

Abstract
Afadin is a novel regulator of epithelial cell junctions assembly. However, its role in the formation of endothelial cell junctions and the regulation of vascular permeability remains obscure. We previously described protective effects of oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (OxPAPC) in the in vitro and in vivo models of lung endothelial barrier dysfunction and acute lung injury, which were mediated by Rac GTPase. This study examined a role of afadin in the OxPAPC-induced enhancement of interactions between adherens junctions and tight junctions as a novel mechanism of endothelial cell (EC) barrier preservation. OxPAPC induced Rap1-dependent afadin accumulation at the cell periphery and Rap1-dependent afadin interaction with adherens junction and tight junction proteins p120-catenin and ZO-1, respectively. Afadin knockdown using siRNA or ectopic expression of afadin mutant lacking Rap1 GTPase binding domain suppressed OxPAPC-induced EC barrier enhancement and abolished barrier protective effects of OxPAPC against thrombin-induced EC permeability. Afadin knockdown also abolished protective effects of OxPAPC against ventilator-induced lung injury in vivo. These results demonstrate for the first time a critical role of afadin in the regulation of vascular barrier function in vitro and in vivo via coordination of adherens junction - tight junction interactions.
Keywords: oxidized phospholipids, endothelium, permeability, cytoskeleton, cell contacts, lung injury
INTRODUCTION
Alterations in vascular permeability are defining feature of diverse processes including inflammation, ischemia/reperfusion and ventilator-induced lung injury, and may increase alveolar flooding, leukocyte infiltration and hypoxemia leading to increased morbidity and mortality (Groeneveld, 2002; Maniatis et al., 2008; Matthay et al., 2002; Matthay et al., 2003; Vandenbroucke et al., 2008). Molecular mechanisms underlying endothelial responses to barrier-protective stimuli involve actin rearrangement and formation of cortical F-actin rim, recruitment of junctional proteins to the cell periphery accompanied by enhancement of adherens junction areas, and increased interactions within junctional protein complexes (Dudek and Garcia, 2001; Mehta and Malik, 2006; Prasain and Stevens, 2009).
In vitro oxidation of 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphatidyl choline, a phospholipid present in the outer leaflet of cell membrane, generates a group of oxidized phospholipid products (OxPAPC) with potent barrier protective and anti-inflammatory effects verified in the in vitro and in vivo models of sepsis, acute lung injury and vascular endothelial barrier dysfunction (Birukov et al., 2004; Bochkov et al., 2002; Ma et al., 2004; Nonas et al., 2008). Barrier protective concentrations of OxPAPC enhanced basal pulmonary endothelial (EC) barrier properties and attenuated EC permeability induced by inflammatory agonists (Birukov et al., 2004; Birukova et al., 2007b; Birukova et al., 2007d). Effects of OxPAPC had been linked to the activation of small GTPases Rac and Cdc42, which mediated enhancement of peripheral actin cytoskeleton and promoted adherens junction (AJ) and focal adhesion (FA) assembly in pulmonary endothelium (Birukov et al., 2004; Birukova et al., 2007c; Birukova et al., 2007d). In addition to the recognized role of AJ in regulation of endothelial permeability, increasing evidence suggests an important role for communication between AJ and tight junction (TJ) structures in control of monolayer integrity (Dejana et al., 2009b; Mehta and Malik, 2006; Miyoshi and Takai, 2005).
Afadin is a scaffold protein with domain structure. It contains actin-binding domain and Ras-binding domains (Hoshino et al., 2005; Kooistra et al., 2007; Sato et al., 2006) and can be activated by small GTPase Rap1, which is shown to control cell adhesion dynamics (Boettner and Van Aelst, 2009). Upon activation afadin promotes assembly of cadherin-based AJ in cancer-derived epithelial cell lines (Fukuyama et al., 2005; Sato et al., 2006). In epithelial cells afadin also interacts with TJ protein ZO-1 and AJ protein α-catenin and regulates epithelial TJ assembly, while knockdown of afadin disrupts the recruitment of TJ proteins to cell-cell contact sites (Kooistra et al., 2007; Sato et al., 2006). Afadin activation is mediated by binding of Ras family Rap1 GTPase (Kooistra et al., 2007) also shown to become activated in OxPAPC-stimulated endothelium (Birukova et al., 2011). Whether afadin controls AJ – TJ interactions in endothelial cells and how these interactions may contribute to EC barrier enhancement induced by barrier protective agonists remains unknown.
This study explored the role of afadin in the OxPAPC-mediated adherens junction and tight junction assembly and interactions, and tested a novel afadin-dependent mechanism of lung vascular endothelial barrier preservation induced by oxidized phospholipids in EC cultures and the animal model of acute lung injury.
MATERIALS AND METHODS
Cell culture and reagents
Human pulmonary artery endothelial cells (HPAEC) were obtained from Lonza (Allendale, NJ). Cells were maintained according to the manufacturer’s recommendations and used for experiments at passages 5–8. Unless specified, biochemical reagents were obtained from Sigma (St. Louis, MO). Non oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (PAPC) was obtained from Avanti Polar Lipids (Alabaster, AL). Oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (OxPAPC) was obtained by exposure of dry lipid to air as previously (Birukov et al., 2004; Watson et al., 1997). The extent of oxidation was measured by positive ion electrospray mass spectrometry (ESI-MS) described elsewhere (Watson et al., 1997). OxPAPC dissolved in chloroform was stored at −70° C and used within 2 weeks after mass spectrometry testing. VE-cadherin, afadin and p120-catenin antibodies were purchased from BD Transduction Laboratories (San Diego, CA); ZO-1 antibody was purchased from Invitrogen (Carlsbad, CA). All reagents for immunofluorescence were purchased from Molecular Probes (Eugene, OR). Plasmids encoding afadin-wild type and afadin-Δ RBD (Ras/Rap1-binding domain) bearing GFP tags were kindly provided by Y. Takai (Kobe University, Kobe, Japan). EC were used for transient transfections according to protocol described previously (Birukov et al., 2004). After 48 hr of transfection cells were treated with either vehicle or OxPAPC were used for measurements of transendothelial electrical resistance (TER) reflecting EC permeability.
Protein depletion using siRNA approach
To deplete endogenous afadin or Rap1, Stealth™ Select 3 RNAi sets were used. Pre-designed human or mouse siRNAs of standard purity were ordered from Invitrogen (Carlsbad, CA). Transfection of EC with siRNA was performed as previously described (Singleton et al., 2009). Nonspecific, non-targeting siRNA was used as a control treatment. After 48 hr of 72 hr of transfection cells were used for experiments or harvested for western blot verification of specific protein depletion. For in vivo afadin depletion, polymer-based administration of non-specific or afadin specific siRNA conjugated with polycation polyethilenimine (PEI-22) was used according to previously developed protocol (Singleton et al., 2009). PEI-22 was kindly provided by A. Klibanov (MIT, Boston, MA). The optimal concentration of siRNA was determined in series of preliminary experiments. In brief, PEI22/siRNA ratio (N/P=10:1) used in these experiments represents ratio of PEI22 nitrogen to RNA phosphate. Required amounts of PEI22 were brought to 200 μl with 5% aqueous glucose and added to the equal volume of the glucose solution containing corresponding amounts of siRNA to reach 2 mg/kg, 4 mg/kg and 6 mg/kg siRNA dosage in vivo. The resulting polyplexes were incubated at room temperature for 10 min. Obtained PEI22-siRNA polyplexes (400 μl) were injected into jugular vein of 8- to 10-week-old C57BL/6 male mice, with weight of 20–25 grams (Jackson Laboratory, Bar Harbor, ME) under anesthesia. SiRNA at 4 mg/kg showed the most significant inhibition of the target gene after 72 hr of transfection, as determined by western blot analysis. Treated mice showed no signs of non-specific siRNA-induced inflammation. Nonspecific, non-targeting siRNA (Dharmacon, Lafayette, CO) was used as a control treatment for both in vitro and in vivo experiments.
Measurement of transendothelial electrical resistance
The cellular barrier properties were analyzed by measurements of transendothelial electrical resistance (TER) across confluent human pulmonary artery endothelial monolayers using an electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY) as previously described (Birukov et al., 2004). In the current studies we did not observe significant effects of non-specific RNA, specific siRNA or DNA constructs on cell viability and monolayer integrity, and initial testing of non-transfected, siRNA- and DNA-transfected EC monolayers did not reveal significant differences in basal TER levels.
Immunofluorescence
Endothelial monolayers plated on glass cover slips were subjected to double immunofluorescence staining with appropriate antibody, as described previously (Birukova et al., 2007d). Texas Red phalloidin was used to visualize F-actin. After immunostaining, slides were analyzed using a Nikon video imaging system (Nikon Instech Co., Tokyo, Japan). Images were processed with Image J software (National Institute of Health, Washington, USA) and Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA) software.
Co-immunoprecipitation, differential fractionation, and immunoblotting
Confluent HPAEC were stimulated with OxPAPC, and cytosolic and membrane fractions were separated using S-PEK kit (EMD Chemicals, Gibbstown, NJ). Co-immunoprecipitation studies and western blot analysis were performed using confluent HPAEC monolayers treated with vehicle or stimulated with OxPAPC, as described previously (Birukova et al., 2007d).
Mechanical ventilation protocol
All animal care and treatment procedures were approved by the University of Chicago Institutional Animal Care and Use Committee and were handled according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Adult male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were randomized to concurrently receive sterile saline solution or OxPAPC (1.5 mg/kg, intravenous injection in the external jugular vein) followed by 4 hours of mechanical ventilation with high tidal volume (30 ml/kg, HTV) ventilation as previously described (Nonas et al., 2008). At the end of experiment, measurements of cell count and protein concentration in bronchoalveolar lavage fluid (BAL) were performed as previously described (Birukova et al., 2007b; Nonas et al., 2008).
Statistical analysis
Results are presented as mean ± SD of four to eight independent experiments. Stimulated samples were compared to controls by unpaired Student’s t-test. For multiple-group comparisons, a one-way analysis of variance (ANOVA), followed by the post hoc Tukey test, were used. P<0.05 was considered statistically significant. Additional data analysis was performed using non-parametric Wilcoxon rank sum test for two groups and Kruskal-Wallis test with Dunn’s post test for multiple comparisons and confirmed significance of differences determined using parametric tests.
RESULTS
Barrier protective concentrations of OxPAPC recruit afadin to the cell-cell adhesion sites
Previous studies determined the range of OxPAPC concentrations (5–20 μg/ml) inducing potent and sustained barrier protective response in pulmonary EC (Birukov et al., 2004; Birukova et al., 2007b). Using OxPAPC at barrier-protective concentrations, we examined intracellular redistribution of afadin in the OxPAPC-treated pulmonary EC. OxPAPC treatment induced rearrangement of actin cytoskeleton with formation of peripheral actin rim accompanied by robust translocation of afadin to the cell periphery (Figure 1A).
Figure 1. Effect of OxPAPC on cytoskeletal remodeling, and Rap1-dependence of OxPAPC-induced afadin translocation.

A - Endothelial cells grown on glass coverslips were stimulated with OxPAPC (10 μg/ml) for 30 min followed by double immunofluorescence staining with Texas Red phalloidin to detect actin filaments and afadin antibody. Inset: control staining of cells without afadin antibody. Arrows depict peripheral actin rims (upper panel) and areas of afadin accumulation (lower panel). B - HPAEC were stimulated with OxPAPC (10 μg/ml) or vehicle for various periods of time. Membrane and cytosolic fractions were isolated as described in Methods section. The content of afadin, p120-catenin, and ZO-1 was determined by western blot analysis of cytosolic and membrane fractions with specific antibodies. Equal protein content in whole cell lysates was confirmed by detection of afadin and β-tubulin in control and OxPAPC-treated samples. Shown are representative results of three independent experiments.
Subcellular fractionation assays showed that OxPAPC induced time-dependent accumulation of afadin in the membrane fraction with concomitant decrease in the cytosolic fraction (Figure 1B). OxPAPC also caused accumulation of adherens junction protein p120-catenin and tight junction protein ZO-1 in the membrane fraction (Figure 1B) suggesting enhancement of adherens and tight junctions upon OxPAPC stimulation.
OxPAPC triggers Rap1 dependent targeting of afadin to the cell-cell adhesion sites
Our recent report showed OxPAPC-induced activation of Rap1 GTPase (Birukova et al., 2011). Depletion of Rap1 using siRNA approach abolished OxPAPC-induced peripheral translocation of afadin (Figure 2A). This finding suggest involvement of afadin in the Rap1-dependent mechanism of OxPAPC-mediated enhancement of AJ-TJ interactions in pulmonary EC. These data were further confirmed using biochemical approach. Rap1 knockdown suppressed OxPAPC-mediated afadin translocation and accumulation in the membrane fraction (Figure 2B).
Figure 2. Effect of Rap1 knockdown on OxPAPC-induced afadin translocation.

HPAEC were transfected with non-specific RNA or Rap1-specific siRNA duplexes followed by treatment with OxPAPC (10 μg/ml) or vehicle for 30 min. A - Endothelial cells grown on glass coverslips were subjected to immunofluorescence staining for afadin. Areas of afadin accumulation are marked by arrows. B - The membrane fraction was isolated, and afadin content was determined by western blotting with specific antibodies. Rap1 depletion was confirmed by western blot analysis of whole cell lysates from control (non-specific RNA) and siRap1-treated EC. Equal protein content in whole cell lysates was confirmed by detection of β-tubulin in control and OxPAPC-treated samples. Shown are representative results of three independent experiments.
OxPAPC promotes afadin colocalization and interaction with p120-catenin and ZO-1
To examine potential interaction between afadin and p120-catenin in OxPAPC-challenged pulmonary EC, we performed immunofluorescence studies followed by image analysis of afadin (green) and p120-catenin (red) labeled cells. Merged images showed a weak association between afadin and p120-catenin in quiescent EC, whereas OxPAPC treatment increased co-localization of afadin and p120-catenin at the cell-cell junctions (Figure 3A). OxPAPC treatment also increased colocalization of afadin (green) with tight junction protein ZO-1 (red) detected by double immunofluorescence staining and depicted in merged images (Figure 3B).
Figure 3. Effect of OxPAPC on adherens junction and tight junction remodeling.

Endothelial cells grown on glass coverslips were stimulated with OxPAPC (10 μg/ml, 30 min) followed by double immunofluorescence staining for: A - afadin (green) and p120-catein (red) and B - afadin (green) and ZO-1 (red). Merged images depict areas of afadin/ZO-1 co-localization which appear in yellow and marked by arrows. Shown are representative results of three independent experiments.
Effects of OxPAPC on afadin interactions with its potential partners p120-catenin and ZO-1 were further investigated in co-immunoprecipitation studies. Pulmonary EC were challenged with OxPAPC for various periods of time followed by precipitation of p120-catenin or ZO-1 under non-denaturing conditions. In agreement with immunofluorescence staining data in Figure 3, OxPAPC treatment caused time-dependent accumulation of VE-cadherin, afadin, and ZO-1 in p120-catenin immunoprecipitates (Figure 4A). OxPAPC also increased amounts of afadin and p120-catenin co-precipitated with ZO-1 (Figure 4B). Association of afadin, ZO-1, p120-catenin, and VE-cadherin was observed after 15 min and remained elevated for at least 60 min after OxPAPC stimulation. Treatment with barrier-disruptive agonist thrombin was used as a negative control.
Figure 4. OxPAPC induces afadin interaction with adherens junction and tight junction proteins.

A and B- Pulmonary EC were stimulated with OxPAPC (10 μg/ml), thrombin (0.1 U/ml, 10 min), or vehicle for various time periods followed by immunoprecipitation of: A - p120-catenin and B - or ZO-1. Afadin, VE-cadherin, p-120 catenin, and ZO-1 content in the immunoprecipitates was detected using specific antibodies. Equal protein loading was confirmed by membrane reprobing with p120-catenin (A) or ZO-1 (B) antibodies. Results are representative of four independent experiments. C - HPAEC transfected with Rap1-specific siRNA or non-specific RNA were treated with OxPAPC (10 μg/ml) or vehicle for 30 min followed by immunoprecipitation of p-120 catenin or ZO-1. Afadin content in the immunoprecipitates was determined using specific antibodies. Results are representative of three independent experiments.
Importantly, association of afadin with p120-catenin and ZO-1 was dependent on Rap1. Co-immunoprecipitation assays showed that siRNA-based Rap1 knockdown inhibited enhanced p120-catenin/afadin and ZO-1/afadin interactions in pulmonary EC stimulated with OxPAPC (Figure 4C).
Downregulation of afadin attenuates OxPAPC-mediated EC barrier preservation
To further delineate the role of afadin in the mediation of barrier-protective effects by oxidized phospholipids, we depleted endogenous afadin pool using EC treatment with afadin-specific siRNA. After 72 hrs of transfection with non-specific or afadin-specific siRNA duplexes, human pulmonary EC were pretreated with OxPAPC or vehicle, and transendothelial electrical resistance (TER) was monitored over the time. Depletion of afadin did not significantly affect the basal TER readings in EC monolayers (data not shown). Importantly, afadin knockdown significantly suppressed OxPAPC-induced enhancement of TER as compared to EC monolayers transfected with non-specific RNA (Figure 5A). Ectopic expression of wild type afadin did not affect barrier protective effects of OxPAPC, as compared to EC monlayers transfected with control vector (Figure 5B). In contrast, ectopic expression of afadin mutant lacking Ras/Rap1 GTPase binding domain (Afadin-ΔRBD), which therefore cannot be activated by Rap1, attenuated barrier protective effects of OxPAPC, as compared to EC monolayers transfected with empty vector (Figure 5B, lower panel).
Figure 5. Involvement of afadin in the OxPAPC-induced EC barrier enhancement.

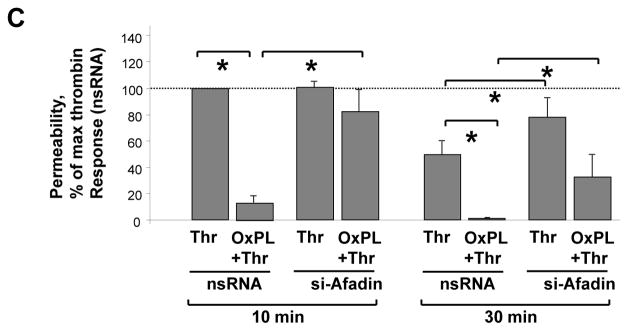
A - Pulmonary EC were transfected with afadin-specific siRNA or non-specific RNA. EC were stimulated with OxPAPC (10 μg/ml) or vehicle at the time indicated by arrow, and TER changes were monitored over 5 hours. Results are representative of five independent experiments. B - EC monolayers were subjected to transfection with full length GFP-tagged afadin (upper panel), or afadin-ΔRBD mutant bearing GFP tag (lower panel). Cells transfected with empty vector served as controls. Inset - expression of recombinant wild type afadin and afadin-ΔRBD mutant in pulmonary EC detected by western blot with GFP antibody. After 48 hrs of transfection, cells were stimulated with OxPAPC. Changes in endothelial permeability were monitored by measurements of TER. Results are representative of four independent experiments. C - HPAEC were transfected with afadin-specific siRNA or non-specific RNA duplexes. EC were pretreated with OxPAPC (10 μg/ml, 30 min) or vehicle prior to thrombin (0.1 U/ml) challenge, and TER was monitored over the time. Permeability increase caused by thrombin stimulation for 10 min (538+/−73 Ohm in comparison to 1164+/−269 Ohm for non-stimulated cells) of EC transfected with non-specific RNA was taken as 100%. Results are representative of six independent experiments.
A role of afadin in protective effects of OxPAPC against thrombin-induced EC barrier dysfunction was examined in the following studies. Control HPAEC transfected with non-specific RNA, or cells transfected with afadin-specific siRNA were pretreated with vehicle or OxPAPC followed by thrombin stimulation. EC barrier function was assessed by TER measurements. OxPAPC markedly attenuated thrombin-induced hyper-permeability in control cells transfected with non-specific RNA. In contrast, OxPAPC protective effects were suppressed in afadin-depleted EC monolayers (Figure 5C). Thrombin-induced maximal permeability increase during acute phase (0–20 min) was considered as 100%. TER values in thrombin-treated cells were 538+/−73 Ohm in comparison to 1164+/−269 Ohm for non-stimulated cells. These results delineate the important role of afadin as a mediator of barrier protective effects elicited by oxidized phospholipids.
Afadin knockdown attenuates OxPAPC-induced interactions between adherens junctions and tight junctions
The results of this study show that OxPAPC-induced barrier enhancement is associated with increased interactions between adherens junction and tight junction proteins. The role of afadin in increased p120-catenin/ZO-1 association induced by oxidized phospholipids was further investigated using siRNA-induced afadin knockdown approach. Afadin was depleted as described above, and OxPAPC-induced p120-catenin/ZO-1 peripheral co-localization was examined by double immunofluorescence staining of ZO-1 (green) and p120-catenin (red) (Figure 6). In contrast to control cells treated with non-specific RNA (Figure 6A), afadin knockdown attenuated junctional accumulation of ZO-1 and p120-catenin (Figure 6B) after OxPAPC stimulation. The merged images showed enhanced junctional p120-catenin/ZO-1 accumulation and colocalization in OxPAPC-stimulated cells treated with non-specific RNA. In contrast, EC transfection with si-Afadin attenuated OxPAPC-induced increase in the peripheral p120-catenin/ZO-1 immunoreactivity and disrupted continuous pattern of p120-catenin and ZO-1 peripheral localization (Figure 6, right panels).
Figure 6. Afadin depletion attenuates OxPAPC-mediated adherens junction and tight junction remodeling.

Pulmonary EC were transfected with: A - non-specific RNA, or B - afadin-specific siRNA. Cells were stimulated with OxPAPC (10 μg/ml, 30 min) followed by double immunofluorescence staining for ZO-1 (green) and p120-catein (red). Merged images depict areas of OxPACP-induced protein co-localization which appear in yellow and marked by arrows. This effect was attenuated in afadin-depleted EC. Shown are representative results of three independent experiments.
To further test a hypothesis about a role of afadin in OxPAPC-induced AJ-TJ interactions, we performed co-immunoprecipitation assays in control and afadin-depleted pulmonary EC using p120-catenin and ZO-1 antibodies. P120-catenin was immunoprecipitated under non-denaturing conditions from vehicle- or OxPAPC-stimulated cells and ZO-1 content was determined in precipitates (Figure 7A). In reverse experiments ZO-1 antibody was used for immunoprecipitation followed by determination of co-precipitated p120-catenin content (Figure 7B). Similar to non-transfected controls (Figure 4), OxPAPC treatment of EC transfected with non-specific RNA induced interactions between p120-catein and ZO-1 (Figure 7AB, left panels). This interaction was suppressed in afadin-depleted EC cultures (Figure 7AB, right panels). Collectively, these data suggest afadin-dependent mechanism of OXPAPC-induced association between p120-catenin and ZO-1 and link these effects with EC barrier enhancement.
Figure 7. Afadin knockdown attenuates OxPAPC-induced interactions between ZO-1 and p120-catenin.

Cells were transfected with non-specific RNA or afadin-specific siRNA followed by OxPAPC stimulation (10 μg/ml, 30 min). Co-immunoprecipitation assays using antibodies to A - p120-catenin or B - ZO-1 were performed, and afadin, p120-catenin, or ZO-1 content in the immunoprecipitates was detected using specific antibodies. Equal protein loading was confirmed by membrane reprobing with antibodies to p120-catenin (A) or ZO-1 (B). Lower panels depict siRNA-based depletion of endogenous afadin. Show are results representative of three independent experiments.
Afadin knockdown inhibits protective effects of OxPAPC in vivo
Our previous studies have demonstrated potent protective effect of OxPAPC in rat and murine models of ventilator induced lung injury (VILI) (Nonas et al., 2008). The role of afadin in the mediation of such protective effects was further investigated in the model of VILI. Endogenous afadin was depleted using siRNA approach according to the protocol established in our group (Singleton et al., 2009). Mice transfected with non-specific or afadin-specific siRNA (72 hrs) were subjected to mechanical ventilation at high tidal volume (HTV, 30 ml/kg, 4 hrs) with or without OxPAPC treatment. Afadin protein depletion was confirmed by western blot analysis of lung tissue (Figure 8A, inset). Bronchoalveolar lavage (BAL) fluid was collected at the end of experiment, and BAL protein and cellular content were analyzed to assess the severity of lung injury. In control mice transfected with non-specific RNA, HTV caused significant increase in BAL cell counts and protein content. These effects were suppressed by intravenous OxPAPC administration (Figure 8AB, left bar graphs). Knockdown of afadin did not significantly affect increases in BAL cell counts and protein concentration caused by HTV, but abolished inhibitory effects of OxPAPC on HTV-induced accumulation of protein and cell elements in BAL fluid (Figure 8AB, right bar graphs). These data indicate that afadin knockdown suppressed protective effects of OxPAPC against ventilator-induced lung injury. Taken together, these data strongly suggest involvement of afadin in the mechanisms of the OxPAPC-induced pulmonary barrier preservation.
Figure 8. Afadin depletion in vivo abolishes protective effects by OxPAPC in the murine model of ventilator induced lung injury.

Mice were transfected with non-specific or afadin-specific siRNA (72 hr) followed by high tidal volume mechanical ventilation (HTV; 30 ml/kg, 4 hr) with or without OxPAPC treatment (1.5 mg/kg, i/v). A - cell count, and B - protein concentration in BAL samples were analyzed. For each bar graph, cell counts or protein content determined in BAL from HTV-ventilated, nsRNA-treated animals were taken as 100%; n=4–6 per condition; *p<0.05. Inset depicts afadin depletion in lung samples assessed by western blot. Probing of the membrane with β-tubulin antibody served as a loading control.
DISCUSSION
Afadin has been described in epithelial cells as an integral protein interacting with transmembrane adhesive proteins nectins and connecting them to the actin cytoskeleton (Takai and Nakanishi, 2003). In epithelial cells, binding of afadin recruits epithelial E-cadherin to the nectin containing cell-cell contact sites and establishes new adherens junctions (Takai et al., 2008; Takai and Nakanishi, 2003). Activated afadin-Rap1 complex has been recently implicated in the mechanisms VEGF and sphingosine 1-phosphate induced endothelial cell migration and angiogenesis (Tawa et al., 2010). However, the role of afadin in regulation of endothelial permeability remains unknown. This study shows for the first time that afadin is critical for pulmonary endothelial monolayer barrier enhancement in vitro and plays essential role in protective effects of oxidized phospholipids against ventilator induced lung injury. We show the novel mechanism of endothelial barrier enhancement via afadin-mediated integration of adherens junction and tight junction complexes.
Prominent effects of OxPAPC on adherens junctions remodeling have been described previously (Birukov et al., 2004; Birukova et al., 2007d). OxPAPC promoted interaction between focal adhesion adaptor protein paxillin and adherens junction protein β-catenin, which was mediated by Rac GTPase and contributed to enhancement of endothelial barrier caused by OxPAPC (Birukova et al., 2007d). The present data show that OxPAPC not only enhances association of AJ components p120-catenin and VE-cadherin, but also promotes association between AJ and TJ proteins, and these interactions are mediated by afadin. The time course of OxPAPC-induced AJ-TJ associations demonstrates their stability even 1 hr after OxPAPC administration, which is consistent with observed sustained EC barrier protective response. In turn, knockdown of afadin abolishes peripheral colocalization and increased association between p120-catenin and ZO-1 induced by OxPAPC. In addition, afadin knockdown suppresses barrier enhancing OxPAPC effects in endothelial cell cultures and barrier protective OxPAPC effects against lung barrier dysfunction caused by high tidal volume mechanical ventilation in vivo. Permeability measurements show that afadin knockdown did not completely inhibit OxPAPC-induced EC barrier enhancement. This may be explained by incomplete depletion of afadin in these experiments. We also do not exclude other alternative signaling mechanisms including PI3-kinase, Src kinase, PKA, and Rac GTPase that may be triggered by OxPAPC (Birukova et al., 2007a; Singleton et al., 2009) and function in parallel to Rap-afadin signaling to enhance EC barrier.
The present results show that in endothelial cells stimulated with barrier protective agonists, activated afadin promotes interactions between AJ and TJ protein complexes. How may these events be possibly orchestrated? Our results suggest that OxPAPC-activated afadin enhances assembly of AJ and TJ complexes and promotes interaction between ZO-1 and p-120 catenin. Direct and reverse coimmunoprecipitation assays show presence of afadin, p120-catenin and ZO-1 in the same complex, which strongly suggests direct associations between AJ and TJ driven by afadin. Regulatory role of afadin in this complex formation is illustrated by experiments with afadin knockdown, which led to dissociation of ZO-1 from p120-catenin. These results strongly suggest that afadin-mediated linkage enforces the integrity of paracellular barrier.
It was noted in several studies that disassembly of adherens junctions leads to weakening of tight junctions and vice versa (Dejana et al., 2009a; Tsukita et al., 2009). The present data suggest that afadin may serve as a critical linker between AJ and TJ coordinating their assembly and enhancement upon stimulation of endothelial monolayers with barrier protective agonists.
Our recent report showed that Ras family small GTPase Rap1 becomes activated by OxPAPC and promotes assembly of adherens junction and tight junction protein complexes (Birukova et al., 2011). In addition, Rap1 stimulated interactions between p120-catenin and ZO-1 in pulmonary EC. However, the mechanism of such interactions remained unclear. This study tested afadin as a potential transducer of Rap1 signal to AJ and TJ in pulmonary endothelium. Afadin interacts with activated form of Rap1 (GTP-Rap1) via Ras/Rap1-association (RBD) domains, and this interaction converts afadin to activated form (Kooistra et al., 2007; Takai et al., 2008). The current results show that molecular inhibition of Rap1 prevented OxPAPC-induced accumulation of afadin and cell-cell junction areas, and afadin depletion abolished OxPAPC-induced formation of p120-catenin – afadin - ZO-1 complex. These results are consistent with our previous report demonstrating the role of Rap1 in assembly of adherens junction and tight junction protein complexes (Birukova et al., 2011). Ectopic expression of afadin with deleted RBD domains performed in this study caused attenuation of EC barrier enhancing response to OxPAPC (Figure 5B). Taken together, our results strongly support Rap1-dependent mechanism of OxPAPC-induced activation of afadin leading to enhancement of interactions between AJ and TJ proteins. However, we do not exclude other mechanisms which may play a role in afadin regulation. Further studies will address this important question. It is also important to note that afadin protein depletion did not significantly affect basal p120-catenin - ZO-1 interactions and intracellular localization detected in non-stimulated EC monolayers. These results suggest that Rap1-afadin signaling may be only critical for remodeling or de novo formation of cell junction complexes in response to various external stimuli, while basal maintenance of EC junctions is regulated by other mechanisms.
In addition to stimulation of afadin functions, Rap1 activation may trigger additional mechanisms of EC barrier enhancement, such as activation of Rac1 cascade via stimulation of Rac-specific guanine nucleotide exchange factors Tiam1 and Vav2 (Arthur et al., 2004; Birukova et al., 2007e) leading to cortical actin enhancement and Rac-mediated peripheral strengthening of AJ and FA (Birukova et al., 2007d). Previous study also showed alternative mechanism of Rac activation independently of Rap through Akt-mediated transactivation of sphingosine 1-phosphate receptor (Singleton et al., 2009). Thus, multiple mechanisms triggered by OxPAPC also may explain partial effect of afadin depletion on the OxPAPC-induced EC barrier protective response.
This study and other reports (Hopkins et al., 2003; Kooistra et al., 2005; Pannekoek et al., 2009; Wittchen et al., 2005) show that coordinated regulation of peripheral actin cytoskeleton, focal adhesive complexes and cell-cell junctions by Rac and Rap GTPases is essential for the maintenance of endothelial monolayer integrity and prevention of vascular leak. On the other hand, Rac- and Rap-induced formation of cell-cell junction protein complexes maintains local Rac and Rap activation in these regions. For example, Rac-PAK1-mediated phosphorylation of paxillin maintains elevated Rac activation in OxPAPC-treated cells, while paxillin mutation at PAK1-specific phosphorylation site Serine-273 reduces OxPAPC-induced Rac activity (Birukova et al., 2008). In turn, expression of afadin lacking Rap1 binding site decreases the pool of activated Rap1. As a consequence, localization of Rac-specific GEF Vav2 and Rac at the leading edge was hardly detected in cells overexpressing ΔRBD afadin mutant (Tawa et al., 2010). These reports illustrate positive feedback mechanisms of Rac and Rap1 GTPase regulation by cell junction complexes. Such mechanisms appear to be a general biological principle of cell-cell and cell-substrate communications aimed at the maintenance of cellular monolayer integrity.
In summary, we propose a hypothetical scheme describing a role of afadin in regulation of cell junctions and endothelial permeability. Barrier protective agents such as OxPAPC activate Rap1, which stimulates Rac GTPase via activation of Tiam1 and Vav2 leading to cytoskeletal remodeling, enhancement of cortical cytoskeleton, peripheral accumulation and assembly of adherens junctions and focal adhesions. In parallel, Rap1 activates its effector afadin, which also promotes AJ and TJ assembly and coordinates AJ-TJ interactions. Altogether, Rap1-Rac and Rap1-afadin mechanisms enhance endothelial monolayer barrier and preserve vascular barrier integrity in pathological settings of acute lung injury. Further investigations are required to explore other afadin functions as a regulator of endothelial permeability and cell junction-controlled small GTPase activity.
Acknowledgments
Supported by National Heart, Lung, and Blood Institutes grants HL87823, HL76259, and HL58064 for KGB; HL89257 and the American Heart Association Midwest Affiliate Grant-in-Aid for AAB
The authors would like to thank Dr. Yoshimi Takai (Kobe University, Japan) for providing afadin constructs and Drs. Alexander Klibanov and Jennifer Fortune (Massachusetts Institute of Technology, Cambridge, MA, USA) for providing PEI carriers for in vivo transfections.
References
- Arthur WT, Quilliam LA, Cooper JA. Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. The Journal of cell biology. 2004;167(1):111–122. doi: 10.1083/jcb.200404068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukov KG, Bochkov VN, Birukova AA, Kawkitinarong K, Rios A, Leitner A, Verin AD, Bokoch GM, Leitinger N, Garcia JG. Epoxycyclopentenone-containing oxidized phospholipids restore endothelial barrier function via Cdc42 and Rac. Circ Res. 2004;95(9):892–901. doi: 10.1161/01.RES.0000147310.18962.06. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Alekseeva E, Cokic I, Turner CE, Birukov KG. Cross talk between paxillin and Rac is critical for mediation of barrier-protective effects by oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol. 2008;295(4):L593–602. doi: 10.1152/ajplung.90257.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Chatchavalvanich S, Oskolkova O, Bochkov VN, Birukov KG. Signaling pathways involved in OxPAPC-induced pulmonary endothelial barrier protection. Microvasc Res. 2007a;73(3):173–181. doi: 10.1016/j.mvr.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Fu P, Chatchavalvanich S, Burdette D, Oskolkova O, Bochkov VN, Birukov KG. Polar head groups are important for barrier protective effects of oxidized phospholipids on pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol. 2007b;292(4):L924–935. doi: 10.1152/ajplung.00395.2006. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Malyukova I, Mikaelyan A, Fu P, Birukov KG. Tiam1 and betaPIX mediate Rac-dependent endothelial barrier protective response to oxidized phospholipids. Journal of cellular physiology. 2007c;211(3):608–617. doi: 10.1002/jcp.20966. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Malyukova I, Poroyko V, Birukov KG. Paxillin - {beta}-catenin interactions are involved in Rac/Cdc42-mediated endothelial barrier-protective response to oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol. 2007d;293(1):L199–211. doi: 10.1152/ajplung.00020.2007. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Zagranichnaya T, Alekseeva E, Fu P, Chen W, Jacobson JR, Birukov KG. Prostaglandins PGE2 and PGI2 promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Experimental cell research. 2007e;313(11):2504–2520. doi: 10.1016/j.yexcr.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Zebda N, Fu P, Poroyko V, Cokic I, Birukov KG. Association between adherens junctions and tight junctions via Rap1 promotes barrier protective effects of oxidized phospholipids. Journal of cellular physiology. 2011;226(8):2052–2062. doi: 10.1002/jcp.22543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkov VN, Kadl A, Huber J, Gruber F, Binder BR, Leitinger N. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature. 2002;419(6902):77–81. doi: 10.1038/nature01023. [DOI] [PubMed] [Google Scholar]
- Boettner B, Van Aelst L. Control of cell adhesion dynamics by Rap1 signaling. Curr Opin Cell Biol. 2009;21(5):684–693. doi: 10.1016/j.ceb.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E, Orsenigo F, Molendini C, Baluk P, McDonald DM. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009a;335(1):17–25. doi: 10.1007/s00441-008-0694-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. 2009b;16(2):209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91(4):1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- Fukuyama T, Ogita H, Kawakatsu T, Fukuhara T, Yamada T, Sato T, Shimizu K, Nakamura T, Matsuda M, Takai Y. Involvement of the c-Src-Crk-C3G-Rap1 signaling in the nectin-induced activation of Cdc42 and formation of adherens junctions. The Journal of biological chemistry. 2005;280(1):815–825. doi: 10.1074/jbc.M411099200. [DOI] [PubMed] [Google Scholar]
- Groeneveld AB. Vascular pharmacology of acute lung injury and acute respiratory distress syndrome. Vascul Pharmacol. 2002;39(4–5):247–256. doi: 10.1016/s1537-1891(03)00013-2. [DOI] [PubMed] [Google Scholar]
- Hopkins AM, Walsh SV, Verkade P, Boquet P, Nusrat A. Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. Journal of cell science. 2003;116(Pt 4):725–742. doi: 10.1242/jcs.00300. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Sakisaka T, Baba T, Yamada T, Kimura T, Takai Y. Regulation of E-cadherin endocytosis by nectin through afadin, Rap1, and p120ctn. The Journal of biological chemistry. 2005;280(25):24095–24103. doi: 10.1074/jbc.M414447200. [DOI] [PubMed] [Google Scholar]
- Kooistra MR, Corada M, Dejana E, Bos JL. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005;579(22):4966–4972. doi: 10.1016/j.febslet.2005.07.080. [DOI] [PubMed] [Google Scholar]
- Kooistra MR, Dube N, Bos JL. Rap1: a key regulator in cell-cell junction formation. Journal of cell science. 2007;120(Pt 1):17–22. doi: 10.1242/jcs.03306. [DOI] [PubMed] [Google Scholar]
- Ma Z, Li J, Yang L, Mu Y, Xie W, Pitt B, Li S. Inhibition of LPS- and CpG DNA-induced TNF-alpha response by oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L808–816. doi: 10.1152/ajplung.00220.2003. [DOI] [PubMed] [Google Scholar]
- Maniatis NA, Kotanidou A, Catravas JD, Orfanos SE. Endothelial pathomechanisms in acute lung injury. Vascul Pharmacol. 2008;49(4–6):119–133. doi: 10.1016/j.vph.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay MA, Bhattacharya S, Gaver D, Ware LB, Lim LH, Syrkina O, Eyal F, Hubmayr R. Ventilator-induced lung injury: in vivo and in vitro mechanisms. Am J Physiol Lung Cell Mol Physiol. 2002;283(4):L678–682. doi: 10.1152/ajplung.00154.2002. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Zimmerman GA, Esmon C, Bhattacharya J, Coller B, Doerschuk CM, Floros J, Gimbrone MA, Jr, Hoffman E, Hubmayr RD, Leppert M, Matalon S, Munford R, Parsons P, Slutsky AS, Tracey KJ, Ward P, Gail DB, Harabin AL. Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med. 2003;167(7):1027–1035. doi: 10.1164/rccm.200208-966WS. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- Miyoshi J, Takai Y. Molecular perspective on tight-junction assembly and epithelial polarity. Adv Drug Deliv Rev. 2005;57(6):815–855. doi: 10.1016/j.addr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Nonas S, Birukova AA, Fu P, Xing J, Chatchavalvanich S, Bochkov VN, Leitinger N, Garcia JG, Birukov KG. Oxidized phospholipids reduce ventilator-induced vascular leak and inflammation in vivo. Crit Care. 2008;12(1):R27. doi: 10.1186/cc6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannekoek WJ, Kooistra MR, Zwartkruis FJ, Bos JL. Cell-cell junction formation: The role of Rap1 and Rap1 guanine nucleotide exchange factors. Biochim Biophys Acta. 2009;1788(4):790–796. doi: 10.1016/j.bbamem.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Prasain N, Stevens T. The actin cytoskeleton in endothelial cell phenotypes. Microvasc Res. 2009;77(1):53–63. doi: 10.1016/j.mvr.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Fujita N, Yamada A, Ooshio T, Okamoto R, Irie K, Takai Y. Regulation of the assembly and adhesion activity of E-cadherin by nectin and afadin for the formation of adherens junctions in Madin-Darby canine kidney cells. The Journal of biological chemistry. 2006;281(8):5288–5299. doi: 10.1074/jbc.M510070200. [DOI] [PubMed] [Google Scholar]
- Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, Klibanov AM, Garcia JG, Birukov KG. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res. 2009;104(8):978–986. doi: 10.1161/CIRCRESAHA.108.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y, Ikeda W, Ogita H, Rikitake Y. The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu Rev Cell Dev Biol. 2008;24:309–342. doi: 10.1146/annurev.cellbio.24.110707.175339. [DOI] [PubMed] [Google Scholar]
- Takai Y, Nakanishi H. Nectin and afadin: novel organizers of intercellular junctions. Journal of cell science. 2003;116(Pt 1):17–27. doi: 10.1242/jcs.00167. [DOI] [PubMed] [Google Scholar]
- Tawa H, Rikitake Y, Takahashi M, Amano H, Miyata M, Satomi-Kobayashi S, Kinugasa M, Nagamatsu Y, Majima T, Ogita H, Miyoshi J, Hirata K, Takai Y. Role of afadin in vascular endothelial growth factor- and sphingosine 1-phosphate-induced angiogenesis. Circ Res. 2010;106(11):1731–1742. doi: 10.1161/CIRCRESAHA.110.216747. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Katsuno T, Yamazaki Y, Umeda K, Tamura A, Tsukita S. Roles of ZO-1 and ZO-2 in establishment of the belt-like adherens and tight junctions with paracellular permselective barrier function. Ann N Y Acad Sci. 2009;1165:44–52. doi: 10.1111/j.1749-6632.2009.04056.x. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. 2008;1123:134–145. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witztum JL, Palinski W, Schwenke D, Salomon RG, Sha W, Subbanagounder G, Fogelman AM, Berliner JA. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. The Journal of biological chemistry. 1997;272(21):13597–13607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- Wittchen ES, Worthylake RA, Kelly P, Casey PJ, Quilliam LA, Burridge K. Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. The Journal of biological chemistry. 2005;280(12):11675–11682. doi: 10.1074/jbc.M412595200. [DOI] [PubMed] [Google Scholar]