may lead to tubulointerstitial fibrosis [25] and atrophy, both of which are common manifestations of CKD [26]. However, only a small percentage of animals that undergo acute ischemic renal insult will progress to chronic failure. Gentamicin, a nephrotoxic antibiotic in high doses, induces tubular cell apoptosis and inhibits cell transport, altering tubular reabsorption and cell viability [27]. We hypothesized that a combination of gentamicin and ischemia-reperfusion injury would prolong renal ischemia and cause chronic kidney disease in rats more efficiently. The advantage of this model is the development of both glomerulosclerosis and tubulointerstitial fibrosis over a relatively short, 10-week period of time. Increased serum creatinine is associated with marked renal fibrosis following IR-GM injury. The model is reproducible; however, approximately 15% of the animals developed severe renal failure and therefore had to be excluded from the study, which resulted in a lower number of animals in some experimental groups.
Hypoxia and toxins induce alterations in renal hemodynamics, energy supply, and the endocrine milieu that may exacerbate kidney injury. The improvement in renal function is dependent in part on the restoration of filtration rate and functional tubular epithelium [28]. The delivery of hPKC(F+) and hPKC significantly improved renal filtration function as early as at 1 week after injection, and this improvement continued for 12 more weeks until the end of the study in both experimental groups. This profound effect on renal function suggests that treatment with hPKC and hPKC(F+) would be highly efficacious for the improvement of renal function. Moreover, the effects on serum creatinine were similar among hPKC, hPKC(F+), and rhEPO groups. The hemoglobin levels were improved only in the rhEPO group but did not change in hPKC and hPKC(F+), suggesting that the effects of cell therapy in hPKC and hPKC(F+) were unrelated to anemia in these animals. However, hPKC(F+) were more beneficial for renal injury than hPKC, implying an improvement in kidney filtration and tubular function. The excretion of renal injury markers, such as albumin and KIM-1, was also significantly reduced in animals receiving hPKC(F+) compared with the control group. Albumin is a marker of altered glomerular filtration, as well as attenuated ability of damaged tubules to reabsorb substances from the tubular fluid. Moreover, albuminuria may be involved in chronic kidney disease progression [29]. KIM-1 is a more specific biomarker of renal injury compared with serum creatinine [30, 31]. Previous studies demonstrated increased expression of KIM-1 in the injured proximal tubular cells in polycystic kidney disease, ischemic acute tubular necrosis, and protein-overload nephropathy [30–34].
Increased free radicals and inflammatory cell infiltration are major mechanisms leading to renal fibrosis [35]. The number of CD68-positive cells as an indicator of macrophage and monocyte infiltration was reduced in both regions of the kidney studied. However, MCP-1, a factor that recruits monocytes to the site of injury, was attenuated only in the renal cortex of the hPKC(F+) group, suggesting that other chemoattractive factor besides MCP-1 may mediate inflammatory cell infiltration in the renal medulla. The levels of MCP-1 were reportedly increased following ischemia/reperfusion injury and mediated by oxidative stress [36, 37]. Interestingly, the levels of oxidative damage marker 8-OHdG were reduced only in the cortex in the hPKC(F+) group.
There were differential effects of studied cell populations on tubulointerstitial fibrosis in cortical and medullary regions of the kidney. hPKC(F+) were similarly effective against interstitial fibrosis in both cortex and medulla; however, hPKC reduced fibrosis only in the medulla. It is possible that EPO-producing cells can migrate toward medulla to induce anti-fibrotic effects. Data from our group showed that human renal cells expanded in culture can migrate toward different growth factors that coordinate kidney regeneration [11].
Cell tracking studies with a human anti-nuclear antibody revealed that there are only a few transplanted cells left in the kidney by the end of the 12-week study, suggesting that there was no transplanted cell propagation or clonal cell expansion [38]. This result is consistent with a study reported by Kelley et al. showing that the relative presence of the SRY gene was estimated to be only one in 33,333 cells 6 months after the delivery of differentiated adult parenchymal cells into the kidney of the 5/6 nephrectomy model animals [14].
Conclusion
In summary, this study demonstrates that magnetic-bead sorting with anti-fibroblast antibody could be used to increase the frequency of renal EPO-producing cells in culture. Moreover, the presented data suggest that the renoprotective effects of hPKC(F+) are due to activation of anti-inflammatory and antioxidant pathways that consequently attenuate renal fibrosis and improve renal architecture. It is possible that by improving renal architecture, the cell population enriched in EPO-positive cells creates a better environment for dedifferentiation of resident cells and cell proliferation needed for the regeneration of functional tissue [2, 25]. Ongoing experiments will determine whether the administration of hPKC(F+) induces cell proliferation or dedifferentiation. Additional experiments include the possibility of achieving therapeutic outcomes in this model with other specific cell subpopulations derived from human CKD kidney tissue.
Acknowledgments
This study was supported by Tengion, Inc., through a sponsored research agreement. We thank Bridgette Jones for assistance with tissue processing and histology, Adam Wilson for assistance with animal surgeries and creatinine assays, and Dr. Jennifer L. Olson for editorial assistance with the manuscript. L.M.Y. is currently affiliated with the Hypertension and Vascular Research Center of the Wake Forest School of Medicine, Winston-Salem, NC. S.A. is currently affiliated with the Department of Urology, National Kidney and Transplant Institute, Quezon City, Philippines.
Author Contributions
L.M.Y.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing and editing, final approval of manuscript; N.K.G.-S.: conception and design, collection of data, data analysis and interpretation, manuscript editing; L.S.K. and K.G.: collection of data; S.A.: collection of data, manuscript editing; A.A.: conception and design, financial support, administrative support; T.A.: manuscript editing, administrative support; J.J.Y.: conception and design, administrative support, manuscript editing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
A.A. and J.J.Y. served as consultants for Tengion, Inc., during the period when this study was conducted.
References
- 1.Al-Awqati Q, Oliver JA. Stem cells in the kidney. Kidney Int. 2002;61:387–395. doi: 10.1046/j.1523-1755.2002.00164.x. [DOI] [PubMed] [Google Scholar]
- 2.Benigni A, Morigi M, Remuzzi G. Kidney regeneration. Lancet. 2010;375:1310–1317. doi: 10.1016/S0140-6736(10)60237-1. [DOI] [PubMed] [Google Scholar]
- 3.Guo JK, Cantley LG. Cellular maintenance and repair of the kidney. Annu Rev Physiol. 2010;72:357–376. doi: 10.1146/annurev.physiol.010908.163245. [DOI] [PubMed] [Google Scholar]
- 4.U.S. Renal Data System. Minneapolis, MN: U.S. Renal Data System Coordinating Center; 2009. U.S. Renal Data System Annual Report. [Google Scholar]
- 5.Zenovich AG, Taylor DA. Cell therapy in kidney disease: Cautious optimism … but optimism nonetheless. Perit Dial Int. 2007;27(suppl 2):S94–S103. [PubMed] [Google Scholar]
- 6.Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int. 1993;44:1149–1162. doi: 10.1038/ki.1993.362. [DOI] [PubMed] [Google Scholar]
- 7.Lipsic E, van der Meer P, Henning RH, et al. Timing of erythropoietin treatment for cardioprotection in ischemia/reperfusion. J Cardiovasc Pharmacol. 2004;44:473–479. doi: 10.1097/01.fjc.0000140209.04675.c3. [DOI] [PubMed] [Google Scholar]
- 8.Dang J, Jia R, Tu Y, et al. Erythropoietin prevents reactive oxygen species generation and renal tubular cell apoptosis at high glucose level. Biomed Pharmacother. 2010;64:681–685. doi: 10.1016/j.biopha.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 9.De Beuf A, D'Haese PC, Verhulst A. Epoetin delta as an antifibrotic agent in the remnant kidney rat: A possible role for transforming growth factor beta and hepatocyte growth factor. Nephron Exp Nephrol. 2010;115:e46–e59. doi: 10.1159/000313830. [DOI] [PubMed] [Google Scholar]
- 10.De Beuf A, Hou XH, D'Haese PC, et al. Epoetin delta reduces oxidative stress in primary human renal tubular cells. J Biomed Biotechnol. 2010;2010:395785. doi: 10.1155/2010/395785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guimaraes-Souza NK, Yamaleyeva LM, Aboushwareb T, et al. In vitro reconstitution of human kidney structures for renal cell therapy. Nephrol Dial Transplant. 2012 doi: 10.1093/ndt/gfr785. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 12.Presnell SC, Bruce AT, Wallace SM, et al. Isolation, characterization, and expansion methods for defined primary renal cell populations from rodent, canine, and human normal and diseased kidneys. Tissue Eng Part C Methods. 2011;17:261–273. doi: 10.1089/ten.TEC.2010.0399. [DOI] [PubMed] [Google Scholar]
- 13.Aboushwareb T, Egydio F, Straker L, et al. Erythropoietin producing cells for potential cell therapy. World J Urol. 2008;26:295–300. doi: 10.1007/s00345-008-0301-0. [DOI] [PubMed] [Google Scholar]
- 14.Kelley R, Werdin ES, Bruce AT, et al. Tubular cell-enriched subpopulation of primary renal cells improves survival and augments kidney function in rodent model of chronic kidney disease. Am J Physiol Renal Physiol. 2010;299:F1026–F1039. doi: 10.1152/ajprenal.00221.2010. [DOI] [PubMed] [Google Scholar]
- 15.Yamaleyeva LM, Gallagher PE, Vinsant S, et al. Discoordinate regulation of renal nitric oxide synthase isoforms in ovariectomized mRen2 Lewis rats. Am J Physiol Regul Integr Comp Physiol. 2007;292:R819–R826. doi: 10.1152/ajpregu.00389.2006. [DOI] [PubMed] [Google Scholar]
- 16.Yamaleyeva LM, Pendergrass KD, Pirro NT, et al. Ovariectomy is protective against renal injury in the high-salt-fed older mRen2 Lewis rat. Am J Physiol Heart Circ Physiol. 2007;293:H2064–H2071. doi: 10.1152/ajpheart.00427.2007. [DOI] [PubMed] [Google Scholar]
- 17.Freed LE, Vunjak-Novakovic G, Biron RJ, et al. Biodegradable polymer scaffolds for tissue engineering. Biotechnology. 1994;12:689–693. doi: 10.1038/nbt0794-689. [DOI] [PubMed] [Google Scholar]
- 18.Tögel F, Hu Z, Weiss K, et al. Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. Am J Physiol Renal Physiol. 2005;289:F31–F42. doi: 10.1152/ajprenal.00007.2005. [DOI] [PubMed] [Google Scholar]
- 19.Lange C, Togel F, Ittrich H, et al. Administered mesenchymal stem cells enhance recovery from ischemia/reperfusion-induced acute renal failure in rats. Kidney Int. 2005;68:1613–1617. doi: 10.1111/j.1523-1755.2005.00573.x. [DOI] [PubMed] [Google Scholar]
- 20.Sagrinati C, Ronconi E, Lazzeri E, et al. Stem-cell approaches for kidney repair: Choosing the right cells. Trends Mol Med. 2008;14:277–285. doi: 10.1016/j.molmed.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 21.Bachmann S, Le Hir M, Eckardt KU. Co-localization of erythropoietin mRNA and ecto-5′-nucleotidase immunoreactivity in peritubular cells of rat renal cortex indicates that fibroblasts produce erythropoietin. J Histochem Cytochem. 1993;41:335–341. doi: 10.1177/41.3.8429197. [DOI] [PubMed] [Google Scholar]
- 22.Bussolati B, Bruno S, Grange C, et al. Isolation of renal progenitor cells from adult human kidney. Am J Pathol. 2005;166:545–555. doi: 10.1016/S0002-9440(10)62276-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matthay MA. Advances and challenges in translating stem cell therapies for clinical diseases. Transl Res. 2010;156:107–111. doi: 10.1016/j.trsl.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Oliver JA. Adult renal stem cells and renal repair. Curr Opin Nephrol Hypertens. 2004;13:17–22. doi: 10.1097/00041552-200401000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Lazzeri E, Mazzinghi B, Romagnani P. Regeneration and the kidney. Curr Opin Nephrol Hypertens. 2010;19:248–253. doi: 10.1097/MNH.0b013e32833680dc. [DOI] [PubMed] [Google Scholar]
- 26.Levey AS, Eckardt KU, Tsukamoto Y, et al. Definition and classification of chronic kidney disease: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO) Kidney Int. 2005;67:2089–2100. doi: 10.1111/j.1523-1755.2005.00365.x. [DOI] [PubMed] [Google Scholar]
- 27.Lopez-Novoa JM, Quiros Y, Vicente L, et al. New insights into the mechanism of aminoglycoside nephrotoxicity: An integrative point of view. Kidney Int. 2011;79:33–45. doi: 10.1038/ki.2010.337. [DOI] [PubMed] [Google Scholar]
- 28.Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 29.Amsellem S, Gburek J, Hamard G, et al. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J Am Soc Nephrol. 2010;21:1859–1867. doi: 10.1681/ASN.2010050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonventre JV. Kidney injury molecule-1 (KIM-1): A urinary biomarker and much more. Nephrol Dial Transplant. 2009;24:3265–3268. doi: 10.1093/ndt/gfp010. [DOI] [PubMed] [Google Scholar]
- 31.Nijboer WN, Schuurs TA, Damman J, et al. Kidney injury molecule-1 is an early noninvasive indicator for donor brain death-induced injury prior to kidney transplantation. Am J Transplant. 2009;9:1752–1759. doi: 10.1111/j.1600-6143.2009.02713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Borst MH, van Timmeren MM, Vaidya VS, et al. Induction of kidney injury molecule-1 in homozygous Ren2 rats is attenuated by blockade of the renin-angiotensin system or p38 MAP kinase. Am J Physiol Renal Physiol. 2007;292:F313–F320. doi: 10.1152/ajprenal.00180.2006. [DOI] [PubMed] [Google Scholar]
- 33.van Timmeren MM, Bakker SJ, Vaidya VS, et al. Tubular kidney injury molecule-1 in protein-overload nephropathy. Am J Physiol Renal Physiol. 2006;291:F456–F464. doi: 10.1152/ajprenal.00403.2005. [DOI] [PubMed] [Google Scholar]
- 34.Kuehn EW, Park KM, Somlo S, et al. Kidney injury molecule-1 expression in murine polycystic kidney disease. Am J Physiol Renal Physiol. 2002;283:F1326–F1336. doi: 10.1152/ajprenal.00166.2002. [DOI] [PubMed] [Google Scholar]
- 35.Anders HJ, Ryu M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011;80:915–925. doi: 10.1038/ki.2011.217. [DOI] [PubMed] [Google Scholar]
- 36.Sung FL, Zhu TY, Au-Yeung KK, et al. Enhanced MCP-1 expression during ischemia/reperfusion injury is mediated by oxidative stress, NF-kappaB. Kidney Int. 2002;62:1160–1170. doi: 10.1111/j.1523-1755.2002.kid577.x. [DOI] [PubMed] [Google Scholar]
- 37.Ysebaert DK, De Greef KE, Vercauteren SR, et al. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol Dial Transplant. 2000;15:1562–1574. doi: 10.1093/ndt/15.10.1562. [DOI] [PubMed] [Google Scholar]
- 38.Held PK, Al-Dhalimy M, Willenbring H, et al. In vivo genetic selection of renal proximal tubules. Mol Ther. 2006;13:49–58. doi: 10.1016/j.ymthe.2005.09.004. [DOI] [PubMed] [Google Scholar]