

Despite the established use of poly-chemotherapy, relapse continues to be the most common cause of death in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS). The therapeutic elimination of all leukemia stem cells (LSC) and pre-LSC will be essential for achieving lasting cures. This article reviews recent and ongoing advances in understanding the roles of pre-LSC, and the aberrations that lead to pre-LSC formation and subsequent LSC transformation.
Keywords: Acute myelogenous leukemia, Hematopoietic stem cells, Cancer stem cells, Stem/progenitor cell
Abstract
Recent experimental evidence has shown that acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) arise from transformed immature hematopoietic cells following the accumulation of multiple stepwise genetic and epigenetic changes in hematopoietic stem cells and committed progenitors. The series of transforming events initially gives rise to preleukemic stem cells (pre-LSC), preceding the formation of fully transformed leukemia stem cells (LSC). Despite the established use of poly-chemotherapy, relapse continues to be the most common cause of death in AML and MDS. The therapeutic elimination of all LSC, as well as pre-LSC, which provide a silent reservoir for the re-formation of LSC, will be essential for achieving lasting cures. Conventional sequencing and next-generation genome sequencing have allowed us to describe many of the recurrent mutations in the bulk cell populations in AML and MDS, and recent work has also focused on identifying the initial molecular changes contributing to leukemogenesis. Here we review recent and ongoing advances in understanding the roles of pre-LSC, and the aberrations that lead to pre-LSC formation and subsequent LSC transformation.
Introduction
Healthy hematopoiesis is hierarchically organized, beginning with multipotent, quiescent stem cells that are able to perpetuate themselves through self-renewal and generate more mature, transit-amplifying progeny through differentiation. Recent experimental evidence has shown that acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) arise from transformed immature hematopoietic cells following the accumulation of multiple stepwise genetic and epigenetic changes in hematopoietic stem cells (HSC) and committed progenitors. The series of transforming events is thought to give rise to preleukemic stem cells (pre-LSC), including the initial leukemia cell-of-origin (LCO), preceding the formation of fully transformed leukemia stem cells (LSC) (illustrated in Fig. 1). Whereas pre-LSC consist of self-renewing hematopoietic stem and progenitor cells that can give rise to leukemia in vivo with variable latency upon gradual accumulation of additional genetic hits, LSC are functionally defined by their capacity to give rise to fully penetrant, short-onset leukemia in murine transplantation models.
Figure 1.
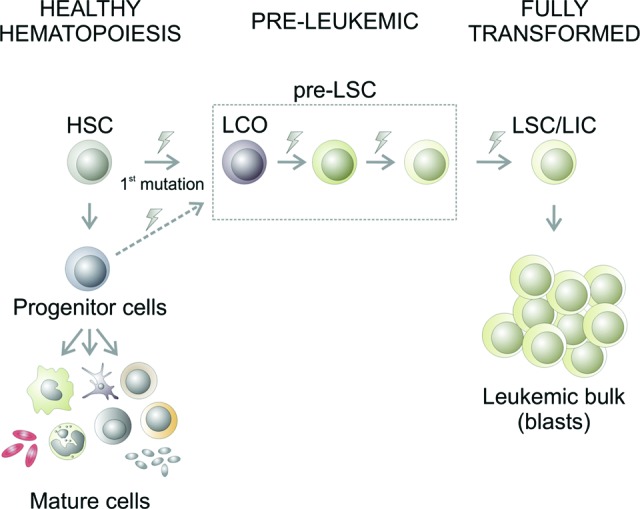
Multistep transformation of leukemia stem cells. During healthy hematopoiesis (displayed on the left), HSC give rise to committed progenitor cells that further differentiate into all mature blood cell types. Leukemia arises from hematopoietic cells that have progressively acquired genetic and/or epigenetic modifications (represented by lightning bolt symbols) and ultimately form self-renewing LSC, also known as LIC, that sustain the disease. An initial (“founding”) mutation in HSC or progenitor cells originates the LCO. During the preleukemic stage, the LCO and subsequent preleukemic stem cell stages (pre-LSC) progressively acquire further aberrations and finally generate fully transformed LSC/LIC, which are functionally defined by their ability to initiate disease upon transplantation. These aberrations, illustrated by lightning bolt symbols, include mutations or deregulation of transcription factors, epigenetic factors, metabolic factors, and proteins involved in signal transduction and cell cycle regulation. LSC/LIC are self-renewing and contain a full set of genetic and epigenetic changes that lead to blocked hematopoietic differentiation and the accumulation of dysfunctional leukemic blasts that form the bulk of the tumor cells but are not capable of initiating/maintaining the disease on their own. Abbreviations: HSC, hematopoietic stem cell; LCO, leukemia cell-of-origin; LIC, leukemia-initiating cell; LSC, leukemia stem cell.
Despite the established use of poly-chemotherapy and the application of newer agents that transiently reduce the tumor burden, relapse continues to be the most common cause of death in AML and MDS. Such failures demonstrate that the functional LSC compartment is not effectively eradicated by current regimens. However, the therapeutic elimination of all LSC, as well as pre-LSC, which provide a silent reservoir for the re-formation of LSC, will be essential for achieving lasting cures. Thus, the identification of specific LSC and pre-LSC populations, as well as the study of their molecular characteristics, is critical for understanding the genesis of leukemia and for developing strategies by which these cells can be eradicated.
Although still a work in progress, recent research has improved our understanding of the molecular pathways and the temporal and spatial order of events involved in the transformation process. According to the current molecular pathogenetic model of AML, at least two molecular events are required for the development of malignant cells [1]. One causes a differentiation arrest, and the second confers proliferative properties to the cells, leading to the formation of a pool of LSC that in turn gives rise to a hierarchy of functionally heterogeneous bulk tumor cells [2]. In addition, some disease alleles also increase stem cell self-renewal, further contributing to leukemogenesis and to leukemia stem cell maintenance. Although conventional sequencing and next-generation genome sequencing of blast populations have allowed us to describe many of these recurrent mutations, our understanding of the initial genetic changes of the pre-LSC contributing to leukemogenesis remains limited. Here we review recent and ongoing advances in understanding the roles of pre-LSC, and the changes that lead to pre-LSC formation and subsequent LSC transformation.
Insights Obtained From Genetic Modeling
As a constantly increasing number of recurrent mutations have been identified in myeloid malignancies, murine models of disease alleles found in human AML patients have been used to investigate how early disruptions in the hematopoietic stem cell compartment contribute to leukemogenesis. Models of leukemogenesis include transplantation models in which leukemia oncogenes are ectopically expressed, as well as transgenic mice carrying mutations or mimicking other molecular changes found in human leukemia. Murine models also offer the opportunity to understand the effects of combining recurring mutations with epigenetic and transcriptional changes and, importantly, allow for specific investigation of preleukemic stem and progenitor cell populations.
Transcription Factors
The process of leukemogenesis is understood to begin at the level of hematopoietic stem and progenitor cells (HSPC). The involvement of several transcriptional regulators that govern normal hematopoietic differentiation and stem cell maintenance in leukemogenesis is suggested by their frequent mutation or altered regulation in patients [3–5]. Properties of increased self-renewal, blocked differentiation, reduced programmed cell death, and altered signaling and metabolism play a role in clonal pre-LSC development and promote the acquisition of subsequent genetic alterations by the pre-LSC, leading in a stepwise fashion to complete transformation to LSC and the full leukemic phenotype.
Mutational profiling of paired diagnosis and relapse bone marrow samples from patients with AML has attempted to identify the molecular drivers of leukemogenesis. Modeling such changes in mice has offered insight into which molecular changes effect development of pre-LSC. For example, there is strong evidence that mutations in CCAAT/enhancer-binding protein-α (CEBPA) may be early events in the generation of leukemic clones. In contrast to mutations in genes such as fms-related tyrosine kinase 3 (FLT3), the rat sarcoma genes (RAS), or stem cell growth factor receptor (KIT), the majority of patients carrying CEBPA mutations undergoing relapse of AML display the same mutations in both CEBPA alleles as they did before achieving complete remission [6–8]. A biallelic knockin mouse model of the most common CEBPA mutation in AML results in myeloid progenitors with increased self-renewal capacity and mice uniformly progress to a transplantable AML. Moreover, these CEBPA mutations also induce pre-LSC development characterized by a cell-intrinsic, premalignant expansion of the HSC and multipotent progenitor (MPP) compartments induced by loss of quiescence prior to overt leukemia development [8, 9]. CEBPA is not alone: other mutations such as the fusion oncogenes AML1 (acute myeloid leukemia-1)-ETO (eight-twenty-one corepressor) and MLL (mixed lineage leukemia)-AF9 (ALL1-fused gene from chromosome 9 protein) similarly cause an increase in HSC numbers followed by accumulation of abnormal myeloid progenitors [10, 11]. CBFB (core binding factor subunit-β)-SMMHC (smooth muscle myosin heavy chain) mutations, in contrast, have been shown to decrease HSC repopulating activity while leading to the accumulation of abnormal myeloid progenitors [12]. These studies demonstrate how preleukemic mutations affecting key transcription factors may allow HSPC to evade normal homeostatic control of their numbers and survival and form pre-LSC primed for acquisition of further changes that contribute to the subsequent generation of fully transformed LSC.
Studies in both mouse and human LSC and preleukemic HSPC have shown that in addition to recurrent mutations, disturbed gene expression not only causes deregulation of homeostatic hematopoiesis but may contribute to leukemogenesis. These factors comprise key transcriptional regulators that are crucial for maintaining basic cell functions and allowing for a sufficient life span of pre-LSC to acquire multiple further hits for full transformation. For example, in a murine model of AML caused by 80% reduced levels of the transcription factor PU.1 (Spi1), the course of disease includes a preleukemic phase with an accumulation of immature myelomonocytic cells in the marrow, eventually followed by a leukemic phase with high numbers of malignant cells in both the blood and marrow [13]. Progressive inactivation of transcription factor jun-B (Junb) in a murine model deregulates cell-cycle machinery and increases the proliferation of long-term repopulating HSC (LT-HSC) by destabilizing a complex network of genes and pathways that normally limit myeloid differentiation leading to the development of a myeloproliferative disorder [14, 15]. In another murine model, GATA-binding protein 1 (Gata1) knockdown leads to the formation of a disease resembling MDS after a long latency, and, importantly, some remaining GATA1 activity (∼5%) seems to be required for MDS formation, as Gata1-null mice have a normal life expectancy [16]. These models, and the paucity of null mutations or deletions in transcription-factor encoding genes in patients, highlight the importance of remaining residual levels of key regulators, as opposed to complete deficiency. A minimal remaining level may be required for maintaining basic cell functions and allowing for a sufficient life span of pre-LSC to acquire multiple further hits for full transformation.
Genome-wide transcriptional profiling of pre-LSC prior to the onset of AML has delineated transcriptional pathways that are functionally essential for formation of LSC that arise later during the course of disease. For instance, animals with low PU.1 have definable preleukemic HSC and develop AML only in combination with subsequently deregulated cofactors such as JunB and after a fairly long latency (median, 6 months) [17]. Downregulation of JUNB expression, correlating with low PU.1 expression, has also been found in the HSC compartment of patients with AML [17]. This strategy of pre-LSC gene expression profiling also led to the discovery of the nonclustered homeobox transcription factor H2.0-like homeobox (Hlx), which is upregulated in PU.1 low pre-LSC. HLX was found to block the differentiation of HSC and lead to formation and accumulation of abnormal myeloid progenitors with unlimited serial clonogenicity [18]. Importantly, HLX upregulation appears to be an early lesion and by itself seems not sufficient for full LSC transformation [18].
These models, together with other genetic studies in mice, have supported the idea of a network of genes that regulate HSC numbers, proliferative activity, resistance to apoptosis, and retention in the bone marrow niche [19] and thereby contribute to formation and maintenance of pre-LSC populations. Other important mediators of these processes include cell-cycle regulators, such as the D cyclins; transcription factors, such as growth factor-independent 1 transcription repressor (Gfi1), homeobox B4 (HoxB4), GATA-binding protein 2 (Gata2), and homeobox A9 (HoxA9); and extrinsic regulatory pathways, including S-phase kinase-associated protein 2 (Skp2), notch (Notch), transforming growth factor-β (Tgfb), and wingless-type MMTV integration site family members (Wnt), which control HSC self-renewal and proliferation [15, 20–28].
Epigenetic Factors
Although the regulation of hematopoietic stem cell self-renewal and lineage differentiation by specific transcription factors has been well-characterized, our understanding of the epigenetic processes upstream of these factors is still in its infancy. DNA cytosine methylation is an epigenetic modification that has an important role in chromatin organization and in regulating gene expression [29, 30]. Significant changes in methylation patterns have been observed when comparing different stages of murine hematopoietic cells [31, 32], indicating that methylation patterns may effect important transcriptional changes during differentiation. Knockout of the maintenance DNA methyltransferase 1 (Dnmt1), for example, has shown that DNA methylation protects HSC from myeloerythroid restriction [33, 34].
Recurring mutations in a number of important epigenetic mediators, including regulators of DNA cytosine methylation, have been identified in patients with myeloid malignancies, and recent studies have shown that aberrant DNA cytosine methylation is a hallmark of patients with AML and MDS [35–38]. Genetic modeling of individual epigenetic factors involved in AML pathogenesis has shown that the majority affect the HSC compartment and alter hematopoietic differentiation, in some cases leading to myelodysplasia, stem cell expansion, or other preleukemic conditions. These include suspected loss-of-function or dominant interfering mutations such as DNA methyltransferase 3α (DNMT3A), tet methylcytosine dioxygenase 2 (TET2), enhancer of zeste homolog 2 (EZH2), and additional sex combs like 1 and 2 (ASXL1/2), as well as neomorphic mutations, such as isocitrate dehydrogenase 1 and 2 (IDH1/2) [36, 39–53].
Loss of DNMT3A impedes HSC differentiation, expanding HSC numbers in the bone marrow [54]. Gene expression analysis showed that DNMT3A loss leads to upregulated expression of HSC multipotency genes and lower expression of genes with a known role in HSC differentiation [54]. However, despite these preleukemic features, no development of overt leukemia was observed in a Dnmt3a knockout model. Alterations that impair the catalytic activity of TET2, which hydroxylates 5mC, lead to increased stem cell self-renewal and progressive stem cell expansion, ultimately causing a myeloproliferative disease-like disorder [55]. Again, no acute leukemias were observed in this model. Likewise, mutations of the IDH1 enzyme, which produces a new metabolite (2-hydroxyglutarate) that competes with α-ketoglutarate to impair the catalytic activity of dioxygenases, including TET2, lead to increased numbers of early hematopoietic progenitors and dysfunctional hematopoiesis, although, like TET2, these mutations do not lead to overt leukemia [56]. Loss of ASXL1 was recently shown to cooperate with NRASG12D mutations in promoting the development of AML, an effect that is mediated through a loss of polycomb repressive complex 2 (PRC2)-mediated gene repression [57]. The MLL-AF9 fusion oncogene was recently reported to require the histone demethylase lysine (K)-specific demethylase 1A (KDM1A) to maintain functional LSC [58]. Together, these findings suggest that these commonly mutated epigenetic factors work in concert to protect HSC from aberrant expansion and myeloid transformation, and that their respective mutations lead to alterations in DNA methylation patterns and other epigenetic modifications in HSC and consequent expansions of pre-LSC populations.
Metabolic Factors
Recent data from various murine models have begun to clarify roles for molecular changes in metabolic pathways in leukemogenesis. Disruption of metabolic pathways in HSC may play a role in the formation of pre-LSC and transformation to LSC. Recent models have shown that deletion of several negative regulators of mammalian target of rapamycin (mTORC1) signaling, including phosphatase and tensin homolog (Pten), tuberous sclerosis 1 (Tsc1), promyelocytic leukemia (Pml), and F-box and WD repeat domain containing 7 (Fbxw7), result in similar stem cell phenotypes characterized by HSC hyperproliferation and subsequent exhaustion and defective repopulating potential [59–64]. Inactivation of PTEN, a negative regulator of the phosphatidylinositol 3-kinase-Akt pathway, for example, causes short-term expansion but long-term decline of HSC, and mice with Pten-mutant bone marrow develop a myeloproliferative disorder [60]. Similarly, deficiency of the tumor suppressor PML induces cycling of HSC and results in short-term increased but long-term reduced HSC repopulation ability [63]. Although these and other regulators of metabolic pathways in HSC, such as hypoxia inducible factor 1, α-subunit (Hif-1a) [65], and the liver kinase B1 (Lkb1) [59, 66, 67], have been shown to alter the size of the hematopoietic stem cell compartment in mouse genetic models, the role of changes in their expression or mutations in the genes encoding these factors in LSC or pre-LSC of patients with myeloid malignancies is still largely unclear.
Cellular Stages of Transformation
A lingering question in the understanding of leukemogenesis is which cellular stages during hematopoiesis acquire the molecular hits leading to formation of pre-LSC and, ultimately, complete transformation to LSC. The initial changes can occur at the level of the HSC, and then their downstream progeny achieve additional mutations until achievement of full leukemic transformation. Alternatively, it is possible that the initial mutations could confer inappropriate self-renewal on downstream progenitor cells and thereby give them the necessary life span to acquire further transforming mutations.
Using mouse genetic modeling, at least two approaches to this question have been taken: to analyze models of leukemogenesis and dissect which cellular population(s) the (transplantable) LSC reside in, and to specifically introduce molecular changes in defined HSC or progenitor compartments and assess whether LSC formation occurs. The examination of different models of myeloid malignancies has provided evidence that in principle both HSC and committed progenitor types can be leukemia cells-of-origin that give rise to LSC, depending on the specific disease allele. For instance, studies using retroviral expression of the MLL-ENL (mixed-lineage leukemia translocated 1) and MOZ (monocytic leukemia zinc finger)-TIF2 (transcriptional intermediary factor 2) fusion genes have shown that both HSC and committed myeloid progenitor cells can be transformed by these fusion oncogenes, although with higher efficiency in HSC populations [68, 69]. In a series of tissue-specific knockout models of Junb, it was shown that loss of JUNB in the HSC compartment is required for leukemia initiation and that Junb deletion in granulocyte/monocyte progenitors (GMP) or downstream progeny only resulted in a transient myelomonocytic expansion [14, 15, 70]. Other studies have shown that retrovirally introduced MLL-AF9 can transform both early HSC and committed myeloid progenitors; however, in an MLL-AF9 transgenic model, where MLL-AF9 is expressed at more physiologic gene dosages, the HSC population, but not the more committed GMP population, was able to be transformed [11, 71, 72]. These findings indicate that the gene dosages in some of the retroviral studies may have allowed for activation of target genes in populations that may not naturally be a receptive cellular environment for transformation. Interestingly, in the CEBPA knockout model, despite preleukemic expansion and elevated cycling activity of the HSC and MPP populations, the LSC responsible for sustaining AML did not exhibit features of HSC or MPP [9]. These findings indicate that pre-LSC can reside in different cellular compartments than LSC and that the final genetic or epigenetic changes that lead to generation of fully transformed and transplantable LSC may occur in more committed myeloid progenitors. Further systematic combinatorial modeling of different disease alleles in future studies will provide more insight into the exact temporal and spatial order of events during multistep transformation of LSC.
Of note, although mouse genetic models have provided important insight into cellular stages that can contribute to leukemogenesis, they cannot definitively answer the question of which cell types actually contribute to leukemogenesis and what characterizes the naturally occurring series of events in human leukemia formation. The analysis of purified stem and progenitor populations from patients with AML and MDS seems to suggest that HSC harbor the leukemia cells-of-origin in many subtypes of these diseases, whereas multipotent or further committed progenitors harbor the majority of fully transformed LSC.
Insights Obtained From the Study of Human Leukemias
Advanced cell sorting techniques and the use of xeno-transplantation models have improved our ability to isolate and study LSC in human AML. However, only recently, studies have begun to attempt to define the human pre-LSC compartment including the LCO, which harbor the founding molecular changes that put in motion the process of leukemogenesis. Understanding the pre-LSC and attempting to distinguish pre-LSC from their healthy neighbors have both prognostic and therapeutic implications for the clinic.
It has been demonstrated that preleukemic mutations are present in the self-renewing HSC after chemotherapy and achievement of long-term complete remissions, suggesting that residual HSC in leukemia patients are preleukemic [73–75]. In fact, very early studies examining X-chromosome-linked glucose-6-phosphate dehydrogenase of de novo AML cells and hematopoietic cells during remission had found that cells can possess identical inactivation patterns [76–79]. This observation indicated that AML cells and normal hematopoietic cells in remission could be derived from a dominant HSPC clone that is itself not fully malignant—thus fulfilling the criteria of a pre-LSC. Studies of t(8,21)-positive AML have demonstrated that patients in long-term complete remission contain apparently normal HSC that produce AML1-ETO transcripts [73, 74]. Similar observations were made in chronic myeloid leukemia [80]. The presence of such cells in patients in long-term remission implies that HSC that carry a clonotypic marker of the leukemic clone but are functionally normal and produce healthy progeny are persisting pre-LSC. Recent studies using genome-wide sequencing in de novo AML patients and at the time of relapse have suggested that subclones of the primary clone at initial diagnosis can drive the relapse [81], and clonal analysis revealed the coexistence of “n minus 1” and “n minus 2” subclones (with n defined as the number of mutations in the fully transformed clone) [82], providing additional evidence for the existence of relevant pre-LSC populations in AML.
One approach to understanding the identity of and characterizing pre-LSC in human AML has been the transcriptional analysis of purified stem cells. It was found that fractionated phenotypic LT-HSC from AML patients (distinct from LSC-enriched populations, which have a short-term HSC/MPP immunophenotype) show deregulated transcription of key regulators such as PU.1 and JUNB in comparison with LT-HSC from healthy controls [17]. These findings demonstrated that transcription is disturbed already in very early HSC (i.e., at a pre-LSC level) in AML and that it is possible, in principle, to delineate the molecular events that distinguish pre-LSC from LSC and from HSC from healthy control persons. Further studies compared more comprehensive expression profiles of normal HSC with those of LSC, and pathways such as Wnt signaling, MAP kinase signaling, and adherens junctions and phenotypic markers such as CD32 and CD25 were shown to distinguish healthy HSC from LSC [83–85]. Owing to the recent discovery that LSC can be contained within different cellular compartments and at relatively low frequencies [86], transcriptional analysis strategies have been further expanded. To identify genes dysregulated early on in AML, we have recently used a strategy of parallel transcriptional profiling of multiple, highly fractionated stem and progenitor populations in individual AML patients and compared them with the respective control populations from age-matched healthy individuals [87]. This led to the identification of a relatively small number of genes that were commonly deregulated in all examined compartments and which may thus be characteristic of early pre-LSC. For instance, we found that the protein interleukin-1 receptor accessory protein (IL1RAP) is overexpressed specifically on the surface of HSC of AML and high-risk MDS patients, that IL1RAP is functionally relevant for the clonogenicity of AML cells, and that high IL1RAP expression is independently associated with poor overall survival of AML patients [87].
Evidence at the genetic level for an involvement of the stem cell compartment in disease initiation in AML was initially obtained by cytogenetic analysis of subpopulations of CD34+ cells [88, 89]. Important insight has also come from the study of MDS, which frequently progresses to AML. In MDS patients with a 5q deletion, almost all CD34+CD38− cells appeared to be involved in the 5q-deleted clone [90], and similar findings were made in MDS with trisomy 8 (+8) aberrations [91]. We have recently found in MDS patients with monosomy 7 (−7) or monosomy 20, and in AML with −7, that the majority of Lin−CD34+CD38− early HSC carry the clonotypic aberrations [87, 92]. Recently, genome-wide sequencing of different types of AML cases and of healthy HSC has demonstrated how different molecular events at a genetic level act as initiating mutations in different forms of AML. Comparison of the genome of patients with PML-RARA (retinoic acid receptor-α) with genomes of other AML patients (FAB M1, normal karyotype AML) suggested that mutations in nucleophosmin (NPM1), DNMT3A, and IDH1 may act as major initiating mutations in M1 AML [82]. Recent work has also compared de novo AML and patient-matched residual nonleukemic HSC, to distinguish “early,” persisting mutations present in residual HSC from “late” mutations absent from residual HSC and only present in leukemic cells [75]. Applying exome sequencing and single-cell assays of residual HSC, the authors mapped the clonal evolution of the AML genomes of patients with FLT3-ITD-positive AML, identifying founding mutations in pre-LSC (e.g., TET2 mutations). Notably, the FLT3-ITD mutation was absent in residual HSC, indicating that it was acquired at later stages during LSC transformation [75]. In another recent report, examination of myeloid skewing in normal elderly individuals revealed the existence of clonal somatic mutations, including TET2, in the hematopoietic compartment without overt hematopoietic malignancies [93].
Attempts have been made to identify core transcriptional signatures of LSC that can be used prognostically. Recently, it has been shown that an LSC-specific transcriptional signature and a core transcriptional program shared by LSC and healthy HSC is an independent predictor of AML patient survival [85, 94]. Correlative evidence has supported the idea that leukemic stem cell frequencies and properties may be prognostic in AML, in particular for the minimal residual (stem cell) disease and relapse setting, linking clinical outcome with either the capacity for a sample to be xenografted or surface expression of LSC-linked markers [92, 95–97]. Other studies have shown that malignant stem cells persist in MDS with 5q− in complete remission [98]. Additionally, the specific expanded compartment has also been linked to prognosis. In MDS, for example, high-risk patients exhibit expansion of the phenotypic LT-HSC and GMP compartments, whereas lower-risk patients demonstrate expansion of the phenotypic common myeloid progenitor compartment [92]. Furthermore, expansion of the phenotypic HSC compartment can precede clinical relapse in MDS by several months [92]. These findings, together with the eventual progression of MDS to AML in approximately 30% of cases, suggest that aberrant MDS stem cells may be a pre-LSC stage with regards to AML.
Interesting insights into the progression of pre-LSC have come from studies of inherited and congenital disorders that predispose individuals to hematopoietic malignancies. In such cases, the first “hit” or cancer-initiating step may be the constitutional genetic abnormality itself. In Down syndrome, Fanconi anemia, and familial platelet disorder, for example, patients are predisposed to myeloid malignancies, indicating such predisposing genetic hits are necessary but insufficient for the development of malignancy. In Down syndrome, including in cases of mosaic Down syndrome, up to 10% of neonates develop a trisomy 21 positive transient myeloproliferative disorder (TMD) that resolves without therapeutic intervention. Between years 1 and 4 of life, in approximately 20% of these children, these TMD clones, which can be considered residual pre-LSC, acquire additional postnatal genetic events leading to acute megakaryoblastic leukemia characterized not only by trisomy 21 but also additional complex cytogenetic abnormalities [99–101]. Fanconi anemia (FA) is a genetic disease characterized by genomic instability, bone marrow failure caused by cell cycle arrest, and a predisposition to malignancies, including MDS and AML. In response to stem cell exhaustion in FA, there is pressure for clonal evolution of hematopoiesis in FA patients, resulting, interestingly, in some cells undergoing spontaneous reverse mutation, leading to somatic mosaicism and allowing genetically corrected cells to repopulate the bone marrow. Strikingly, however, such reversion is not capable of protecting against transformation to MDS and leukemia, and “nonreverted” HSC still clonally evolve to leukemia [102–104]. Such findings indicate that the congenitally aberrant HSC of patients with such disorders are actually pre-LSC, “predisposed” to the progression of further steps leading to MDS/AML.
Conclusion
Data obtained from both mouse and human AML and MDS have provided evidence for the existence of functionally relevant preleukemic stem cell populations, and mouse genetic models have shown that these pre-LSC are starting points for the multistep pathogenesis of AML and formation of fully transformed LSC. Data from patients with AML and MDS indicate the persistence of pre-LSC in remission and strongly suggest their involvement in minimal residual disease that can cause relapse. Further refined sorting strategies and separation of pre-LSC (including the leukemia cells-of-origin), LSC, and normal HSC and progenitor populations, combined with current genome-wide sequencing technologies and functional testing in the context of clinical studies for the treatment of patients with AML and MDS, will be instrumental to target all relevant (pre-)LSC populations for both diagnostic and therapeutic purposes. Further combinatorial genetic modeling will be helpful to determine early and late aberrations in the pathogenesis of AML and may thus ultimately allow for more specific targeting of pre-LSC to achieve more long-term remissions and possibly a lasting cure of the disease.
Acknowledgments
The important work of many has contributed to shaping the current model of leukemogenesis. We apologize to all scientists whose work could not be cited because of space constraints. We thank Britta Will for helpful discussion and critiques, and the reviewers for highly insightful comments and suggestions in preparation of this review. U.S. is the Diane and Arthur B. Belfer Faculty Scholar in Cancer Research of the Albert Einstein College of Medicine.
Author Contributions
A.P. and L.B.: manuscript writing; U.S.: manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Gilliland DG, Tallman MS. Focus on acute leukemias. Cancer Cell. 2002;1:417–420. doi: 10.1016/s1535-6108(02)00081-8. [DOI] [PubMed] [Google Scholar]
- 2.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–743. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 3.Friedman AD. Transcriptional control of granulocyte and monocyte development. Oncogene. 2007;26:6816–6828. doi: 10.1038/sj.onc.1210764. [DOI] [PubMed] [Google Scholar]
- 4.Laiosa CV, Stadtfeld M, Graf T. Determinants of lymphoid-myeloid lineage diversification. Annu Rev Immunol. 2006;24:705–738. doi: 10.1146/annurev.immunol.24.021605.090742. [DOI] [PubMed] [Google Scholar]
- 5.Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003;3:89–101. doi: 10.1038/nrc989. [DOI] [PubMed] [Google Scholar]
- 6.Shih LY, Liang DC, Huang CF, et al. AML patients with CEBPalpha mutations mostly retain identical mutant patterns but frequently change in allelic distribution at relapse: A comparative analysis on paired diagnosis and relapse samples. Leukemia. 2006;20:604–609. doi: 10.1038/sj.leu.2404124. [DOI] [PubMed] [Google Scholar]
- 7.Tiesmeier J, Czwalinna A, Muller-Tidow C, et al. Evidence for allelic evolution of C/EBPalpha mutations in acute myeloid leukaemia. Br J Haematol. 2003;123:413–419. doi: 10.1046/j.1365-2141.2003.04618.x. [DOI] [PubMed] [Google Scholar]
- 8.Kirstetter P, Schuster MB, Bereshchenko O, et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell. 2008;13:299–310. doi: 10.1016/j.ccr.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 9.Bereshchenko O, Mancini E, Moore S, et al. Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPalpha mutant AML. Cancer Cell. 2009;16:390–400. doi: 10.1016/j.ccr.2009.09.036. [DOI] [PubMed] [Google Scholar]
- 10.de Guzman CG, Warren AJ, Zhang Z, et al. Hematopoietic stem cell expansion and distinct myeloid developmental abnormalities in a murine model of the AML1-ETO translocation. Mol Cell Biol. 2002;22:5506–5517. doi: 10.1128/MCB.22.15.5506-5517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W, Kumar AR, Hudson WA, et al. Malignant transformation initiated by Mll-AF9: Gene dosage and critical target cells. Cancer Cell. 2008;13:432–440. doi: 10.1016/j.ccr.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuo YH, Landrette SF, Heilman SA, et al. Cbf beta-SMMHC induces distinct abnormal myeloid progenitors able to develop acute myeloid leukemia. Cancer Cell. 2006;9:57–68. doi: 10.1016/j.ccr.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 13.Rosenbauer F, Wagner K, Kutok JL, et al. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, Pu 1. Nat Genet. 2004;36:624–630. doi: 10.1038/ng1361. [DOI] [PubMed] [Google Scholar]
- 14.Passegué E, Wagner EF, Weissman IL. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell. 2004;119:431–443. doi: 10.1016/j.cell.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 15.Santaguida M, Schepers K, King B, et al. JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell. 2009;15:341–352. doi: 10.1016/j.ccr.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimizu R, Kuroha T, Ohneda O, et al. Leukemogenesis caused by incapacitated GATA-1 function. Mol Cell Biol. 2004;24:10814–10825. doi: 10.1128/MCB.24.24.10814-10825.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steidl U, Rosenbauer F, Verhaak RG, et al. Essential role of Jun family transcription factors in PU.1 knockdown-induced leukemic stem cells. Nat Genet. 2006;38:1269–1277. doi: 10.1038/ng1898. [DOI] [PubMed] [Google Scholar]
- 18.Kawahara M, Pandolfi A, Bartholdy B, et al. H2.0-like homeobox regulates early hematopoiesis and promotes acute myeloid leukemia. Cancer Cell. 2012;22:194–208. doi: 10.1016/j.ccr.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orkin SH, Zon LI. Hematopoiesis: An evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khandanpour C, Kosan C, Gaudreau MC, et al. Growth factor independence 1 protects hematopoietic stem cells against apoptosis but also prevents the development of a myeloproliferative-like disease. Stem Cells. 2011;29:376–385. doi: 10.1002/stem.575. [DOI] [PubMed] [Google Scholar]
- 21.Smith LL, Yeung J, Zeisig BB, et al. Functional crosstalk between Bmi1 and MLL/Hoxa9 axis in establishment of normal hematopoietic and leukemic stem cells. Cell Stem Cell. 2011;8:649–662. doi: 10.1016/j.stem.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 22.Antonchuk J, Sauvageau G, Humphries RK. HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell. 2002;109:39–45. doi: 10.1016/s0092-8674(02)00697-9. [DOI] [PubMed] [Google Scholar]
- 23.Ruiz-Herguido C, Guiu J, D'Altri T, et al. Hematopoietic stem cell development requires transient Wnt/beta-catenin activity. J Exp Med. 2012;209:1457–1468. doi: 10.1084/jem.20120225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodrigues NP, Tipping AJ, Wang Z, et al. GATA-2 mediated regulation of normal hematopoietic stem/progenitor cell function, myelodysplasia and myeloid leukemia. Int J Biochem Cell Biol. 2012;44:457–460. doi: 10.1016/j.biocel.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akala OO, Clarke MF. Hematopoietic stem cell self-renewal. Curr Opin Genet Dev. 2006;16:496–501. doi: 10.1016/j.gde.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 26.Blank U, Karlsson G, Karlsson S. Signaling pathways governing stem-cell fate. Blood. 2008;111:492–503. doi: 10.1182/blood-2007-07-075168. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Han F, Wu J, et al. The role of Skp2 in hematopoietic stem cell quiescence, pool size, and self-renewal. Blood. 2011;118:5429–5438. doi: 10.1182/blood-2010-10-312785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu JQ, Seay M, Schulz VP, et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS Genet. 2012;8:e1002565. doi: 10.1371/journal.pgen.1002565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ooi SK, O'Donnell AH, Bestor TH. Mammalian cytosine methylation at a glance. J Cell Sci. 2009;122:2787–2791. doi: 10.1242/jcs.015123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Illingworth RS, Bird AP. CpG islands: “A rough guide.”. FEBS Lett. 2009;583:1713–1720. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 31.Ji H, Ehrlich LI, Seita J, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–342. doi: 10.1038/nature09367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bocker MT, Hellwig I, Breiling A, et al. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood. 2011;117:e182–e189. doi: 10.1182/blood-2011-01-331926. [DOI] [PubMed] [Google Scholar]
- 33.Bröske AM, Vockentanz L, Kharazi S, et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet. 2009;41:1207–1215. doi: 10.1038/ng.463. [DOI] [PubMed] [Google Scholar]
- 34.Trowbridge JJ, Snow JW, Kim J, et al. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell. 2009;5:442–449. doi: 10.1016/j.stem.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Figueroa ME, Lugthart S, Li Y, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akalin A, Garrett-Bakelman FE, Kormaksson M, et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 2012;8:e1002781. doi: 10.1371/journal.pgen.1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou L, Opalinska J, Sohal D, et al. Aberrant epigenetic and genetic marks are seen in myelodysplastic leukocytes and reveal Dock4 as a candidate pathogenic gene on chromosome 7q. J Biol Chem. 2011;286:25211–25223. doi: 10.1074/jbc.M111.235028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan XJ, Xu J, Gu ZH, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309–315. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 41.Hollink IH, Feng Q, Danen-van Oorschot AA, et al. Low frequency of DNMT3A mutations in pediatric AML, and the identification of the OCI-AML3 cell line as an in vitro model. Leukemia. 2012;26:371–373. doi: 10.1038/leu.2011.210. [DOI] [PubMed] [Google Scholar]
- 42.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka S, Miyagi S, Sashida G, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood. 2012;120:1107–1117. doi: 10.1182/blood-2011-11-394932. [DOI] [PubMed] [Google Scholar]
- 44.Ko M, Huang Y, Jankowska AM, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chou WC, Chou SC, Liu CY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118:3803–3810. doi: 10.1182/blood-2011-02-339747. [DOI] [PubMed] [Google Scholar]
- 46.Langemeijer SM, Kuiper RP, Berends M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 47.Tefferi A, Lim KH, Abdel-Wahab O, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009;23:1343–1345. doi: 10.1038/leu.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Couronné L, Lippert E, Andrieux J, et al. Analyses of TET2 mutations in post-myeloproliferative neoplasm acute myeloid leukemias. Leukemia. 2010;24:201–203. doi: 10.1038/leu.2009.169. [DOI] [PubMed] [Google Scholar]
- 49.Kosmider O, Gelsi-Boyer V, Ciudad M, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. 2009;94:1676–1681. doi: 10.3324/haematol.2009.011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pratcorona M, Abbas S, Sanders MA, et al. Acquired mutations in ASXL1 in acute myeloid leukemia: Prevalence and prognostic value. Haematologica. 2012;97:388–392. doi: 10.3324/haematol.2011.051532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144–147. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paschka P, Schlenk RF, Gaidzik VI, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636–3643. doi: 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- 53.Schnittger S, Haferlach C, Ulke M, et al. IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood. 2010;116:5486–5496. doi: 10.1182/blood-2010-02-267955. [DOI] [PubMed] [Google Scholar]
- 54.Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sasaki M, Knobbe CB, Munger JC, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abdel-Wahab O, Adli M, Lafave LM, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22:180–193. doi: 10.1016/j.ccr.2012.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harris WJ, Huang X, Lynch JT, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 59.Gan B, Hu J, Jiang S, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468:701–704. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J, Grindley JC, Yin T, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 61.Iriuchishima H, Takubo K, Matsuoka S, et al. Ex vivo maintenance of hematopoietic stem cells by quiescence induction through Fbxw7α overexpression. Blood. 2011;117:2373–2377. doi: 10.1182/blood-2010-07-294801. [DOI] [PubMed] [Google Scholar]
- 62.Grignani F, Valtieri M, Gabbianelli M, et al. PML/RAR alpha fusion protein expression in normal human hematopoietic progenitors dictates myeloid commitment and the promyelocytic phenotype. Blood. 2000;96:1531–1537. [PubMed] [Google Scholar]
- 63.Ito K, Bernardi R, Morotti A, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072–1078. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen C, Liu Y, Liu R, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205:2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takubo K, Goda N, Yamada W, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7:391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 66.Gurumurthy S, Xie SZ, Alagesan B, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468:659–663. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cozzio A, Passegue E, Ayton PM, et al. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17:3029–3035. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huntly BJ, Shigematsu H, Deguchi K, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6:587–596. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 70.Passegué E, Jochum W, Schorpp-Kistner M, et al. Chronic myeloid leukemia with increased granulocyte progenitors in mice lacking junB expression in the myeloid lineage. Cell. 2001;104:21–32. doi: 10.1016/s0092-8674(01)00188-x. [DOI] [PubMed] [Google Scholar]
- 71.Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10:257–268. doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 72.Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 73.Miyamoto T, Nagafuji K, Akashi K, et al. Persistence of multipotent progenitors expressing AML1/ETO transcripts in long-term remission patients with t(8;21) acute myelogenous leukemia. Blood. 1996;87:4789–4796. [PubMed] [Google Scholar]
- 74.Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci USA. 2000;97:7521–7526. doi: 10.1073/pnas.97.13.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jan M, Snyder T, Corces-Zimmerman M, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fialkow PJ, Martin PJ, Najfeld V, et al. Evidence for a multistep pathogenesis of chronic myelogenous leukemia. Blood. 1981;58:158–163. [PubMed] [Google Scholar]
- 77.Ferraris AM, Raskind WH, Bjornson BH, et al. Heterogeneity of B cell involvement in acute nonlymphocytic leukemia. Blood. 1985;66:342–344. [PubMed] [Google Scholar]
- 78.Fialkow PJ, Singer JW, Raskind WH, et al. Clonal development, stem-cell differentiation, and clinical remissions in acute nonlymphocytic leukemia. N Engl J Med. 1987;317:468–473. doi: 10.1056/NEJM198708203170802. [DOI] [PubMed] [Google Scholar]
- 79.Fialkow PJ, Janssen JW, Bartram CR. Clonal remissions in acute nonlymphocytic leukemia: Evidence for a multistep pathogenesis of the malignancy. Blood. 1991;77:1415–1417. [PubMed] [Google Scholar]
- 80.Chomel JC, Brizard F, Veinstein A, et al. Persistence of BCR-ABL genomic rearrangement in chronic myeloid leukemia patients in complete and sustained cytogenetic remission after interferon-alpha therapy or allogeneic bone marrow transplantation. Blood. 2000;95:404–408. [PubMed] [Google Scholar]
- 81.Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Majeti R, Becker MW, Tian Q, et al. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc Natl Acad Sci USA. 2009;106:3396–3401. doi: 10.1073/pnas.0900089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Saito Y, Kitamura H, Hijikata A, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med. 2010;2:17ra9. doi: 10.1126/scitranslmed.3000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gentles AJ, Plevritis SK, Majeti R, et al. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA. 2010;304:2706–2715. doi: 10.1001/jama.2010.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sarry JE, Murphy K, Perry R, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J Clin Invest. 2011;121:384–395. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barreyro L, Will B, Bartholdy B, et al. Overexpression of interleukin 1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood. 2012;120:1290–1298. doi: 10.1182/blood-2012-01-404699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haase D, Feuring-Buske M, Konemann S, et al. Evidence for malignant transformation in acute myeloid leukemia at the level of early hematopoietic stem cells by cytogenetic analysis of CD34+ subpopulations. Blood. 1995;86:2906–2912. [PubMed] [Google Scholar]
- 89.Mehrotra B, George TI, Kavanau K, et al. Cytogenetically aberrant cells in the stem cell compartment (CD34+lin-) in acute myeloid leukemia. Blood. 1995;86:1139–1147. [PubMed] [Google Scholar]
- 90.Nilsson L, Astrand-Grundstrom I, Arvidsson I, et al. Isolation and characterization of hematopoietic progenitor/stem cells in 5q-deleted myelodysplastic syndromes: Evidence for involvement at the hematopoietic stem cell level. Blood. 2000;96:2012–2021. [PubMed] [Google Scholar]
- 91.Nilsson L, Astrand-Grundstrom I, Anderson K, et al. Involvement and functional impairment of the CD34(+)CD38(-)Thy-1(+) hematopoietic stem cell pool in myelodysplastic syndromes with trisomy 8. Blood. 2002;100:259–267. doi: 10.1182/blood-2001-12-0188. [DOI] [PubMed] [Google Scholar]
- 92.Will B, Zhou L, Vogler T, et al. Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood. 2012;120:2076–2086. doi: 10.1182/blood-2011-12-399683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Busque L, Patel JP, Figueroa ME, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012 doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 95.van Rhenen A, Feller N, Kelder A, et al. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res. 2005;11:6520–6527. doi: 10.1158/1078-0432.CCR-05-0468. [DOI] [PubMed] [Google Scholar]
- 96.Pearce DJ, Taussig D, Zibara K, et al. AML engraftment in the NOD/SCID assay reflects the outcome of AML: Implications for our understanding of the heterogeneity of AML. Blood. 2006;107:1166–1173. doi: 10.1182/blood-2005-06-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Terwijn M, Kelder A, Snel AN, et al. Minimal residual disease detection defined as the malignant fraction of the total primitive stem cell compartment offers additional prognostic information in acute myeloid leukaemia. Int J Lab Hematol. 2012;34:432–441. doi: 10.1111/j.1751-553X.2012.01416.x. [DOI] [PubMed] [Google Scholar]
- 98.Tehranchi R, Woll PS, Anderson K, et al. Persistent malignant stem cells in del(5q) myelodysplasia in remission. N Engl J Med. 2010;363:1025–1037. doi: 10.1056/NEJMoa0912228. [DOI] [PubMed] [Google Scholar]
- 99.Zipursky A. Transient leukaemia: A benign form of leukaemia in newborn infants with trisomy 21. Br J Haematol. 2003;120:930–938. doi: 10.1046/j.1365-2141.2003.04229.x. [DOI] [PubMed] [Google Scholar]
- 100.Wong KY, Jones MM, Srivastava AK, et al. Transient myeloproliferative disorder and acute nonlymphoblastic leukemia in Down syndrome. J Pediatr. 1988;112:18–22. doi: 10.1016/s0022-3476(88)80112-4. [DOI] [PubMed] [Google Scholar]
- 101.Roy A, Roberts I, Norton A, et al. Acute megakaryoblastic leukaemia (AMKL) and transient myeloproliferative disorder (TMD) in Down syndrome: A multi-step model of myeloid leukaemogenesis. Br J Haematol. 2009;147:3–12. doi: 10.1111/j.1365-2141.2009.07789.x. [DOI] [PubMed] [Google Scholar]
- 102.Gregory JJ, Jr., Wagner JE, Verlander PC, et al. Somatic mosaicism in Fanconi anemia: Evidence of genotypic reversion in lymphohematopoietic stem cells. Proc Natl Acad Sci USA. 2001;98:2532–2537. doi: 10.1073/pnas.051609898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li J, Sejas DP, Zhang X, et al. TNF-alpha induces leukemic clonal evolution ex vivo in Fanconi anemia group C murine stem cells. J Clin Invest. 2007;117:3283–3295. doi: 10.1172/JCI31772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ceccaldi R, Briot D, Larghero J, et al. Spontaneous abrogation of the G(2)DNA damage checkpoint has clinical benefits but promotes leukemogenesis in Fanconi anemia patients. J Clin Invest. 2011;121:184–194. doi: 10.1172/JCI43836. [DOI] [PMC free article] [PubMed] [Google Scholar]