

This study demonstrated the synergistic antitumor efficacy of PF-03084014 in combination with docetaxel in triple-negative breast cancer models. Mechanistic evaluations showed that PF-03084014 reduced the survival of cancer stem cell (CSC) and non-CSC populations. This work therefore provides a strong preclinical rationale for the clinical utility of PF-03084014 to improve taxane therapy.
Keywords: Breast cancer, FACS, Stem cell, Stem cell transplantation
Abstract
Notch signaling mediates breast cancer cell survival and chemoresistance. In this report, we aimed to evaluate the antitumor efficacy of PF-03084014 in combination with docetaxel in triple-negative breast cancer models. The mechanism of action was investigated. PF-03084014 significantly enhanced the antitumor activity of docetaxel in multiple xenograft models including HCC1599, MDA-MB-231Luc, and AA1077. Docetaxel activated the Notch pathway by increasing the cleaved Notch1 intracellular domain and suppressing the endogenous Notch inhibitor NUMB. PF-03084014 used in combination with docetaxel reversed these effects and demonstrated early-stage synergistic apoptosis. Docetaxel elicited chemoresistance by elevating cytokine release and expression of survivin and induced an endothelial mesenchymal transition (EMT) phenotype by increasing the expressions of Snail, Slug, and N-cadherin. When reimplanted, the docetaxel-residual cells not only became much more tumorigenic, as evidenced by a higher fraction of tumor-initiating cells (TICs), but also showed higher metastatic potential compared with nontreated cells, leading to significantly shortened survival. In contrast, PF-03084014 was able to suppress expression of survivin and MCL1, reduce ABCB1 and ABCC2, upregulate BIM, reverse the EMT phenotype, and diminish the TICs. Additionally, the changes to the ALDH+ and CD133+/CD44+ subpopulations following therapy corresponded with the TIC self-renewal assay outcome. In summary, PF-03084014 demonstrated synergistic effects with docetaxel through multiple mechanisms. This work provides a strong preclinical rationale for the clinical utility of PF-03084014 to improve taxane therapy.
Introduction
Notch signaling is linked to the progression of various solid tumor diseases including breast cancer [1]. Upon activation of the pathway, γ-secretase cleaves Notch and releases the Notch intracellular domain (NICD), which translocates to the nucleus and activates a cascade of transcriptional events that mediate cellular proliferation, differentiation, and apoptosis. Notch signaling is mechanistically associated with chemoresistance [2, 3] through its regulation of ABC transporters [4], apoptotic proteins [5], and the p53 pathway [6]. Notch is also involved in autocrine production by tumor cells, which has been abrogated by the γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT; LY-374973) [7]. The interleukin (IL)-6 and IL-8 cytokine networks promote chemoresistance by upregulating antiapoptotic proteins [8] and multidrug-resistant genes [9] in tumor cells. In metastatic breast cancer, circulating IL-6 predicts the severity of metastases and patient survival [10] and maintains breast cancer stem cell (CSC) self-renewal through mitogen-activated protein kinase (MAPK)-dependent upregulation of Notch 3 [11], which is essential for stem cell regulation and differentiation in a variety of tissues.
Notch signaling confers chemoresistance in part by mediating CSC self-renewal and survival [12]. Elevated Notch signaling has been reported in breast cancer-initiating populations compared with bulk tumor cells [13–15]. The CSC theory proposes that standard chemotherapy [16] and radiation [17] primarily targets differentiated cells or the majority of bulk tumor cells, leaving a small population of dormant CSCs behind. These CSCs exhibit higher tumorigenic potential and often acquire an endothelial mesenchymal transition (EMT) phenotype, leading to therapy-resistant relapse and metastasis. In breast cancer, tumor-initiating cells (TICs) were first isolated from a patient with breast carcinoma and identified as CD44+/CD24− cells that demonstrated a high tumor-forming ability [18]. Clinically, a higher fraction of CD44+/CD24−/low cells was found in the basal-like breast cancer subtype [19, 20] and was linked to decreased survival. An elevated CD44+/CD24− subpopulation has been observed in disseminated tumor cells [21] and after chemotherapy [16, 22]. Over the past few years, therapeutic strategies to target CSCs are emerging in cancer drug discovery [2, 23]. Notch signaling has been extensively studied for its role in the self-renewal and survival of CSCs [12]. The pharmacological inhibition of Notch signaling by the blockade of Notch ligands Dll4 [24] and the γ-secretase inhibitor (GSI) [14, 25] results in reduced TICs in experimental models. Multiple reports have demonstrated that Notch inhibitors enhance chemotherapy in solid tumor models [24, 26, 27].
Identifying a reliable CSC biomarker would enable implementation of the CSC concept in a clinical setting. Although CD44+/CD24−/low breast cancer cells showed tumor-imitating characteristics, additional evidence revealed a disconnect between the percentage of CD44+/CD24− cells and tumorigenicity [19, 28]. A further report identified other markers for breast CSCs. ALDH1 activity alone [29] or in conjunction with CD44+/CD24− was recently reported to identify breast CSC-like cells [30]. CD133+ cells isolated from BRCA1 breast tumors also displayed CSC characteristics [13]. These studies demonstrated the phenotypic heterogeneity of breast CSCs and the technical challenges of identifying a robust CSC biomarker.
PF-03084014 is a γ-secretase inhibitor that exhibits antitumor efficacy in hematological and breast xenograft models via pleiotropic mechanisms [31, 32]. In this report, we evaluated the antitumor efficacy of PF-03084014 alone and in combination with docetaxel against triple-negative breast cancer. The mechanisms of action were explored by evaluating their combined effects against differentiated tumor cells, as well as against the self-renewal ability of the TICs. The breast CSC markers were profiled for their correlation with tumor-imitating ability. This work provides insights into the antitumor mechanisms of PF-03084014 in combination with docetaxel and offers a potential clinical strategy to enhance taxane therapy against breast cancer.
Materials and Methods
In Vivo Combination Studies and Drug Administration
All experimental animal procedures complied with the Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Research, 1996) and were approved by the Pfizer Global Research and Development Institutional Animal Care and Use Committee. AA1077 is a patient-derived xenograft model, and the primary human breast tumor tissue was collected under a University of California San Diego institutional review board-approved protocol with prospective consent. Tumor cells or trocar fragments from patient-derived tumors were implanted in the dorsal region of SCID Beige mice (Charles River Laboratories, Wilmington, MA, http://www.criver.com). To evaluate drug efficacy, the mice were randomly assigned to groups so that the mean value of tumor size was identical between groups. The mice were assigned to four groups (10 each) and treated with: (a) vehicle; (b) PF-03084014 (p.o.) twice daily at 90 mg/kg on days 1–7 and 15–21; (c) docetaxel (i.p.) on days 1 and 15; and (d) PF-03084014 and docetaxel using the same regimen as for (b) and (c), respectively. To evaluate the combinatorial effect on tumor growth, caliper measurements of tumor sizes were performed twice a week until the mean tumor volume in each group reached 1,000 mm3. For pharmacodynamic analysis, the tumors were snap-frozen and pulverized in a liquid nitrogen-cooled mortar prior to lysis.
To perform the CSC functional and marker profiling analysis, a separate study was conducted using the same design. On day 19 after dosing commencement, tumors were harvested from each group. Fresh tumors were dissociated into single-cell suspensions before CSC characterization.
Pharmacodynamic Endpoint Assessment
Serum IL-6 and IL-8 levels were assessed using enzyme-linked immunosorbent assay (ELISA) kits (Invitrogen, Carlsbad, CA, http://www.invitrogen.com). Tumors were preserved frozen for Western blot or in RNAlater RNA stabilization reagent (Qiagen, Hilden, Germany, http://www.qiagen.com) for RNA analyses. All antibodies for Western blot were purchased from Cell Signaling and applied according to the manufacturer's instructions. β-Actin (Abcam, Cambridge, U.K., http://www.abcam.com) was used as an internal standard. The mRNA expression levels of the human Notch pathway genes (Life Technologies, Rockville, MD, http://www.lifetech.com) were determined via TaqMan real-time PCR (Life Technologies). One microgram of RNA was converted to cDNA in a 20-μl reaction using a high capacity RNA-to-cDNA kit (Life Technologies). Real-time PCR was performed in duplicate on a ViiaTM7 real time PCR system (Applied Biosystems, Foster City, CA, http://www.appliedbiosystems.com). The expression levels were normalized to endogenous glyceraldehyde-3-phosphate dehydrogenase.
Dissociation of Tumor
The collected tumor tissue was minced into small pieces (2–3 mm3) with a scalpel and then incubated in medium containing collagenase/hyaluronidase (StemCell Technologies, Vancouver, BC, Canada, http://www.stemcell.com) at 37°C for 1–4 hours based on the tumor type [33]. After centrifugation, the cell pellet was collected and further dissociated in 0.25% trypsin-EDTA for 5 minutes. The cells were then passed through a 40-μm nylon mesh to produce a single-cell suspension. Mouse cell depletion was performed by incubating the dissociated tumor cells with biotinylated mouse antibodies (H2Kd and CD45; BD Biosciences, San Diego, CA, http://www.bdbiosciences.com) for 15 minutes followed by the addition of Streptavidin MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany, http://www.miltenyibiotec.com) for an additional 15 minutes. The unbound, negatively selected cells were collected using an LD column (Miltenyi Biotec). The isolated cells were then subjected to functional and stem cell marker profiling analyses.
Flow Cytometry
The cell surface marker and aldehyde dehydrogenase (ALDH) analyses were performed on a BD FACSCanto flow cytometer (BD Biosciences). Antibody staining was performed using freshly dispersed cell suspensions at a density of 1 × 106 cells per milliliter in phosphate-buffered saline (PBS). All antibodies for cell surface markers were obtained from BD Biosciences, including fluorescein isothiocyanate anti-human CD44, phycoerythrin (PE) anti-human CD24, and PE anti-human CD133. The corresponding isotype control was used to exclude nonspecific background staining. Nonviable cells were excluded using 7-aminoactinomycin D. The cells were incubated with the antibodies at their respective optimal concentrations at 4°C for 30 minutes and washed twice with PBS prior to fluorescence-activated cell sorting (FACS) analyses.
An ALDH assay was performed using the ALDEFLUOR Kit (StemCell Technologies) according to the manufacturer's instructions. Briefly, the isolated tumor cells were incubated with an ALDH substrate, bodipy-aminoacetaldehyde (5 μl of 300 μM per 1 × 106 cells) in an assay buffer at 37°C for 45 minutes. As a negative control, an aliquot from each sample was incubated under identical conditions in the presence of the ALDH inhibitor diethylaminobenzaldehyde. Following incubation, all of the test tubes were spun and resuspended in ALDEFLUOR buffer and then analyzed on a BD FACSCanto flow cytometer (BD Biosciences).
Secondary Tumor Implant for In Vivo Limiting Dilution Analysis
The in vivo self-renewal ability of the tumor cells was determined after the therapeutic treatment. The dissociated tumor cells were prepared in serial dilutions and injected s.c. into the right flank of SCID Beige mice. To test the tumorigenicity of the sorted CD133+/CD44+ cells, MDA-MB-231Luc tumor-bearing mice received docetaxel (15 mg/kg) i.p. once each week for 3 weeks. When the treatment ended, the tumors were collected and dissociated. CD133+/CD44+ and CD133−/CD44− subpopulations were sorted on a BD FACSAria system, and the cells were prepared in serial dilutions for subsequent transplantation into the recipient SCID Beige mice. Tumor growth was monitored once per week via bioluminescence imaging (BLI) on an IVIS200 system (PerkinElmer, Waltham, MA, http://www.perkinelmer.com) as previously described [34]. Before imaging, the mice were anesthetized with 2.5% isoflurane and received 150 mg/kg D-luciferin firefly potassium salt (Caliper Life Sciences) i.p. BLI of the tumor burden was performed 10 minutes following injection.
Mammosphere Formation Assay
A mammosphere formation assay was performed using Mammo Cult complete medium (StemCell Technologies) supplemented with 4 μg/ml heparin and 0.5 μg/ml hydrocortisone. Dissociated tumor cells were plated in 24-well ultralow attachment plates (Corning) at varying densities for 6 days prior to analysis. The quantitation and imaging were performed using an Olympus 1X51 inverted microscope system (Olympus, Center Valley, PA, http://www.olympusamerica.com). Mammospheres greater than 60 μm in diameter were counted.
Data Analysis
Statistical analyses were conducted using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA, http://www.graphpad.com). The tumor sizes of single-agent and combination-treated groups were compared using Student's t test (two-sided p value). The p values for the Kaplan-Meier survival curves were determined using the GraphPad log-rank test.
Results
The Residual Cancer Cells Surviving Docetaxel Therapy Demonstrated Higher Tumorigenic and Metastatic Potential
Chemotherapy is known to induce secondary malignancies through induction of epithelial-mesenchymal transition and CSCs. To understand the biological properties of the residual tumors after docetaxel therapy, MDA-MB-231Luc tumor-bearing mice were treated with an efficacious dose of docetaxel (15 mg/kg, i.p.) once per week for three cycles prior to tumor harvest. After the “tumor debulking” by docetaxel, Western blot analysis (Fig. 1A) was performed. The residual tumor showed a decrease in NUMB, an endogenous Notch inhibitor, suggesting the activation of Notch signaling. Concurrently, a downregulation of E-cadherin and an upregulation of Snail were also observed in the residual tumor, indicating the possible acquisition of EMT.
Figure 1.

The residual tumor cells display higher tumorigenic and metastatic potential after docetaxel therapy. MDA-MB-231Luc tumor-bearing mice were treated with an efficacious dose of docetaxel (15 mg/kg, i.p.) once per week for three cycles prior to harvesting tumors. Tumor cells were dissociated from the vehicle- and docetaxel-residual tumors (n = 10 each group) for fluorescence-activated cell sorting (FACS) analyses and secondary reimplant. The values are the means ± SEM. (A): Western blot analysis of NUMB and the endothelial mesenchymal transition markers. (B): Representative images of ALDH+ cells via FACS. (C): The percentages of CD133+/CD44+, ALDH+, and CD44+/CD24− subpopulations. For FACS analysis, the isotype control was used for each individual sample. (D, E): The secondary reimplant experiment was performed by implanting cells subcutaneously (D) or intravenously (E) in SCID Beige mice (n = 10 mice per group). A Kaplan-Meier survival plot showed disease progression of the mice with intravenous tumor implant (E). The representative bioluminescence images were collected on day 30 after the reimplant. Abbreviations: ALDH, aldehyde dehydrogenase; DEAB, diethylaminobenzaldehyde; Doce, docetaxel; E-Cad, E-cadherin; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; SSC, side scatter.
Tumor cells were isolated for FACS analyses. Docetaxel treatment enriched the ALDH+ subpopulation from 8.7% (control) to 16.7% (Fig. 1B, 1C). In addition, an elevated percentage of the CD133+/CD44+ subpopulation (28%) was observed in docetaxel-treated tumors compared with vehicle control (8%). On the other hand, the staining pattern of CD44+/CD24−, one of the most studied breast CSC markers, was not altered following docetaxel therapy (Fig. 1C).
A secondary implant was performed using the isolated tumor cells. The tumorigenicity and metastatic potential of the residual tumor cells after docetaxel treatment were compared with the nontreated tumor cells. MDA-MB-231Luc tumors from vehicle or docetaxel-treated mice were then harvested, and the cells were inoculated either subcutaneously (1 × 104 cells per site) or intravenously (1 × 105 cells per mouse) in SCID Beige mice. Mice bearing docetaxel-residual cells exhibited more rapid disease relapse compared with those with naïve tumor cells (Fig. 1D). In experimental metastasis settings, tumor cell colonization occurred primarily in the lungs as shown in the representative bioluminescence images (Fig. 1E). Kaplan-Meier curves indicated a significantly shortened survival (p < .05) in mice bearing docetaxel-residual tumor cells compared with the mice implanted with parental tumor cells. Overall, we observed an increased tumorigenic and metastatic potential of the docetaxel-residual cells.
PF-03084014 Enhanced the Antitumor Efficacy of Docetaxel in Triple-Negative Breast Cancer Xenograft Models
The effect of PF-03084014 in combination with docetaxel was assessed in different tumor models as described in Materials and Methods. The dosing and sampling schedules for the efficacy and pharmacodynamic assessment are illustrated in Figure 2A. For each model, the dosage of docetaxel was preoptimized to induce a moderate antitumor response so that we could measure the genuine enhancement of efficacy in a combinatorial setting. The dosages of docetaxel were 15, 15, and 25 mg/kg for AA1077, MDA-MB-231Luc, and HCC1599, respectively. Among these models, HCC1599 cells harbor a Notch1 fusion and strongly respond to γ-secretase inhibitors in an antiproliferation assay [32].
Figure 2.
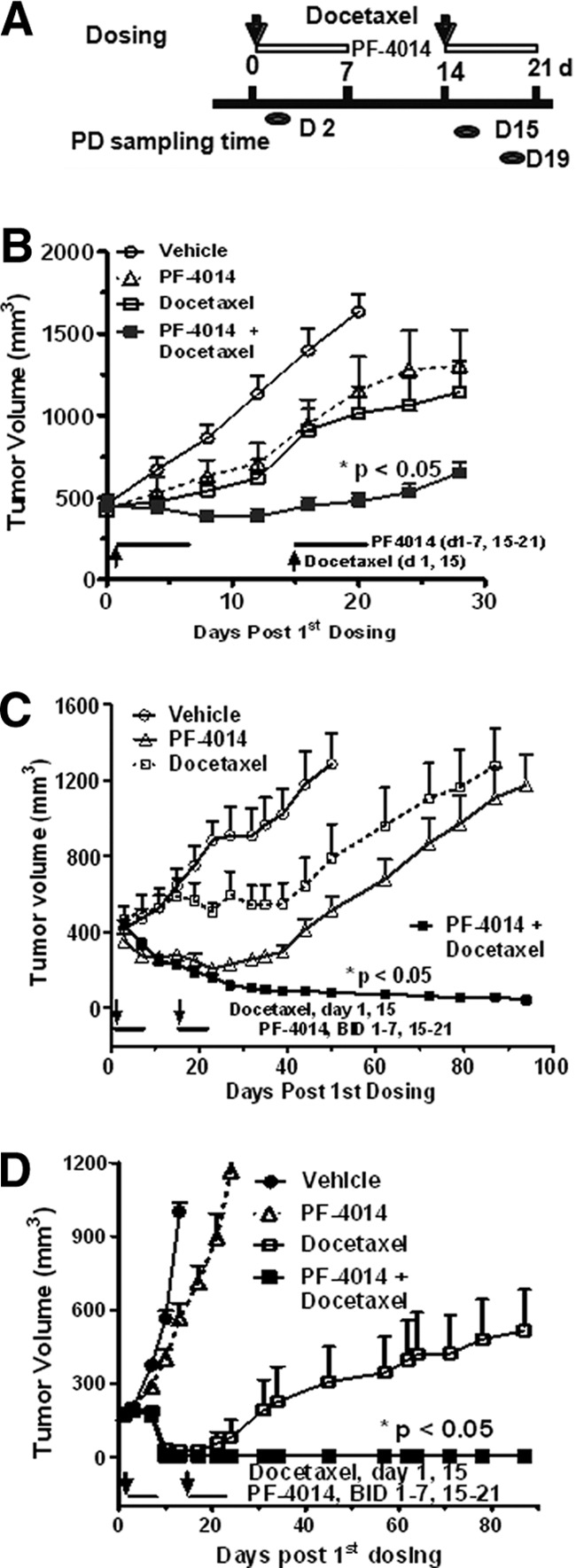
PF-03084014 exhibits synergistic effects with docetaxel in breast xenograft models. Size-matched tumor-bearing mice were treated as described in Materials and Methods. The docetaxel dose was 15 mg/kg (MDA-MB-231Luc and AA1077) or 25 mg/kg (HCC1599). (A): The dosing schedule for the efficacy and pharmacodynamic assessments. (B–D): PF-03084014 significantly improved the efficacies of docetaxel in the MBA-MB-231Luc (B), HCC1599 (C), and AA1077 (D) tumor models (n = 10 mice per group). The values are the means ± SEM. *, p < .05 versus the treatment group of PF-03084014 or docetaxel alone. Abbreviations: d, days; PD, pharmacodynamic endpoint.
In the MDA-MB-231Luc model, the combination of PF-03084014 with docetaxel showed significantly enhanced efficacy compared with docetaxel (p < .05) or PF-03084014 (p < .05) alone (Fig. 2B). The most significant synergy of these two agents was observed in the HCC1599 and AA1077 models (Fig. 2C, 2D). With HCC1599, PF-03084014 at 90 mg/kg (p.o., b.i.d.) induced moderate tumor regression, but all tumors eventually relapsed after the treatment ended. Weak antitumor activity was observed in the docetaxel-treated group, whereas the concurrent treatment of these two agents led to complete tumor regression over the 120-day study period. A similar outcome was found in the AA1077 model. In all of the tested models, no adverse effects were observed in any treatment groups.
PF-03084014 Impaired the Notch Pathway and Resulted in a Synergistic Cytotoxic Effect With Docetaxel in Bulk Tumor Cells
To determine whether the combined effect correlates with a change in the Notch pathway, pharmacodynamic assessments were performed at an early stage of treatment. On the second day after the dosing was initiated, tumors were harvested and subjected to Western blot analysis (Fig. 3A). An increased level of NICD1 was shown in docetaxel-treated tumors, suggesting Notch1 pathway activation, whereas PF-03084014 alone or in combination with docetaxel suppressed the NICD1 level. In addition, decreased expression of NUMB, an endogenous Notch inhibitor, was observed in docetaxel-treated tumors, and this effect was reversed by cotreatment with PF-03084014. Concurrently, docetaxel plus PF-03084014 treatment resulted in increased apoptosis compared with either single-agent-treated groups as evidenced by caspase 3 cleavage. Increased apoptosis in the combination treatment group was accompanied by an elevated BIM level. Notch target genes including NRARP, BIRC5, MYC, CCND1, HES4, Hes2, HEY2, HES6, HES1, CDKN1A, and NOXA were significantly affected by PF-03084014 alone or in a cotreatment setting by the blockade of pan-Notch signaling (Fig. 3B). These results indicate that the synergistic cell killing observed in the early stages of therapy correlates with impairment of the Notch pathway.
Figure 3.

Pharmacodynamic assessment of PF-03084014 alone and in combination with docetaxel in breast xenograft models (n = 5 mice per group). The values are the means ± SEM. (A–C): The data were generated using HCC1599 tumors. (A, C): Western blot analysis of biomarkers on day 2 (A) and day 19 (C) after dosing commencement. (B): Notch target gene modulations on day 19 after treatment. (D): Serum levels of IL-6 or IL-8 on day 15 after dosing commencement in multiple models. (E): Changes in the endothelial mesenchymal transition markers and ABC transporters were observed in MDA-MB-231Luc tumor on day 19. Abbreviations: Doce, docetaxel; E-Cad, E-cadherin; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PF, PF-03084014; Veh, vehicle.
PF-03084014 Treatment Led to the Reversal of Chemoresistance and Endothelial-Mesenchymal Transition
Notch signaling promotes chemoresistance in part through activation of the NF-κB pathway and subsequently induces cytokine release to upregulate antiapoptotic proteins. We investigated the biomarkers in this pathway by Western blot (Fig. 3C) and ELISA (Fig. 3D). Blood serum and tumors were collected on days 15 and 19, respectively, after the commencement of dosing. In all three tested models, docetaxel treatment promoted serum IL-8 release as measured by ELISA, and cotreatment with PF-03084014 reversed this effect. The same trend was observed with IL-6 levels in the MDA-MB-231Luc model. HCC1599 and AA1077 tumor-bearing mice produced no detectable IL-6 in the serum.
In HCC1599 tumors, docetaxel caused increased levels of both survivin and ABCB1, and these effects were abrogated by PF-03084014. In addition, we observed a marked decrease in phospho-survivin (Thr34), MCL-1, and ABCC2 protein levels in PF-03084014 alone and PF-03084014 plus docetaxel groups when compared with vehicle or docetaxel treatment alone. These results were reproducible in the MDA-MB-231Luc tumor model.
Notch pathway activation is involved in the acquisition of EMT [3]. To explore the effect of GSI on EMT, MDA-MB-231Luc tumors were harvested on day 19 post-treatment and subjected to Western blot analysis (Fig. 3E). Compared with docetaxel-treated tumors, the cotreatment of PF-03084014 at 90 mg/kg resulted in significantly decreased ABCB1 and ABCC2 levels. We also observed changes in EMT markers including E-cadherin, Snail, and Slug. The difference between vehicle and single-agent PF-03084014-treated tumors followed the same trend. These results strongly suggested the reversal of EMT by PF-03084014.
PF-03084014 Diminishes Tumor-Initiating Cells in a Notch1Mut HCC1599 Xenograft Model
We next evaluated the effect of PF-03084014 against TICs using the same dose and treatment schedule as was used previously (Fig. 2A). All tumors were collected from HCC1599 tumor-bearing mice for analysis on day 19 after dosing commencement. The tumor cells were dissociated to perform an ex vivo mammosphere formation assay under serum-free and stem cell selective culture conditions (Fig. 4A) and for FACS analysis (Fig. 4B). PF-03084014 alone or in combination with docetaxel suppressed mammosphere formation as measured by the mammosphere forming efficiency (MMFE). Moreover, in vivo self-renewal was assessed by a serial transplantation assay. After in vivo treatment, HCC1599 tumor cells were isolated and reimplanted with 5 × 106 tumor cells per mouse. The mice received no further treatment. Between the vehicle and docetaxel treatments, we observed marginal differences in the secondary tumor-initiating abilities, whereas PF-03084014 treatment led to a 63% reduction in tumor size (Fig. 4C). The most significant inhibition against tumor cell self-renewal (97%) was observed in mice treated with docetaxel in combination with PF-03084014. This result is in good agreement with the mammosphere formation test results.
Figure 4.
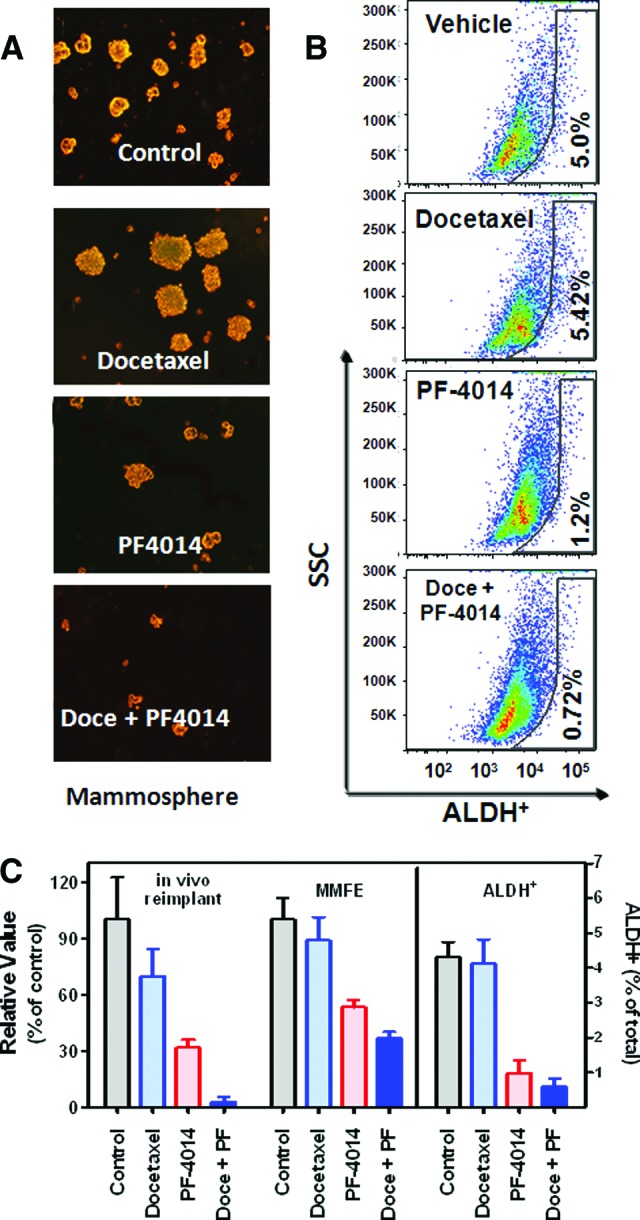
PF-03084014 reduces cancer stem cell frequency in the Notch1mut HCC1599 model (n = 5 mice per group). The values are the means ± SEM. To perform the analyses, tumor cells were dissociated on day 19 after dosing commencement. (A): The representative images in the MMFE test. The tumor-derived cells were seeded at a density of 3,000 cells per well in a 24-well plate for 6 days before analysis. (B): The percentage of ALDH-positive cells. ALDH activity was analyzed by flow cytometry. An aliquot from each sample was tested under identical conditions in the presence of the ALDH inhibitor diethylaminobenzaldehyde. All gates in each sample were created using the DEAB-treated corresponding sample. (C): Quantitative measurement of MMFE, tumor size by serial in vivo transplant, and ALDH+ subpopulation. In vivo self-renewal analysis was performed by implanting 5 × 106 tumor-derived cells per SCID Beige mouse. Abbreviations: ALDH, aldehyde dehydrogenase; Doce, docetaxel; MMFE, mammosphere forming efficiency; PF, PF-03084014; SSC, side scatter.
Observations of the ALDH+ subpopulation corresponded with the functional test outcomes (Fig. 4B, 4C). Compared with the vehicle-treated tumors, the changes in the percentage of ALDH+ cells within the tumor were minimal after docetaxel treatment, whereas PF-03084014 alone or in combination with docetaxel induced a drastic reduction in the ALDH+ cell populations.
PF-03084014 Reduces the Tumor-Imitating Subpopulation in MDA-MB-231Luc Model
Using the same study design as described above, a tumor cell self-renewal assay was performed in the MDA-MB-231Luc model. After the treatment ended, tumor cells from the four treated groups were harvested and dissociated for FACS and functional analyses. Treatment with docetaxel resulted in an increased prevalence (55%) of mammosphere formation (MMFE) compared with vehicle alone (Fig. 5A), indicating that the antitumor activity (45%) of docetaxel caused the relative increase in the number of TICs. The combination of PF-03084014 and docetaxel substantially decreased the number of tumor cell colonies.
Figure 5.

PF-03084014 alone and in combination with docetaxel diminishes the cancer stem cells in MBA-MB-231Luc xenografts. Size-matched tumor-bearing mice were treated as described in Materials and Methods. Tumors were harvested and dissociated on day 19 after dosing commencement to perform fluorescence-activated cell sorting (FACS) and functional analysis (n = 5 mice per group). The values are the means ± SEM. (A): The representative images in the MMFE test. Tumor-derived cells were seeded at a density of 1,500 cells per well in a 24-well plate for 6 days before analysis. (B): The representative bioluminescence imaging images of tumor growth after the secondary implant on day 45. In vivo self-renewal analysis was done by implanting 10 × 104 tumor-derived cells per SCID Beige mouse. (C): Quantitative assessment of the stem cell self-renewal ability. (D): Representative FACS analysis of CD133+/CD44+ cells from each group. Each sample was stained with an isotype control to ensure that nonspecific staining was excluded. (E): Percentage of ALDH+ and CD133+/CD44+ cells in each treatment group. Abbreviations: Doce, docetaxel; Max, maximum; Min, minimum; MMFE, mammosphere forming efficiency.
An in vivo self-renewal test was also conducted after the treatment of MDA-MB-231Luc tumor-bearing mice. Serial dilutions of tumor cells isolated from these treated tumors were implanted s.c. into secondary mice, which received no further treatment. Visualization and quantitation of the tumor-initiating capacity of the cells was monitored by in vivo bioluminescence imaging (Fig. 5B). The results were in agreement with the ex vivo mammosphere assay (Fig. 5C) as tumors that arose from docetaxel residual cells exhibited a 50% increase in tumor size over those from vehicle-treated tumors. In contrast, PF-03084014 inhibited the tumor regrowth rate by 65%. A more substantial decrease in the tumorigenicity was observed (93%) in mice treated with the combination of PF-03084014 and docetaxel compared with the control group.
FACS analyses were performed using the dissociated MDA-MB-231Luc tumor cells after therapy. Compared with vehicle treatment, docetaxel caused an increase in the percentage of CD133+/CD44+ (Fig. 5D) and ALDH+ cells (Fig. 5E), whereas PF-03084014 in combination with docetaxel produced a significant (p < .05) decrease in CD133+/CD44+ and ALDH+ cells. Interestingly, the change in the CD44+/CD24− subpopulation was minimal in all treated groups.
CD133+/CD44+ Is a Tumorigenic Subpopulation in MDA-MB-231Luc Cells
CD133+/CD44+ has not been reported as a breast CSC surface marker. We performed a limiting dilution analysis to understand the self-renewal capability of the CD133+/CD44+ cell subpopulation. To enrich this population, MDA-MB-231Luc tumors were collected and dissociated after 3 weeks of treatment with docetaxel (Fig. 6A). The CD133+/CD44+ and CD133−/CD44− subpopulations were sorted on the FACSAria and prepared in serial dilutions for immediate transplant (Fig. 6B). Each SCID Beige mouse was implanted with CD133+/CD44+ and CD133−/CD44− cells on the left and right flank, respectively. Tumor growth was tracked weekly by bioluminescence imaging. Dose dependence of the tumor formation rate was observed in mice transplanted with CD133+/CD44+ cells (Fig. 6C). As few as 10 CD133+/CD44+ cells were able to give rise to tumors in 4 out of 10 mice. Figure 6D depicts the representative bioluminescence images of mice implanted with 10,000 cells on day 30 postimplantation. When 1,000 or 10,000 cells were implanted, 95% (19 of 20) of mice developed tumors. In contrast, the counterphenotype CD133−/CD44− cells failed to form tumors in most of the implanted mice except one mouse with 10,000 transplanted cells.
Figure 6.

The tumorigenicity of CD133+/CD44+ cells derived from MDA-MB-231Luc tumors. (A): FACS analysis demonstrated that the CD133+/CD44+ subpopulation was enriched after a 3-week treatment of docetaxel (15 mg/kg). (B): Study design for the isolation and reimplant of CD133+/CD44+ and CD133−/CD44− cells. The cells were isolated and sorted after docetaxel treatment. The cell doses for reimplant were 10, 100, 1,000, and 10,000 cells for each of the experimental groups (10 mice per group). (C): The tumorigenic frequency of CD133+/CD44+ or CD133−/CD44− subpopulations. (D): The representative bioluminescence imaging images of the tumor-bearing mice transplanted with 10,000 cells. Abbreviations: FACS, fluorescence-activated cell sorting; Tx, treatment; SOC, standard of care agent.
Discussion
In this study, we report the synergistic antitumor effects of the γ-secretase inhibitor PF-03084014 used in combination with docetaxel in breast cancer models. PF-03084014 not only induced synergistic apoptosis with docetaxel in the bulk tumor cells but also eliminated CSCs that were refractory to docetaxel treatment. Notch plays a critical role for the survival of the bulk tumor cells and TICs upon docetaxel treatment; thus, impairing Notch signaling with PF-03084014 provides a rational combination strategy to enhance docetaxel efficacy.
In multiple breast xenograft models, including MDA-MB-231Luc, AA1077, and HCC1599, PF-03084014 combined with docetaxel significantly enhanced antitumor efficacy compared with either agent alone. In all tested models, we observed less than 10% weight loss, suggesting that the combination produces synergistic antitumor efficacy without increasing Notch-associated gut toxicity [31]. Robust synergy was observed in the AA1077 model, a triple-negative breast PDX xenograft, which is believed to be more reflective of the cellular heterogeneity of tumors in patients. When comparable dosages were applied, one of the models that generated dramatic combinatorial effects was an HCC1599 tumor harboring a Notch1 fusion gene. In mice treated with PF-03084014 or docetaxel alone, all of the tumors eventually relapsed; however, complete tumor regression for more than 4 months was observed in mice receiving the combination treatment. In HCC1599 xenografts, a significant change was observed in a panel of 11 Notch target genes after the treatment of PF-03084014 alone or in combination with docetaxel. Several of these genes, including NRARP, MYC, CCND1, HES4, HEY2, and HES1, were previously reported for correlating with the biological effects of other γ-secretase inhibitors [35, 36]. These findings suggest that the blockade of pan-Notch signaling leads to a Notch-dependent synergy of PF-03084014 plus docetaxel.
Shortly after dosing, docetaxel promoted Notch pathway activation in the HCC1599 tumor by increasing the level of NICD1 and downregulating NUMB, an endogenous Notch inhibitor. The diminished NUMB protein is perhaps a consequence of hyperactivated Notch signaling, as was observed by Chapman et al. [37]. These effects were significantly reversed by cotreatment with PF-03084014. Consequently, a more rapid and abundant onset of apoptosis was observed in combination therapy compared with either single-agent alone. Increased apoptosis in the cotreatment group was accompanied by the elevation of BIM, a Notch3-regulated proapoptotic protein [38]. A similar outcome was observed in the MDA-MD-231Luc model. These results strongly suggest that activation of the Notch pathway by docetaxel and the blockade by PF-03084014 led to synergistic cell killing. The early synergistic cytotoxicity of PF-03084014 and docetaxel occurred in the rapidly proliferating or nontumorigenic cells that constitute the majority of the bulk tumor.
In response to chemotherapy, Notch mediates the NF-κB pathway [7] and the expression of ABC transporters [9], which allow tumor cells to gradually acquire resistance. The IL-6 and IL-8 cytokine networks are highly associated with chemoresistance by regulating both pro- and antiapoptotic proteins [8]. Consistent with these observations, docetaxel treatment resulted in elevated serum levels of IL-6 or IL-8 in all three of the tested models after two cycles of treatment. Coadministration of PF-03084014 abolished these effects and allowed the serum cytokines to return to baseline levels, as was reported with the γ-secretase inhibitor DAPT [7]. Accordingly, the changes in the antiapoptotic protein survivin levels exhibited the same pattern. In MDA-MB-231Luc and HCC1599 tumors, docetaxel caused an upregulation of survivin, whereas in the combination-treated tumors, PF-03084014 significantly downregulated the survivin level, particularly in the Notch1-mutated HCC1599 model. Concurrently, expression of the antiapoptotic protein MCL1, as well as that of the ABC multidrug transporters ABCB1 and ABCC2, was altered with the combination treatment. These phenotypes are highly consistent with the blockade of Notch signaling [5, 9, 39]. PF-03084014 is likely to enable resistant cells to regain sensitivity to docetaxel by manipulating apoptosis pathways and multidrug transporters. This notion is further supported by an increase in BIM1 levels in mice treated with PF-03084014 with docetaxel because BIM1 has been reported to play an important role in docetaxel resistance [40]. Collectively, these results provide molecular evidence that Notch pathway activation is mechanistically linked with a chemoresistance phenotype, and the enhancement of docetaxel efficacy can be partially attributed to the ability of PF-03084014 to overcome chemoresistance.
Ample evidence has indicated the critical role of Notch signaling in CSC-mediated chemoresistance [41]. Considering the PF-03084014-mediated changes in the expression of antiapoptotic proteins and ABC transporters, we further tested whether the synergistic effect with docetaxel derives from the ability of PF-03084014 to eliminate CSCs. In the treated MDA-MB-231Luc and HCC1599 xenografts, limiting dilution assays including mammosphere formation and serial tumor cell reimplant demonstrated that PF-03084014 alone or in the combination settings reduced the proportion of CSCs. In contrast, docetaxel either minimally impacted or promoted tumor cell self-renewal in these models. These findings are highly consistent with previous reports that under preclinical [42] and clinical [16, 43] settings, CSCs can survive radiation or chemotherapy despite the robust early response. In addition, the treatment-induced changes in IL-6 and IL-8 levels further support the function of PF-03084014 against CSCs. IL-6 maintains breast CSC self-renewal through MAPK-dependent upregulation of Notch3 [11], which is essential for stem cell regulation and differentiation in a variety of tissues. The overexpression of IL-8 was found in the ALDH+ tumor-initiating subpopulation [44]. When docetaxel effectively reduced the tumor burden, the fraction of CSCs was elevated in the residual tumor. Eradicating CSCs with PF-03084014 by modulating the Notch pathway led to the enhanced efficacy of docetaxel against a broad spectrum of heterogeneous cancer cells.
As CSC biology is evolving, a significant research effort has been made to identify CSC markers in order to seek more effective therapy. ALDH was one of these reported markers [30]. Indeed, our data showed that the treatment-associated changes in the ALDH+ subpopulation greatly corresponded with the CSC functional analyses assayed by mammosphere formation and secondary tumor reimplant experiments. In the MDA-MB-231Luc and HCC1599 tumor models, docetaxel either increased or minimally affected the ALDH+ cells in the residual tumor, respectively, whereas PF-03084014 diminished this subpopulation. Using MDA-MB-231Luc tumors, we further demonstrated that CD133+/CD44+ is a breast CSC marker, because this subpopulation exhibited much higher tumorigenic potential (1,000-fold) compared with CD133−/CD44− cells, which is in agreement with previous observations in other tumor types [45]. The therapeutic effect of PF-03084014 and docetaxel on the CD133+/CD44+ subpopulation corresponded to the changes in CSC self-renewal ability. Unfortunately, the CD133+/CD44+ subpopulation was below measurable levels in the HCC1599 xenografts despite containing TICs. In addition, we found that the CD44+/CD24− classification was insufficient to identify the tumorigenic cells in our tested models, which is in line with previous reports [28, 30]. The ALDH+ and CD133+/CD44+ subpopulations showed minimal overlap, and both subpopulations were diminished after treatment with PF-03084014, suggesting the heterogeneous nature of the TICs. These results highlight the fact that one of the key challenges for implementing the concept of CSCs in the clinic is lacking reliable surrogate markers that can identify TICs across a broad range of tumors [46].
In addition to its role in regulating CSC self-renewal pathways, Notch signaling is also known to mediate CSC differentiation [47]. Cell dormancy is a crucial mechanism for CSCs to escape from antiproliferative chemotherapy, which may lead to local recurrence or distant metastasis after long latency [42]. After docetaxel therapy, the residual MDA-MB-231Luc cells not only demonstrated higher tumorigenic ability but also showed substantially higher metastatic potential compared with naïve tumor cells. In response to docetaxel, the protein level changes of Snail and E-cadherin were highly consistent with the EMT phenotype. PF-03084014 abolished these effects with a concurrent modulation of NICD1, suggesting that the inactivation of Notch signaling resulted in the reversal of EMT. In the presence of PF-03084014, some proliferating cells may not be able to enter docetaxel-induced dormancy and thus maintain their sensitivity for cell death upon docetaxel treatment. This may be one of the causes for the enhanced antitumor efficacy in PF-03084014 plus docetaxel.
Conclusion
We provide evidence that PF-03084014 reduced the survival of CSC and non-CSC populations, thereby leading to synergistic antitumor efficacy with docetaxel. Mechanistic insights into the synergy revealed the pleotropic effects of PF-03084014, including the activation of apoptotic pathway, abrogation of chemoresistance, promotion of cell differentiation, and impairment of tumor cell self-renewal. This work provides the basis for a rational combination strategy using PF-03084014 to improve taxane-based therapies against breast cancer disease progression. Future work is warranted to identify the patient populations that can most benefit from therapeutic intervention with PF-03084014 and how to best implement this combination strategy in the clinical setting.
Acknowledgments
We acknowledge many Pfizer colleagues for their contributions to this work, including the PF-03084014 early development team and the Pfizer Worldwide Comparative Medicine La Jolla team. We also thank Dr. Xinjian Lin of the University of California San Diego Moores Cancer Center for proofreading the manuscript and providing valuable feedback.
Author Contributions
C.C.Z.: conception and design, data analysis and interpretation, manuscript writing, final approval of manuscript; Z.Y. and Q.Z.: conception and design, collection and/or assembly of data, data analysis and interpretation; D.D.F.: conception and design, manuscript writing; C.P.: collection and/or assembly of data, manuscript writing; Q.Z.: collection and/or assembly of data, data analysis and interpretation; E.C. and M.E.L.: collection and/or assembly of data; A.J.-B.: conception and design; J.G.C.: conception and design, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
All authors are either current or former employees of Pfizer. Q.Z., Z.Y., M.E.L., and J.G.C. also have ownership interests in Pfizer, and J.G.C. has held intellectual property rights with Pfizer.
References
- 1.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 2.Cheng L, Alexander R, Zhang S, et al. The clinical and therapeutic implications of cancer stem cell biology. Expert Rev Anticancer Ther. 2011;11:1131–1143. doi: 10.1586/era.11.82. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z, Li Y, Kong D, et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the Notch signaling pathway. Cancer Res. 2009;69:2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho S, Lu M, He X, et al. Notch1 regulates the expression of the multidrug resistance gene ABCC1/MRP1 in cultured cancer cells. Proc Natl Acad Sci USA. 2011;108:20778–20783. doi: 10.1073/pnas.1019452108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Y, Li D, Liu H, et al. Notch-1 signaling facilitates survivin expression in human non-small cell lung cancer cells. Cancer Biol Ther. 2011;11:14–21. doi: 10.4161/cbt.11.1.13730. [DOI] [PubMed] [Google Scholar]
- 6.Mungamuri SK, Yang X, Thor AD, et al. Survival signaling by Notch1: Mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer Res. 2006;66:4715–4724. doi: 10.1158/0008-5472.CAN-05-3830. [DOI] [PubMed] [Google Scholar]
- 7.Jiao Z, Wang W, Ma J, et al. Notch signaling mediates TNF-α-induced IL-6 production in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Clin Dev Immunol. 2012;2012:350209. doi: 10.1155/2012/350209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H, Holloway MP, Ma L, et al. Acetylation directs survivin nuclear localization to repress STAT3 oncogenic activity. J Biol Chem. 2010;285:36129–36137. doi: 10.1074/jbc.M110.152777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conze D, Weiss L, Regen PS, et al. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 2001;61:8851–8858. [PubMed] [Google Scholar]
- 10.Salgado R, Junius S, Benoy I, et al. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int J Cancer. 2003;103:642–646. doi: 10.1002/ijc.10833. [DOI] [PubMed] [Google Scholar]
- 11.Sansone P, Storci G, Tavolari S, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117:3988–4002. doi: 10.1172/JCI32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takebe N, Harris PJ, Warren RQ, et al. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8:97–106. doi: 10.1038/nrclinonc.2010.196. [DOI] [PubMed] [Google Scholar]
- 13.Wright M, Calcagno A, Salcido C, et al. Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008;10:R10. doi: 10.1186/bcr1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grudzien P, Lo S, Albain KS, et al. Inhibition of notch signaling reduces the stem-like population of breast cancer cells and prevents mammosphere formation. Anticancer Res. 2010;30:3853–3867. [PubMed] [Google Scholar]
- 15.Farnie G, Clarke RB. Mammary stem cells and breast cancer: Role of notch signalling. Stem Cell Rev. 2007;3:169–175. doi: 10.1007/s12015-007-0023-5. [DOI] [PubMed] [Google Scholar]
- 16.Li X, Lewis MT, Huang J, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 17.Phillips TM, McBride WH, Pajonk F. The response of CD24−/low/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 18.Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Honeth G, Bendahl PO, Ringnér M, et al. The CD44+/CD24− phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008;10:R53. doi: 10.1186/bcr2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheridan C, Kishimoto H, Fuchs RK, et al. CD44+/CD24− breast cancer cells exhibit enhanced invasive properties: An early step necessary for metastasis. Breast Cancer Res. 2006;8:R59. doi: 10.1186/bcr1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balic M, Lin H, Young L, et al. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin Cancer Res. 2006;12:5615–5621. doi: 10.1158/1078-0432.CCR-06-0169. [DOI] [PubMed] [Google Scholar]
- 22.Ricardo S, Vieira AF, Gerhard R, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: Expression distribution within intrinsic molecular subtype. J Clin Pathol. 2011;64:937–946. doi: 10.1136/jcp.2011.090456. [DOI] [PubMed] [Google Scholar]
- 23.Zhou B-BS, Zhang H, Damelin M, et al. Tumour-initiating cells: Challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov. 2009;8:806–823. doi: 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]
- 24.Hoey T, Yen WC, Axelrod F, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell. 2009;5:168–177. doi: 10.1016/j.stem.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 25.Kondratyev M, Kreso A, Hallett RM, et al. Gamma-secretase inhibitors target tumor-initiating cells in a mouse model of ERBB2 breast cancer. Oncogene. 2012;31:93–103. doi: 10.1038/onc.2011.212. [DOI] [PubMed] [Google Scholar]
- 26.Gilbert CA, Daou M-C, Moser RP, et al. γ-Secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res. 2010;70:6870–6879. doi: 10.1158/0008-5472.CAN-10-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meng RD, Shelton CC, Li YM, et al. γ-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. 2009;69:573–582. doi: 10.1158/0008-5472.CAN-08-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fillmore C, Kuperwasser C. Human breast cancer stem cell markers CD44 and CD24: Enriching for cells with functional properties in mice or in man? Breast Cancer Res. 2007;9:303. doi: 10.1186/bcr1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Croker AK, Goodale D, Chu J, et al. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med. 2009;13:2236–2252. doi: 10.1111/j.1582-4934.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei P, Walls M, Qiu M, et al. Evaluation of selective γ-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther. 2010;9:1618–1628. doi: 10.1158/1535-7163.MCT-10-0034. [DOI] [PubMed] [Google Scholar]
- 32.Zhang CC, Pavlicek A, Zhang Q, et al. Biomarker and pharmacological evaluation of the γ-secretase inhibitor PF-03084014 in breast cancer models. Clin Cancer Res. 2012;18:5008–5019. doi: 10.1158/1078-0432.CCR-12-1379. [DOI] [PubMed] [Google Scholar]
- 33.Hwang-Verslues WW, Kuo WH, Chang PH, et al. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers. PLoS One. 2009;4:e8377. doi: 10.1371/journal.pone.0008377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang C, Yan Z, Arango ME, et al. Advancing bioluminescence imaging technology for the evaluation of anticancer agents in the MDA-MB-435-HAL-Luc mammary fat pad and subrenal capsule tumor models. Clin Cancer Res. 2009;15:238–246. doi: 10.1158/1078-0432.CCR-08-0897. [DOI] [PubMed] [Google Scholar]
- 35.Rao SS, O’Neil J, Liberator CD, et al. Inhibition of NOTCH signaling by gamma secretase inhibitor engages the RB pathway and elicits cell cycle exit in T-cell acute lymphoblastic leukemia cells. Cancer Res. 2009;69:3060–3068. doi: 10.1158/0008-5472.CAN-08-4295. [DOI] [PubMed] [Google Scholar]
- 36.Luistro L, He W, Smith M, et al. Preclinical profile of a potent γ-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res. 2009;69:7672–7680. doi: 10.1158/0008-5472.CAN-09-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chapman G, Liu L, Sahlgren C, et al. High levels of Notch signaling down-regulate Numb and Numblike. J Cell Biol. 2006;175:535–540. doi: 10.1083/jcb.200602009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konishi J, Yi F, Chen X, et al. Notch3 cooperates with the EGFR pathway to modulate apoptosis through the induction of bim. Oncogene. 2010;29:589–596. doi: 10.1038/onc.2009.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee CW, Simin K, Liu Q, et al. A functional Notch-survivin gene signature in basal breast cancer. Breast Cancer Res. 2008;10:R97. doi: 10.1186/bcr2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crea F, Duhagon-Serrat MA, Hurt EM, et al. BMI1 silencing enhances docetaxel activity and impairs antioxidant response in prostate cancer. Int J Cancer. 2011;128:1946–1954. doi: 10.1002/ijc.25522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pannuti A, Foreman K, Rizzo P, et al. Targeting Notch to target cancer stem cells. Clin Cancer Res. 2010;16:3141–3152. doi: 10.1158/1078-0432.CCR-09-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Naumov GN, Townson JL, MacDonald IC, et al. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res Treat. 2003;82:199–206. doi: 10.1023/B:BREA.0000004377.12288.3c. [DOI] [PubMed] [Google Scholar]
- 43.Creighton CJ, Li X, Landis M, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. 2009;106:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ginestier C, Liu S, Diebel ME, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010;120:485–497. doi: 10.1172/JCI39397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi C, Tian R, Wang M, et al. CD44+ CD133+ population exhibits cancer stem cell-like characteristics in human gallbladder carcinoma. Cancer Biol Ther. 2010;10:1182–1190. doi: 10.4161/cbt.10.11.13664. [DOI] [PubMed] [Google Scholar]
- 46.Nguyen LV, Vanner R, Dirks P, et al. Cancer stem cells: An evolving concept. Nat Rev Cancer. 2012;12:133–143. doi: 10.1038/nrc3184. [DOI] [PubMed] [Google Scholar]
- 47.Schafer ZT, Brugge JS. IL-6 involvement in epithelial cancers. J Clin Invest. 2007;117:3660–3663. doi: 10.1172/JCI34237. [DOI] [PMC free article] [PubMed] [Google Scholar]