

Abstract
Few technologies are more widespread in modern biological laboratories than imaging. Recent advances in optical technologies and instrumentation are providing hitherto unimagined capabilities. Almost all these advances have required the development of software to enable the acquisition, management, analysis, and visualization of the imaging data. We review each computational step that biologists encounter when dealing with digital images, the challenges in that domain, and the overall status of available software for bioimage informatics, focusing on open source options.
Introduction
The last twenty years have seen great advances in optical imaging with the ability to monitor biological phenomena with unprecedented resolution, specificity, dimensionality, complexity, and scale, all while maintaining viability and biological relevance. These novel imaging modalities, which are increasingly multi-parametric, rely heavily on computational approaches. In fact, in many cases the computational technology is just as important as the optics; not just for the digital capture that all systems now use, but in many cases also for visualizing and properly interpreting the data. In the quest for breakthroughs of biological significance, biologists are often confronted with the challenge of processing digital data of increasing complexity and richness which demands an informatics infrastructure with tools to collect, store, manipulate, analyze, interpret, and visualize vast amounts of imaging data in a reproducible way with the flexibility to refine aspects of their experimental and imaging techniques in a tight iterative loop
Despite this great need, the bioimage informatics field is still a fairly nascent community compared to the more established hardware development side of the optical microscopy community. But with the increased mainstream adoption of advanced optical imaging approaches by biologists and the commitment by funding agencies to prioritize bioimage informatics, there has been a great increase in the number of bioimage informatics 1tools over the last five years.
In this article, representative members of the bioimage informatics community have collaborated to review each computational step that biologists encounter when dealing with digital images, the challenges in that domain, and the overall status of available software for bioimage informatics, focusing on open source options. Our goal is to provide an overview of how open source imaging software can be utilized to provide an end-to-end laboratory solution from acquisition and data storage, to data analysis and data mining (Table 1).
Table 1.
Summary of open-source software discussed in this review
| Software Name | Primary Function | Website |
|---|---|---|
| µManager | Microscope Acquisition | http://www.micro-manager.org |
| ScanImage | Microscope Acquisition | http://www.scanimage.org |
| OMERO | Image Database | http://www.openmicroscopy.org |
| Bisque | Image Database | http://www.bioimage.ucsb.org/bisque |
| OMERO.searcher | Image Content Search | http://murphylab.web.cmu.edu/software/searcher |
| Bio-Formats | Image Format Conversion | http://www.openmicroscopy.org |
| ImageJ | Image Analysis | http://www.imagej.nih.gov |
| Fiji | Image Analysis | http://www.fiji.sc |
| BioImageXD | Image Analysis | http://www.bioimagexd.net |
| Icy | Image Analysis | http://icy.bioimageanalysis.org |
| CellProfiler | Image Analysis | http://www.cellprofiler.org |
| Vaa3D | Visualization and Image Analysis | http://www.vaa3d.org |
| FarSight | Visualization | http://www.farsight-toolkit.org/ |
| Visualization Tool Kit (VTK) | Bioimaging library | http://www.vtk.org |
| Insight Tool Kit (ITK) | Bioimaging library | http://www.itk.org |
| OpenCV | Bioimaging library | http://opencv.willowgarage.com/wiki/ |
| WND-CHARM | Machine learning | http://code.google.com/p/wnd-charm/ |
| PSLID | Machine learning | http://pslid.org |
| Ilastik | Machine learning | http://www.ilastik.org/ |
| CellProfiler Analyst | Machine learning and data analysis | http://www.cellprofiler.org |
| PatternUnmixer | Machine learning | http://murphylab.web.cmu.edu/software |
| CellOrganizer | Machine learning, modeling, visualization | http://CellOrganizer.org |
| KNIME | Workflow system | http://www.knime.org/ |
Image acquisition
Biological laboratories usually acquire images by measuring photon flux in parallel (using a camera) or sequentially (using a point detector and equipment that scans the area of interest). Although capturing an image from a camera into the computer is straightforward, in most cases image acquisition needs to be tightly synchronized with other computer-controllable equipment such as shutters, filter wheels, XY stages, Z-axis focus drives, and autofocus mechanisms (implemented in software or hardware). This automation is necessary in order to gather the appropriate information from a sample or to allow the unattended acquisition of large numbers of images in time-lapse series, z-stacks, multiple spatial locations in a large sample, or multiple samples in a large-scale experiment. Image acquisition software is therefore needed to communicate with these various components and coordinate their actions such that the hardware functions as quickly and flawlessly as possible, while permitting the researcher to easily design and execute the desired sequence of acquisition events (Fig. 1).
Figure 1. Image Acquisition.
Legend: Image acquisition spans a range of complexity and variation.
Confocal microscope systems that require computer control to acquire an image are almost always bundled with control software whereas wide-field microscope systems usually aren’t. Instead, several different independent software packages are available for wide-field microscope control and automation. But not all software packages support all microscopy-related hardware. Because the cost to write code to support hardware is high, the choice of which hardware a given software package will support is typically driven by commercial interests, and support for a hardware component can rarely be added by third parties. Hence, software compatibility with hardware is an essential consideration when planning a new microscope system.
Several commercial software packages combine image acquisition and analysis. These include Metamorph (Molecular Devices), SlideBook (3i), Image-Pro (MediaCybernetics) and Volocity (Perkin-Elmer). These software packages were often started in individual research labs and only later were commercialized. In addition, each of the major microscope companies have their own software packages, such as AxioVision (Zeiss), NIS-Elements (Nikon) and cellSens (Olympus).
The obvious advantage of commercial image acquisition packages is that they provide a turnkey solution to all “standard” image analysis strategies (snapping individual images, taking time-lapse series, three-dimensional (3-D) stacks at multiple XY positions, etc.) and when purchased as part of a system there is less danger they won’t be compatible with the system’s configuration. It is virtually impossible, however, for individual researchers to substantially extend any of these software packages or make substantial custom hardware changes to the imaging system. This reduces the rate at which novel imaging techniques can be transferred to laboratories outside that of the inventor.
Researchers that have non-standard or frequently changing needs and equipment must often write their own code. Software development is facilitated by toolkit environments such as LabVIEW (National Instruments) and MATLAB (Mathworks), which provide interfaces to a subset of available equipment and can be used to create a graphical user interface. Developing novel imaging technologies necessitates writing instrument control code, as the needs for these novel techniques simply could not be anticipated in existing software packages. Examples of novel developments enabled by software written in research laboratories include structured illumination microscopy2, super resolution microscopy3–5 and Bessel beam microscopy6. Although these toolkit environments provide high flexibility and are well suited for tools intended for the group that wrote them, they are less appropriate for distributing these tools to others primarily because the infrastructure back-ends are costly and the distribution channels are not sufficiently developed.
Two open-source software projects, µManager and ScanImage, whose development is driven by researchers, are intended to provide tools with more flexibility than commercial tools and greater ease of use than the toolkit environments. µManager mainly targets camera-based imaging, although it is also used with scanning systems 7 (http://micro-manager.org). It includes an easy-to-use interface that runs as an ImageJ plugin and enables researchers to design and execute common microscopy functions as well as customized image acquisition routines. The solutions can be easily distributed as scripts or plugins. µManager’s hardware abstraction layer can also be used without its ImageJ user interface in environments such as Icy, MATLAB, LabVIEW and Python, further facilitating development and transfer of software for new imaging approaches. The software framework enables any entity, academic or commercial, to write and contribute their own device adapters for hardware components. µManager provides full control of the components of the light microscope such as cameras, stages, and filter wheels. The program can be used to collect multichannel data over space and time, such as tracking fluorescently tagged cell fusion events in live cells in a multiwell plate overnight8.
Another open source package, ScanImage, provides a software framework to control laser scanning microscopes and is used extensively for two-photon excitation microscopy 9 (http://scanimage.org). It implements most standard modes of image acquisition and basic automation and supports continuous image acquisition synchronized to behavioral or physiological data, which is particularly useful for imaging in intact animals. The software framework is object-oriented and event-driven to promote extensibility, online analysis, and plugin development. ScanImage complements µManager in that it can control laser-scanning microscopes such as home built confocal systems and allow for complex recordings where high signal to noise is needed such as tracking axon signaling in neuron cultures.
These projects exemplify certain benefits that can arise organically from open source bioimaging software. Open source software in this domain aids the scientific community by facilitating the rapid dissemination of novel optical techniques from laboratories focused on instrumentation development, without requiring them to run in parallel major software efforts. Furthermore, in the case of µManager, it now supports so many different manufacturers’ cameras that its interface has essentially become a de facto standard for controlling scientific grade cameras, providing benefits to the scientific community and to commercial vendors in this arena.
Image storage
The vast increase in data volume and the complexity of bioimaging experiments and acquisition protocols has rapidly made a paper lab notebook an unsuitable solution for keeping track of information about imaging experiments. In fact, finding, viewing, or analyzing the data with everyday computer software is typically infeasible. Many laboratories routinely generate tens to hundreds of gigabytes of image data per day. New sophisticated automation techniques 10 promise to increase and accelerate this trend. “Enterprise-level” data generation must therefore be matched with software applications that can properly manage, view, share, process and analyze this data (Fig. 2).
Figure 2. Image Storage.
Legend: Options for image storage span a range of complexity and variation.
In the last ten years a number of groups have developed applications for managing large collections of scientific images. These are often referred to as ‘image databases’. These image databases provide integrated platforms to organize, search, process, analyze, share and visualize data associated with biological experiments—including both images and metadata (i.e., information about the images). Such systems for image storage and retrieval are quite dependent on suitable annotation of images, both in terms of describing the experimental details that created the image as well as the automatic or human interpretation of the content of the image (see Box 2: Image Annotation). The usefulness of image annotation is greatly enhanced by ontologies that formalize the names and relationships among image metadata (Box 3: Ontologies).
Box 2: Image annotation.
In order to be analyzed, retrieved, visualized, and/or shared, biological images require annotation, the process of associating images with metadata (i.e., information about the images, including information about how the images and the samples therein were created, as well as information about the content of the image itself). Whether using formal ontologies (see Box 3: Ontologies) or informal means of annotation, describing images in a systematic and machine-computable manner is often critical for making full use of them, either due to the scale of the bioimaging experiment or the reuse of image data for a new purpose. Unlike some other readouts, biological images can contain a vast amount of information that is often not fully extracted in the initial analysis.
Despite its recognized value, image metadata capture and annotation has not yet become widespread for bioimaging experiments 57, though efforts are underway in certain areas. A major challenge is that capturing all types of metadata for a bioimaging experiment, whether by manual or automated means, is currently time consuming and impractical. This is in part because the inherent amount of annotation required for all conceivable downstream uses of imaging data is overwhelming, causing most researchers to record just the information necessary for their own purposes. The challenge also lies in the lack of user-friendly annotation tools to entice biologists to regularly record metadata.
Some tools are in development to ease the annotation process for researchers. For image acquisition, microscopes usually automatically store information about image acquisition in the header of the resulting image files. Unfortunately, this metadata and even the pixel data itself is often stored in a proprietary form making it very difficult for other software packages to read and parse it. The Open Microscopy Environment (http://www.openmicroscopy.org) has two solutions to help address the challenge of proprietary image formats and allow for easy and robust harvesting of acquisition metadata. The Bio-Formats project 57 is a library used by many open-source and commercial imaging software tools that allows for the full parsing of more than 120 proprietary image formats and the accurate conversion of the proprietary metadata to the OME-XML data model. The OME-TIFF project is a container format for the OME-XML data model and serves as the main export format of Bio-Formats. OME-TIFF is therefore a practical choice for software tools that wish to record their metadata in an open image format. The OMERO data system of the Open Microscopy Environment also offers image annotation with current support for text and graphical annotation of regions of interest for images stored in the database. The CCDB’s Web Image Browser tool is in development to enable manual annotation of the organism type and anatomical region where the image or volume is found (http://openccdb.org/software/index.shtm#wib). To encourage biologists to record experimental details leading to the capture of images, especially for complex experiments, ProtocolNavigator captures protocols as a visual map (http://vizbi.org/Posters/2011/D06).
Although annotation of images must often be done through manual curation, approaches for automated annotation of image content are in development (see “Machine learning” section) and are especially needed given the sheer number and volume of images that are now routinely produced. These systems, whether providing automatic harvesting of metadata at acquisition or manual annotation post acquisition, are in the early stages of implementation and adoption. As the field increases its demand for quantitative analysis and robust curation and sharing of the image data, the need for full ontologies and annotations will increase. Annotation is needed at every stage of the biological imaging workflow, but systems covering both manual and automated annotation of the full workflow still need to be developed and implemented.
Box 3: Ontologies.
Annotation of images, in order to be useful for image retrieval and analysis across experiments and laboratories, is greatly enhanced through the use of ontologies. Ontologies are formal expressions of human knowledge about a domain in machine readable form 58. Ontologies enable the consistent description of image metadata (e.g., the type of microscopy or stains used, the cell or tissue type imaged). They can also enable consistent description of visual annotations by defining a set of classes (e.g., cell, organelle) and the relationships between them (e.g., an organelle is part of a cell).
There are a number of valuable benefits to image annotation with ontologies. First, at the most basic level, ontologies provide an effective and flexible means to apply controlled vocabularies to annotation. This allows multiple terms (synonyms, plurals and lexical variants) to be resolved as describing the same item and disambiguates multiple items being described using the same single term (e.g., the nucleus of cell, the nucleus of the brain, and the nucleus of an atom). Second, ontologies are computable. A well-constructed ontology names each class via a unique identifier, ideally structured in the form of a uniform resource identifier (URI), which serves as an easily computable “handle”. Third, when constructed using formal logical languages like OWL (Web Ontology Language), ontologies can perform categorization through similar reasoning processes to a human. For example, ontologies enable the generation of new hierarchies based on rules and axioms (e.g., a Purkinje cell is a type of GABAergic neuron because it is both a type of neuron and uses GABA as a neurotransmitter). By expressing image annotation in machine computable form as a formal ontology, human knowledge can be brought to bear on effective search and interpretation of image data, especially across multiple disciplines, scales, and modalities.
Ontologies and ontology-based services that can be built into databases and tools exist for certain metadata relevant to bioimaging experiments and are in development for others, through efforts such as the Open Biological Ontologies project, the Gene Ontology and the National Center for Biomedical Ontologies Bioportal and the Neuroscience Information Framework (http://neuinfo.org). These ontologies cover major biological entities, such as cell components (Gene Ontology), chemicals of biological interest (Chebi), anatomical entities (Foundational Model of Anatomy, NIFSTD) and cell types (Cell Ontology). Other ontologies cover entities describing experimental acquisition (OME-XML), experimental techniques and protocols (Ontology of Biomedical Investigation) and data elements (Information Artifact Ontology). However, the process of formal ontology construction is slow and may not keep pace with rapidly evolving imaging technologies. Systems have also been proposed by which highly granular vocabulary terms can be developed and evolved locally into an ontology by the experimentalist, with guidance provided that encourages concordance with existing ontologies (59 and http://xpdb.nist.gov/bioroot/bioroot.pl).
Two examples of open source bioimaging database projects are the Open Microscopy Environment’s (OME) Remote Objects (OMERO) platform 11 (http://www.openmicroscopy.org) and the Bio-Image Semantic Query User Environment (BISQUE) project 12 (http://www.bioimage.ucsb.edu/BISQUE). Both are web-based open source projects that allow users to extend both metadata and workflow models for their individual applications. Both also invoke the idea of “remote access”-- a scientist can work with his or her data using a standard Internet connection. This type of technology is referred to as a “client-server application” where a server application holds and manages the data and delivers a view of the images, metadata, annotations, and analytic output in a client such as a web browser. This architecture provides a framework where researchers use tools hosted on a server to process and analyze large image data collections, taking advantage of the increasing power of computational resources in clusters and in the cloud.
Image databases can be searched using annotations, and relevant images can also be found by content-based image retrieval, also known as query-by-image-content. This involves searching for images that are similar to a query by some measure of distance using features or model parameters (these are discussed below under Machine Learning). As an example, the open source software OMERO.searcher provides this capability, building on the Feedback Adaptive Loop for Content-Based Retrieval (FALCON) algorithm 13 used in the Protein Subcellular Location Image Database (PSLID) (http://pslid.org/).
BISQUE and OMERO are just two examples of a growing ecology of software systems to help scientists manage image collections. Commercial solutions like the Columbus Image Data Management system (PerkinElmer) based on OMERO, and the PCI Image Database (Quartz), are also available. The BISQUE platform is integrated with the iPlant cyberinfrastructure to provide a scalable image management and analysis platform for plant biologists 14. Given the vast variety of applications and contexts for bioimaging experiments, it is nearly impossible to create a universal solution for all scientists’ needs. Rather, scientific image databases must be flexible and able to integrate applications and functionality demanded by the scientific application even if unanticipated by the database developers. This is why BISQUE and OMERO provide adaptable frameworks and not complete, monolithic applications. BISQUE and OMERO leverage the latest enterprise database and file server technology and couple that with the unique requirements of the imaging community. Together they represent a class of imaging database solutions that can be deployed by an individual lab, a network of labs, or publicly available repositories (see Box 1: Public image repositories).
Box 1: Public image repositories.
Public bioimage data repositories have been available for a number of years, and the number is growing 56. Some repositories are geared towards education and outreach, such as the The Cell: An Image Library (http://www.cellimagelibrary.org) and The Cell Centered Database (CCDB) (http://ccdb.ucsd.edu), which will be merging in 2012. Others gather sets of images that accompany published papers, such as the Journal of Cell Biology (JCB) DataViewer (http://jcb-dataviewer.rupress.org), which provides full access to original multi-dimensional image data associated with articles published in JCB. Still others are more focused on providing research resources, often focused on particular domains. These include the Human Protein Atlas (http://proteinatlas.org), the Allen Brain Atlas (http://www.brain-map.org), the e-Mouse Atlas Project (http://www.emouseatlas.org), the Protein Subcellular Location Image Database (http://pslid.org), the Worm Atlas (http://www.wormatlas.org), Drosophila Image Repositories (http://flybase.org) and LOCATE (http://locate.imb.uq.edu.au). The methods for searching, analyzing and distributing images from these databases are still developing. Using standard methods of tracking access and downloads will help identify which data are most valuable for the community and which applications and functionality are most scientifically useful.
This flexibility stems in part from the use of open source development practices that allow the community to review, contribute, and participate in the development project and also facilitates connections to other open-source bioimaging software. For example, both OMERO and BISQUE can work with CellProfiler and ImageJ, and implementations exist for interfacing with MATLAB. Further, BISQUE and OMERO share data models. BISQUE supports the OME XML data model and uses the OME Bio-Formats to import proprietary microscopy image data. Given the rapid development of analysis and visualization tools for imaging, this type of collaborative use of code and integration promises to provide very powerful tools for biologists now and in the near future.
Image analysis
Biologists are increasingly interested in using image analysis to convert microscopy images into quantitative data 15, 16. In particular, image analysis is a necessary step for experiments where hundreds or thousands of images are collected by automated microscopy, whether for screening multiple samples, collecting time or z-series data, or other technologies that generate vast volumes of image data. In addition to image analysis in a high throughout context, image processing is important for many biological studies such as quantifying the amount and localization of a signaling protein, measuring changes in structures over time, tracking invading cancer cells or looking at non-spatial data such as fluorescence lifetime data17. Image analysis can help ensure that results are accurate, objective and reproducible (Fig. 3).
Figure 3. Image Analysis and Visualization.
Legend: Image analysis and visualization span a range of complexity and variation.
A vast number of image analysis algorithms and software packages have been developed for biological applications, especially in the past decade. The software packages differ in their intended application areas, the level of usability, openness of the source code, and the cost. Whereas in the past proprietary file formats often necessitated the use of only the commercial software bundled with the microscopes, third-party software programs can now be used for most images generated by most instruments, either directly or through file format readers. Given the number of open source and commercial solutions it is often difficult to choose which tool is appropriate for a given task.
Below we highlight some of the more prominent and widely used examples that have been proven useful for light microscopy across many biomedical research areas. We have focused on image analysis and visualization tools that support interoperability with each other and with software for other steps of the bioimaging workflow (such as acquisition and data storage).
Niche image analysis tools
Most image analysis software packages developed in academia are written to accomplish very specific tasks relevant to a research problem at hand. Software exists that is designed solely for particular cell types (especially neurons), particular organisms, particular assay readouts, and particular imaging modalities. For example, the “Fluorescence Association Rules for Multi-Dimensional Insight” (FARSIGHT) toolkit grew out of the need to map the glio-vascular substrate of brain tissue surrounding neuro-prosthetic devices. In addition to finding appropriate software via web search engines, such tools can be found at online listings, such as the Neuroimaging Tools and Resource Clearinghouse (NITRC), which focuses on image analysis for neuroscience (http://nitrc.org).
Generalist image analysis tools
The second category of image analysis software packages are those that can address a more general set of problems. They are typically modular and thus offer greater flexibility to multiple applications. Some commercial tools in this category include MetaMorph, Amira (Visage Imaging), Volocity, Imaris (Bitplane Scientific Software), NIS-Elements, SlideBook, ImagePro Plus (Media Cybernetics) and ZEN (Zeiss), and are often offered by microscopy companies and sold together with imaging instrumentation. There are many open-source image analysis solutions originally developed to solve the needs of a particular community but later used or expanded to other purposes, such as BioImageXD18, Icy19, Fiji20, Vaa3D21, CellProfiler22, 3D Slicer23, Image Slicer, Reconstruct24, FluoRender25, ImageSurfer26, OsiriX27, and IMOD28.
As with the niche image analysis software, even the generalist open source platforms are developed by researchers involved in particular biology projects and therefore although they are usable for most tasks they emphasize aspects of image analysis that are most relevant for the work they are doing themselves. For example Fiji would currently be the tool of choice in analysis of electron microscopy data, Icy offers unique features for behavioral analysis and cell segmentation and tracking, Vaa3D is heavily biased towards neurobiology and together with BioimageXD offers the best facilities for 3-D visualization (see next section). The extensive online resources available for most platforms are an excellent place to find out what the strengths of the software are. Similarly, the active and communicative mailing lists and chat-room channels run by the projects can be used to engage in direct interaction with the software’s dedicated developers. Many innovative solutions are published and extensively cited which offers traditional means of evaluating the impact of the tools.
ImageJ (originally called NIH Image) occupies a unique position in the landscape of open source tools because it has been in use for the longest period of time and, importantly, has always been free, each of which make it the most popular and widespread multi-purpose image analysis tool (29–31 and http://imagej.nih.gov/ij/). Researchers have written hundreds of plugins and macros within this software to accomplish various image processing and analysis tasks in different application areas. One of the main reasons for the success of this tool is that scientists can leverage ImageJ’s infrastructure and dissemination while focusing on developing just the application-specific algorithm at hand. This extensibility has made it a favorite among both developers and end-users and the general architecture has been later adopted by the more contemporary platforms such as Icy and Vaa3D. Due to its rich history and pioneering status ImageJ can perform a wide variety of common (and many specialized) image processing and analysis tasks, particularly within the life sciences, and the user community has grown very large. ImageJ is constantly evolving to meet the needs of the scientific community as evidenced by the community driven ImageJ2project (http://developer.imagej.net), which is developing the next generation of ImageJ to include support for a myriad of features such as large multidimensional image support, a more flexible data model, and improved developer resources.
One challenge in extensible, interoperable, community-driven software projects is the proliferation of features, options, plugins, and macros. In some contexts, having too many options is just as difficult as having too few, because selecting among them can be overwhelming. A major challenge for the scientist is not only picking which tool to use but within a tool that offers many solutions, where to begin. To address this challenge, the Fiji ImageJ distribution (Fiji = Fiji Is Just ImageJ, http://fiji.sc) 20 was developed to offer ImageJ bundled together with plugins and features specifically tailored to the microscopy community and offering new functionality for microscopy analysis. The plugins are distributed through an integrated updater system that facilitates rapid feedback between plugin users and authors. Additionally, Fiji bundles several scripting languages that can be used in combination with algorithmic libraries written in Java (ImgLib, see next section), to rapidly prototype new algorithms and facilitate productive interdisciplinary collaboration between the biology and computer science research communities.
CellProfiler is a flexible multipurpose open source image analysis tool for the life sciences that has a relatively long history and track record of utility and success (22 and http://www.cellprofiler.org). It contains highly curated modules that can be mixed and matched to create customized image analysis pipelines for a variety of biological systems including cells, colonies, and C. elegans. Designed to accommodate high-throughput analysis, it is used to address a number of application areas including intensity and morphology measurements, phenotype scoring by machine learning, and object tracking.
More recently comprehensive image analysis tools focusing on 3-D or high-dimensional image data have emerged. BioImageXD and Icy both based on the Visualization Toolkit (VTK) (http://www.vtk.org) and the Insight Toolkit (ITK) (http://www.itk.org) offer many options for two-dimensional (2-D) and 3-D analysis and are extensible via plugins and macros. BioImageXD offers generation of immersive visualizations by recording fly paths through 3-D renderings of multidimensional image datasets. Icy is the youngest project in the open source bioimage analysis field and as such aims to combine the very best features of the existing tools as documented by the integrated pipeline building tools, interactions with microscopy hardware and seamless update system. Vaa3D [19] (3-D visualization-assisted-analysis) (http://vaa3d.org), provides state-of-art algorithms for registration, visualization and analysis of large-scale multi-dimensional imagery of complex biological systems (embryos and brains) to anatomical atlases for further integrative analysis.
Extensibility, interoperability and code sharing
Image analysis software is ideally extensible and interoperable, as no one tool can offer every function a researcher needs. For example, in the case of time-lapse cell analysis, a researcher may need to run a myriad of analysis routines including image denoising and deconvolution, cell identification and tracking, and measurement using advanced machine-learning analysis. The biological community has benefited from many recently constructed interfaces between image analysis software packages. It is now possible, for example, to use ImageJ for image processing and CellProfiler for cell tracking due to a link between them 32, enabling a more automated workflow. Fiji is closely collaborating with the ImageJ2 project to provide a next generation ImageJ with improved low and high-level functionality. The benefits of this coupling are numerous particularly in the areas of performance, multidimensional support and modularity. The current data model of ImageJ is largely limited to 2-D and 3-D making multidimensional analysis difficult. The n-dimensional model of ImageJ2 and Fiji will better support multidimensional image analysis including new modalities such as fluorescence lifetime microscopy (FLIM) in ImageJ2 or selective plane illumination microscopy (SPIM) uniquely available in Fiji33. Similarly improvements in the ImageJ2 core will allow for code that is more centralized, easier to repackage and share with other packages in Fiji and other platforms using Java. It is encouraging to note that all the open source platforms discussed in this section are explicitly developing ways to share data and code (see commentary Carpenter and Eliceiri and Cardona and Tomancak in this issue) as only interoperability will enable biologists to mix and match the best features of each platform to solve the daunting image analysis challenges.
Choosing an image analysis platform
All these options for image analysis and image informatics in general (Table 1 and many more not listed) bring up a key challenge for the user: how to even choose which tool to use. Both within commercial and open source software and across these categories there is a daunting array of options with substantial feature overlap. With some aspects of image informatics workflow other than image analysis, this decision is much easier and can be based on feature sets alone as often only one tool will have the features a user would want for data acquisition or storage. However in image analysis choosing between tools often comes down to preference for familiar interface, ease of use and other intangible criteria much in the same way a computer user chooses an operating system. The developer community fully realizes this challenge, which is why there is a big movement to develop software applications to make the choice easier or unnecessary. All the open source software applications described above utilize code from other applications in the form of libraries (see below) and in some cases one platform can be even run inside another. For example all of the programs discussed use Bio-Formats to open (and in some cases save) their data and many, such as Icy and CellProfiler, directly call ImageJ plugins. It is however not possible to run Icy inside ImageJ and so it is important that the projects develop their software architecture in a way that makes integration mutual. Moving beyond a typical monolithic approach means the choice for a user gets easier if they don’t have to decide between Icy and ImageJ; they can use both. Workflow systems (discussed below) which are just beginning to emerge in the biological image analysis community offer even more flexibility. They enable calling each application as components in the analysis pipeline allowing users to build their own virtual systems picking feature sets from any applications.
Image visualization
Although visualization of most two-dimensional microscopy images is trivial using a variety of software packages, modern microscopy methods enable direct capture of n-dimensional data (several channels with data across three spatial dimensions and time) (Fig. 3). The spatial dimensions alone can be very large as the addition of a computer-controlled stage allows the researcher to perform step-and-repeat microscopy, imaging large regions of tissue (millimeters to centimeters) as a montage with sub-micron resolution 34, 35. Techniques such as serial section microscopy enable this montaging to extend to the axial dimension36 In SPIM the dimensionality expansion becomes multiplied as multichannel, multi-view datasets are recorded over time for observing dynamics of protein expression and localization in a live developing embryo. Below we review some of the visualization approaches currently used in microscopy. We also direct the reader to the prior Nature Methods supplement on visualizing biological data, which included an article specifically devoted to the visualization of image data37.
Multi-dimensional imaging techniques allow direct comparison of labeled biological entities in full spatial and temporal context avoiding piecing together separate observations. However, the benefits come at the cost of more complex data storage, visualization, and analysis needs. With modern software tools and contemporary desktop graphics hardware, a three-dimensional multi-channel image cube can be projected onto the screen at interactive speeds, allowing the user to examine the data from any chosen angle and zoom factor. The speed usually results from the exploitation of hardware graphics processing engines, and careful software optimization such as utilized in the Vaa3D software 21. When the entire image series can be loaded in computer memory, Vaa3D can be used to produce real-time 5D rendering. An alternative strategy employed, for example, by ImageJ’s View5D uses cross sectional views that display only the data currently viewed at the appropriate scale (http://www.nanoimaging.uni-jena.de/View5D/View5D.html). This strategy can also be easily realized on the web allowing “Google Maps” style browsing through massive multidimensional image volumes38. Interactive 3-D visualization of large data remains a challenge. One obvious approach is hierarchical visualization, a method that combines both global and local 3-D rendering windows in memory and additional navigation window for large image files for current desktop computers (e.g. > 8–10 Gbytes per image stack to terabytes of image datasets).
Another source of increased dimensionality is systematic imaging of large numbers of reporters in independent samples, for example, dozens of antibody-probes or thousands of 3-D registered image stacks organized as different channels. For such cases, software systems 21 now allow users to use a spread-sheet based color manager to efficiently blend and map data channels into the RGB space of computer monitors for 3-D rendering.
A whole new set of visualization needs emerge after automated image analysis, for example, segmentation (Fig. 4a), tracking (Fig. 4b), feature extraction (Fig. 4c), and modeling. Segmentation and tracking operations produce massive amounts of multivariate data, commonly termed features. The subsequent analysis, visualization, and interpretation of the extracted image features from multidimensional data itself requires specially adapted tools, including scatter plots, histograms, dendrograms, bi-clustering panels, database query forms, and progression displays. The FARSIGHT toolkit actively links these displays with each other and with the multidimensional image data, enabling exploration of relationships among objects and among their derived numerical data. It also enables editing of segmentation and tracking results and quantitative analysis techniques, such as the automatic identification of outliers or groups among the features. Vaa3D is able to act as a platform for further analysis of the feature spaces. For instance, a simultaneous segmentation and recognition approach built upon Vaa3D 39, which deforms the atlas of C. elegans 40 to best fit to a newly acquired 3-D image, allows more robust identification of single cell identities at a lower rate of segmentation error, compared to the widely used 3-D watershed-type segmentation. Vaa3D (http://www.vaa3d.org) handles various image analysis tasks using a plugin interface, with which many modules such as image registration and segmentation, image input and output, and others can be glued together easily.
Figure 4. Segmentation, tracking and feature extraction in multidimensional images.
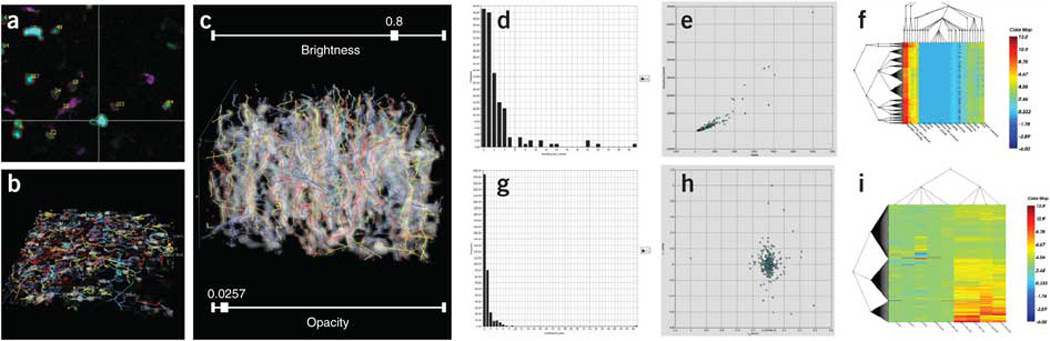
Actual screen view illustrating the series of image analysis steps starting from a multi-channel multi-photon time-lapse movie culminating in a bio-informatic profiling of the extracted spatio-temporal data, using the FARSIGHT toolkit. This movie (Courtesy Dr. Ellen Robey, UC Berkeley) recorded the three-dimensional (3-D) movements of thymocytes in an ex-vivo preparation of a live developing mouse thymus at two-minute intervals, with wild-type thymocytes displayed in cyan, F5 thymocytes in green, and dendritic cells in violet. (a) The first step is cell segmentation, shown as an orthogonal (x, y, z, t) view. Cells are delineated, and identified with numbers that correspond to rows of a table of cell measurements (not shown). The cell tracking results are displayed in multiple ways in panels B and C. (b) "Beads on strings" view showing the 3-D movement paths of cells for detecting anomalies. (c) "3-D kymograph view" showing the same movement paths overlaid on a spatio-temporal (x, y, t) projection for convenience of assessing cell tracking accuracy. (d) Histogram of cell morphological measurements (size). (e) Scatter plots provide a visual cytometric summary of pairs of measurements. (f) Coifman bi-cluster plots organize the cell data into groups based on the cytometric data. (g) Histogram of cell tracking measurements (track tortuosity).(h) Scatter plot view of pairs of cell-track measurements. (i) Coifman bi-cluster plot organizing the cell tracks into groups based on the track-based measurements. The bi-cluster modules are courtesy of Drs. Ronald Coifman (Yale University), and Lawrence Carin (Duke University).
Multidimensional imaging is in a rapid phase of development, both from the perspective of the imaging hardware technology and the visualization and analysis software. The challenges stemming from growth in data volume to terabytes and beyond has motivated major extensions to image processing toolkits discussed in the next section.
Bioimaging libraries and toolkits
The image informatics community needs not only robust image processing software but also imaging libraries, or toolkits (Fig. 5). A library is a collection of low-level algorithms that can be used, by those with programming experience, either directly or as pieces upon which end-user software applications can be built. These toolkits serve a critical role by allowing new approaches to be rapidly and flexibly tested prior to incorporation into end-user software. They tend to be modular and enable adding functionality to end-user applications.
Figure 5. BioImaging Libraries and Toolkits.
Legend: Bioimaging libraries and toolkits are available to cover a range of functionalities.
Although some commercial image-oriented libraries exist (e.g., the Image Processing toolbox within MATLAB), the majority of bioimaging libraries are free and open source. Open source code is particularly helpful for libraries because of their potential to iterate and evolve rapidly with input from the scientific community.
A variety of image analysis libraries exist, differing in their programming language compatibility, memory requirements, types of images supported, algorithmic focus, speed, and level of computer science background required (Table 2). When evaluating in practice whether a particular library or toolkit provides the functionalities needed for a given image processing workflow, it is convenient to start by visiting the introductory tutorials that most of these software projects host. They typically showcase the common uses of the library, and can provide an overview of the library’s capabilities. Online forums and mailing lists are another valuable resource. They tend to be quite active and are an effective mechanism for getting feedback on the capabilities of the software, practical advice on how to apply it to specific problems, and in some cases even ideas or assistance in modifying the source code to support new functionality. In addition to those described in more detail, below, some examples of actively used libraries include VIGRA (Vision with Generic Algorithms) (http://hci.iwr.uni-heidelberg.de/vigra/), a computer vision library with a emphasis on customizable algorithms and data structures, and EBImage41, a modular package that leverages the R environment (http://www.r-project.org/) to segment cells and extract quantitative cellular descriptors.
Table 2.
Summary of Image Analysis Libraries
| Library | Language | compatible with: |
Image Dimensions |
Computer Science Level for users |
Algorithmic Focus |
|---|---|---|---|---|---|
| VTK | C++ | Tcl / Python / Java | 2-D / 3-D | Medium | Filtering / Visualization |
| ITK | C++ | Python / Java | n-D | Advanced | Segmentation / Registration |
| OpenCV | C++ | Python / Java | 2-D+Time | Medium | Feature Extraction / Tracking / Visualization |
| ImgLib | Java | Java | n-D | Advanced | Segmentation / Registration |
| VIGRA | C++ | Python | n-D | Medium | Filtering |
| EBImage | R | R | 2-D / 3-D | Basic | Analysis / Segmentation |
Among open source bioimaging libraries, VTK (http://www.vtk.org) and ITK (http://www.itk.org) play a prominent role. Both VTK and ITK are designed as a collection of data processing units, filters that take input data and produce output data. These filters can be combined into processing pipelines that provide the flexibility and adaptability required by unexpected processing needs. Both ITK and VTK are written in C++, and then are wrapped into other languages, in particular Python, Tcl and Java. These two toolkits provide support for managing datasets that are too large to fit in the main computer memory by partitioning the input data into smaller segments that are then processed one by one. VTK’s main focus is on visualizing 2-D and 3-D images and geometrical meshes with a large variety of rendering techniques, together with 3-D widgets that facilitate user interactions with the objects being visualized. It also provides a collection of methods for performing information visualization, such as charts, plots, trees, and clusters. ITK is a complementary library focused on actual data processing rather than visualization; it is common to find ITK and VTK being used together by application developers. ITK provides one of the largest collections of image analysis algorithms, in particular for image segmentation, image registration, image stitching, and feature extraction. The toolkit supports N-dimensional images, with particular emphasis in 2-D, 3-D and 4-D. ITK also provides support for a large variety of image file formats, including JPEG 2000, HDF5, and TIFF images larger than 4 Gigabytes. Among the many bioimaging applications that are based on ITK or VTK are: Icy, BioImageXD, Slicer (http://slicer.org), Go-Figure (http://gofigure2.org), Vaa3D (http://www.vaa2d.org), and FARSIGHT (http://www.farsight-toolkit.org). When dealing with very large datasets that demand the use of distributed parallel computation platforms, such as clusters and supercomputers, it is common as well to take advantage of ParaView (http://paraview.org), an open source application built on top of VTK, which provides a client-server architecture.
OpenCV (http://opencv.willowgarage.com/wiki) is an open source library that provides a rich set of image analysis algorithms in the domain of computer vision, for native languages (C++, C, Python). OpenCV offers, for example, feature extraction algorithms that can identify notable structures from images, feature matching and tracking algorithms that can follow moving objects in video sequences, and calibration algorithms for correlating objects from 3-D space with features that they project into the 2-D plane of an imaging sensor. It has been used especially in the automated monitoring of phenotype behavior of animal models, which often requires analysis of hundreds of hours of video.
Because many of the widely used generalist platforms for bioimage analysis use or are written in Java, a Java library called ImgLib was developed, primarily under the Fiji project. ImgLib 20 enables the software developer to concentrate on the essence of the image analysis algorithm, transforming a mathematical formulation into a single piece of code that will run on images of any dimensionality (1-D, 2-D, 3-D... n-D), type (8bit, 12bit, 16bit, or complex types) or storage strategy (memory, disc, or internet server). Importantly, several major Java-based software projects (Fiji, ImageJ2, Konstanz Information Miner (KNIME), OMERO and Icy) are in the process of migrating towards using ImgLib as the primary or at least alternative means of image data representation, which will in the future substantially improve their interoperability.
Indeed, using a common library for representing image data enables easily moving algorithmic solutions between platforms or running different parts of the processing pipeline using different software on the same data. The issue of interoperability is crucial for long-term progress in bioimage informatics and general software libraries are one mechanism to achieve maximum integration among diverse platforms geared towards particular use cases (for example, software designed for automated processing of hundreds of thousands of images in high-throughput screens, like CellProfiler, vs. software that emphasizes processing very large images from multi-dimensional microscopy, like Fiji). As well, many bioimaging libraries have recently been linked by bridges, for example between OpenCV and ITK, and between VTK and ITK. This empowers application developers to build upon the functionalities provided by both libraries.
Another way to achieve interoperability even among platforms that rely on fundamentally incompatible programming languages (Java versus C) is to agree on common formats to store the image data and the results of computational analysis so that one software output can be seamlessly used as input for another software package. The Bio-Formats library enables usage of diverse software packages by enabling each one of them to import any proprietary format and to rely on the standardized OME-TIFF format for data exchange. As the results of computational analysis on bioimage data become increasingly complex (i.e. multi-view, multi-channel 4-D SPIM), there is a need for further development of agreed-upon data structures capable of efficiently capturing the analysis results and presenting them for downstream analysis. Integrative platforms for image data management such as OMERO or BISQUE or workflow tools such as KNIME will be indispensable for creating diverse ecosystems of cooperating bioimage analysis tools.
Machine learning
Machine learning has been powerfully applied to experiments involving microscope images 42, 43, and is typically defined as a field concerned with creating programs whose performance improves with experience. Although non-experts may be intimidated by the concepts of machine learning, in reality these tools often require less expertise than tools that use manually configured algorithms (Fig. 6).
Figure 6. Machine Learning.
Legend: Application areas where machine learning is used in bioimaging.
Machine learning in bioimaging is mainly used for classification – classification of either individual pixels, regions of interest in images (e.g. cells), or whole images. Whereas model-based image processing algorithms are often used to directly identify regions of interest (for example, nucleus, cytoplasm, and background), machine learning can be an alternative approach to automatically classify pixels as belonging to each class, particularly in challenging cases. Ilastik (http://www.ilastik.org/) is one open-source tool that enables researchers to train a machine learning algorithm to identify which pixels of an image belong to which class of interest, based on the researcher providing example regions of each. At the image level (whether a whole field of view or a portion thereof showing a biological entity, such as a cell), biologists often need to decide to which class a particular image belongs, such as whether a protein is in one organelle or another, or whether a cell has undergone differentiation or transformation. The classes can be defined based on the biologist’s expertise or manual inspection, but they can also be defined using other information, such as which protein was tagged or what experimental manipulations were done.
To address many such problems, an experimenter provides two or more collections of images and specifies quantities (called features) that are thought or expected to be relevant to the problem (such as the number, size or shape of cell nuclei). Image processing methods are then used to calculate the features and machine learning methods are used to decide the values of the features that distinguish between the classes. However, it is often the case that the quantities to measure are unclear a priori, are difficult for an experimenter to define sufficiently to get robust measurements, or do not in practice achieve the desired goal. In these cases, an alternative is to extract a large set of features and let the computational methods determine, or “learn”, an optimal set to use. In either case, the output is a trained classifier that can be used to sort new images into the pre-defined classes, as well as a set of statistics about the classification accuracy the system achieved. It has been demonstrated that machine learning methods can outperform human vision at recognizing patterns in microscopy images [23b]44, 45.
The features calculated as the basis for applying machine learning algorithms to images can be derived by first identifying structures of biological interest (for example, nuclei and cell boundaries), but they can also report on properties of the image as a whole like textures, pixel statistics, and factors in polynomial equations that approximate the image. Features can also be calculated on transforms of the image. The variety of image content these features represent directly determines the types of image changes to which the machine learning program will be sensitive.
Three general categories of tasks can be carried out using the features: statistical comparisons, supervised learning, and unsupervised learning. Statistical comparisons operate on the feature distributions directly, such as to determine whether two sets of images are statistically distinguishable or which image is most representative of a set, for example, to choose an image for publication, as can be done using the typical image chooser functionality in PSLID (http://pslid.org).
In supervised learning, the biologist defines relationships between images, such as by grouping together example images for different classes (a classification problem), or specifying the concentration of a drug that each sample received (a regression problem). In both cases, the program determines automatically which of the features are informative for distinguishing the classes or estimating the extent of response. Although many machine-learning programs for microscopy rely on some image pre-processing to first identify and isolate (i.e. segment) regions of interest such as cells or nuclei, successful classification has also been achieved without segmentation 46, 47. Thus, machine learning can be applied to biological images with little or no input other than the initial sorting of images into classes. When instead the desired output is a continuous variable, regression methods, which share many concepts with classification, are used. The training images are grouped by their known outputs to construct a regression function (a “standard curve”). Examples include dose-response curves (responses to a drug at increasing concentrations) 48 and timepoints in a time series 49.
When using supervised machine learning, it is important to ensure that the trained system can generalize to new images. Different subsets of the training images are used so that the system does not become too customized to the training images, a phenomenon known as overtraining. Problems are sometimes encountered if the training set does not adequately represent the variability present in the experimental samples (such as if all of the training images are acquired on one day and there is variation from day to day). Elimination of selection bias and systematic bias in the training set is in the hands of the experimenter, and is crucial in machine learning because of the absence of an a priori model.
Sometimes, the classes into which images should be grouped are unknown (or uncertain). In this case, cluster analysis, a form of unsupervised machine learning, can be used to find groups within data. For example, clustering has been used to group drugs by their effects 50 and proteins by their subcellular patterns 51.
Whether to use supervised or unsupervised machine learning for a particular bioimaging problem is often a difficult choice [27]. It hinges mainly on the extent to which the user believes that all important classes (patterns) are already known, but also on the confidence with which the user believes that the training images assigned to each class are representative of that class for all other images that will be analyzed. If labeled images are available (or can be obtained), a frequent approach is to first use supervised learning to determine how well the features can distinguish at least the major classes, and then use unsupervised learning to attempt to discover new classes, subclasses within known classes, or variation within a class over time.
Supervised and unsupervised methods can be combined, an approach referred to as semi-supervised learning. The idea is to use some information about classes, and then extend it using the data. For example, images might be grouped based on the identity of a treatment (supervised), and these samples can then be clustered into morphologically defined classes (unsupervised). An example of this used WND-CHARM (weighted neighbor distance using compound hierarchy of algorithms representing morphology) and timepoints throughout the lifespan to train a classifier to assign individual C. elegans a physiological age score. The distribution of age scores in these chronologically-defined populations revealed that aging is not a gradual process, but progresses through three distinct physiological states 49.
There are also a number of approaches in which input can be introduced during the learning process. For example, in CellProfiler Analyst 52, the biologist is presented with machine classifications of new cells based on their previous input, and given the opportunity to correct errors. This new information is used to re-train the classifier. If, rather than having the user choose which labels to correct, the machine learner can ask for labels for specific examples (e.g., because they have a high degree of uncertainty) it is said to be doing active learning 43. This process can potentially be made fully automatic, as demonstrated in 49 where a classifier is used to control an acquisition system for optimally acquiring additional examples to increase its accuracy.
Machine learning has been proven useful for a number of purposes in bioimaging, but there are some limitations. Classifiers typically do not typically transfer well between cell types or between different imaging systems; re-training must often be performed. A related problem is that classification and clustering systems can only “represent” the patterns they are trained on. An alternative is to try to represent a subcellular pattern or cell morphology using image-derived generative models. Given multiple images of a given pattern, a model can be trained to capture the essence and the variation in that pattern and can synthesize new images that in a statistical sense are drawn from the same distribution as the training images. This approach has only recently been applied in bioimaging; however, reasonably accurate methods for building models for nuclear and cell shape and some organelles are available (http://CellOrganizer.org). A final limitation of machine learning approaches is that they are not typically used to handle mixtures of classes. Take for example the application of machine learning to identify to which organelle a particular protein localizes, having been trained on example images showing localization in each distinct compartment. A protein can be anywhere on a continuum between fully in one compartment to fully in one or more other compartments. This leads classifiers to produce arbitrary results when presented with images of mixed patterns, and can lead clustering algorithms to form “chained” clusters (or to artificially divide the continuum into pieces). A solution to this problem is to directly estimate how much of a given protein is in each of multiple “fundamental” compartments (e.g., 10% is in lysosomes, 90% is in the Golgi complex). This can be done given training images of proteins that are known to be in just one compartment (such as the “marker” proteins used in training classifiers), representing each image by the amount of fluorescence in distinct object types, and using various standard unmixing methods. This “supervised unmixing” approach has been successful [31a] and can be done using the open source PatternUnmixer software (http://murphylab.cbi.cmu.edu/software/PatternUnmixer2.0). It is even possible to do “unsupervised unmixing” by estimating both the fundamental patterns and the mixing fractions [31b].
Workflow systems
Workflow systems for data processing and analysis have begun to serve an important role across many fields in biology, including bioimaging. Workflow based tools allow researchers to flexibly and intuitively model data processing and analysis protocols and integrate a diverse array of tools without writing complex scripts or being constrained to application-focused, monolithic tools (Fig. 7). As such, they often address a complementary niche, where researchers are not repeating the same analysis day after day but instead are exploring and testing solutions or need to adapt to a broad array of applications. These workflow tools have become increasingly attractive because the need to process and analyze data in a sophisticated way is spreading from specialists to the majority of modern biologists. Being able to reproducibly and intuitively read, transform, process, and analyze data has become a necessary skill, very much like word processing twenty years ago.
Figure 7. Workflow Systems.
Legend: Benefits of using a workflow system.
Workflow systems can enable a field to transition from requiring that experts manually string together a series of many single-purpose software tools, to enabling non-experts to create a seamless workflow using a single integrated tool. Workflow tools have begun to serve the bioimaging community well: both expert users who know how to tweak every parameter of an algorithm, to the broader user base needing an intuitive user interface. “Visual programming” is the most common approach to user-friendliness in this domain, enabling access to a variety of functionality (e.g., image processing, data mining) while also giving access to multiple data sources (e.g., chemical, biological, textual). Further advantages are the ability to readily document and disseminate an analysis, supporting reproducible research and rapid sharing of novel approaches.
A number of workflow systems have emerged over the past decade or so. Several companies offer workflow tools, such as SAS’ Enterprise Miner and IBM/SPSS Clementine, offering tools for more general data mining and statistical analysis. More focused on the life science market is Accelrys’ Pipeline Pilot, which offers a combination of tools addressing chemoinformatics, image analysis, and also some data analysis capabilities. These tools are proprietary but have been offered at a steep discount for academics and are thus often used for teaching and academic research. Still, the rapid spread and continual evolution of bioimaging-related software has made it challenging for a single commercial entity to offer solutions that cover the spectrum of needs in this domain and integrate all of the relevant technology. Given the availability of state-of-the-art open source tools for individual steps of the bioimaging workflow, it is therefore no surprise that in recent years open source workflow systems have gained popularity. The workflow system itself has taken a back stage role as a data and tool integration backbone and – much like is the case for Linux – companies are more willing to invest in open standards for such critical infrastructure pieces. Open-source workflow tools such as Taverna (http://www.taverna.org.uk/) and Galaxy (http://galaxy.psu.edu/) focus on bioinformatics, while KNIME (http://www.knime.org/) is an option that serves an even broader set of domains, including business intelligence and predictive analytics. It also has connections to other bioimaging tools, such as ImageJ, OMERO, and CellProfiler, among others, allowing the creation of complex image processing and analysis workflows. The use of workflow software in biological imaging is still very new and yet to be described in any large-scale biological research publications. However these approaches are routinely used in pharmaceutical research and have great potential to be applied to large scale, multistep analysis problems.
When considering what workflow system to use or whether a workflow approach is even necessary it’s important to first define if a monolithic approach or a framework is needed. Many of the previous tools discussed in the image analysis section allow for robust workflows to be created and run, such as a series of image processing tasks in Icy or CellProfiler. The main difference between Icy and other similar applications and tools such as Taverna and KNIME is that the former are trying to do everything by themselves (sometimes incorporating other tools but not necessarily in a very flexible way). The latter are open frameworks that allow the easy integration of various other libraries and tools - in such a way that you can easily swap one tool for another one. Taverna does this by adopting a standard (web services), KNIME has its own open API and data-type format. Both are quite effective but the advantage of the open API approach is that web services are a lot harder to "archive" as web services can vary over time. In contrast if you ran a workflow in KNIME 2.4.3 in 2008 you can still run it in 2018 using that version of KNIME. When thinking about the steps involved in a workflow there are two important steps, the steps in the image analysis pipeline itself and the steps of using such image analysis in a sustainable way. For the steps in the pipeline it is important to emphasize the importance of being able to swap tools out easily and use a different image loading processing, analysis or whatever other library for different tasks (also for any one user who can for different tasks rely on different packages for different jobs). As well the ability to visually document what one has done is important. In thinking about the steps of using such image analysis workflows in a sustainable way, the key issues addressed by a workflow system are reproducibility, archivability and in particular the ability to share workflows.
It is important to note that a workflow system is not needed for many image processing and analysis tasks, and many workflow needs can be addressed by the more monolithic approach in current image analysis applications. One of the strengths of a platform such as KNIME is that it is a separate piece of technology from the software pieces that actually do the work. So KNIME concentrates on the modeling of the analysis "pipeline" and allows the user to integrate whatever software libraries for the image loading, processing or analysis one wants to use (or other routines such as chemical modeling, text analysis). A user can even launch their own in-house toolbox for their preferred way to do an arbitrary image processing process.
The ability to access cutting-edge technology as different modules within a workflow system also yields a challenge: version control. The ability to reproduce an analysis precisely is critical in most scientific domains. Proprietary tools have the ability, at least in theory, to ensure that workflows continue to produce the same results with progressive updates to the software but this is not always the case. Further, those without licenses for the software cannot reproduce another researcher’s analysis, and the closed-source nature of the software limits the ability of others to rely on the software being available in perpetuity. For open-source tools, version control and reproducibility are at least feasible. This is commonly addressed by taking snapshots of the particular version of the code bases on which an end-user application depends, and storing those code bases in a combined repository, or by simply referring to a specific tagged version in the official repository of the code bases. For example, the end-user application Slicer 4.0 is built by pointing to a specific version of ITK, VTK, Python, and many other tools. Vaa3D and Icy are also built by pointing to specific versions of ITK. The multi-platform configuration tool CMake makes this process straightforward by providing the functionality of “superbuilds” and “external projects”. Tools that heavily rely on external toolkits (in the extreme case, via WebService calls) allow a workflow itself to be archived, although the tools it calls may cease to function or may produce different results over time. Tools like KNIME offer users a choice in this respect by offering to call webservices or alternately to freeze a certain state, ensuring workflow reproducibility in the future.
Conclusions
As technology progresses, modern biologists must become increasingly familiar with computational techniques and software tools. Researchers using microscopy are no exception to this rule; fortunately bioimaging software has recently rapidly developed in terms of both functionality and usability. Emphasis on functionality stems from the fact that bioimaging software developers are typically embedded in, or have strong ties to, experimental biology labs. Thus the software produced by this developer community is usually in direct answer to current biology needs and often in response to new emerging problems outside the scope of commercial interest. Usability has often lagged behind, but there is increasing recognition of its value to biomedical research 53.
While initially the focus of each software project in this domain was solely to address a single step in the bioimaging workflow, the bioimaging software community has recently begun to address an important aspect of usability: interoperability. The open-source bioimaging software community has begun to communicate and collaborate, assisted and reflected by conferences such as BioImage Informatics and the Cold Spring Harbor Laboratory’s Automated Imaging and High-Throughput Phenotyping meeting. As the community gains momentum, connections among independent software projects have begun to be prioritized, and some have already been completed, as highlighted in this review. These connections greatly ease researchers’ work by reducing the need to tediously transfer data between multiple software packages.
As described in commentaries in this issue 53, 54, there is a great need to not only collaborate on bioimaging ideas and approaches but also on software coding itself, including consensus on best practices and standards, software quality control, documentation, training, maintainability, and sharing of modular code. The many activities where the developers and users interact and develop their skills-for example, tutorials, conferences and hackathons-promote the progress of the software by fostering rapid innovation and interdisciplinary thinking. In fact, the community of researchers surrounding most open-source bioimaging software projects is usually much more important than the software itself. Software without community is a static resource of limited lifespan, while an active community adapts continuously to new problems. These highly networked collaborations are a common property of open-source software projects, multiplying the value of a simple software resource and enabling the attack of scientific problems too large or interdisciplinary for individual laboratories to address 55. There are many challenges facing the imaging community, such as tracking and annotating large multidimensional datasets and retaining and sharing complex heterogeneous datasets; but also opportunities for pooling resources, such as in software coding or in testing and validation using crowd sourcing. All of these challenges necessitate the development of a robust bioimaging informatics platform.
The last twenty years have seen extraordinary biological advances driven by novel biological imaging tools, many of which directly relied on computational methods. The future of biological imaging innovation depends even more squarely on developing image informatics solutions, from acquisition to storage, analysis to mining, and visualization to dissemination. Continued advances in bioimaging computational approaches will serve not only as the foundation for new imaging methods but as the catalyst for new biological discovery that would not otherwise be possible. Resources invested in the development and maintenance of important bioimaging software applications as well as connections among them promise to yield great dividends to the thousands of biologists relying on bioimaging.
Acknowledgements
The authors wish to acknowledge their respective funding sources and members of their lab for feedback and useful comments. In particular we gratefully acknowledge A. Merouane and A. Narayanaswam of the Roysam Laboratory for their assistance in preparing figures and L. Kamentsky and M. Bray of the Carpenter lab for useful input and edits on the manuscript.
References
- 1.Peng H. Bioimage informatics: a new area of engineering biology. Bioinformatics. 2008;24:1827–1836. doi: 10.1093/bioinformatics/btn346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gustafsson MG. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA. 2005;102:13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones SA, Shim SH, He J, Zhuang X. Fast, three-dimensional super-resolution imaging of live cells. Nat. Methods. 2011;8:499–508. doi: 10.1038/nmeth.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Planchon TA, et al. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat. Methods. 2011;8:417–423. doi: 10.1038/nmeth.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Computer control of microscopes using µManager. Curr. Protoc. Mol. Biol. 2010;92:14.20.11–14.20.17. doi: 10.1002/0471142727.mb1420s92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin HP, Vincenz C, Eliceiri KW, Kerppola TK, Ogle BM. Bimolecular fluorescence complementation analysis of eukaryotic fusion products. Biol. Cell. 2010;102:525–537. doi: 10.1042/BC20100033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pologruto TA, Sabatini BL, Svoboda K. ScanImage: flexible software for operating laser scanning microscopes. Biomed. Eng. Online. 2003;2:13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conrad C, et al. Micropilot: automation of fluorescence microscopy-based imaging for systems biology. Nat. Methods. 2011;8:246–249. doi: 10.1038/nmeth.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allan C, et al. OMERO: flexible, model-driven data management for experimental biology. Nat. Methods. 2012;9:245–253. doi: 10.1038/nmeth.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kvilekval K, Fedorov D, Obara B, Singh A, Manjunath BS. Bisque: a platform for bioimage analysis and management. Bioinformatics. 2010;26:544–552. doi: 10.1093/bioinformatics/btp699. [DOI] [PubMed] [Google Scholar]
- 13.Wu L, Faloutsos C, Sycara KP, Payne TR. Proceedings of the 26th International Conference on Very Large Data Bases. Morgan Kaufmann Publishers Inc.; 2000. [Google Scholar]
- 14.Goff SA, et al. The iPlant Collaborative: Cyberinfrastructure for plant biology. Frontiers in Plant Science. 2011;2 doi: 10.3389/fpls.2011.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glory E, Murphy RF. Automated subcellular location determination and high-throughput microscopy. Dev. Cell. 2007;12:7–16. doi: 10.1016/j.devcel.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 16.Ljosa V, Carpenter AE. Introduction to the quantitative analysis of two-dimensional fluorescence microscopy images for cell-based screening. PLoS Comput. Biol. 2009;5:e1000603. doi: 10.1371/journal.pcbi.1000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lakowicz JR. Principals of Fluorescence Spectroscopy. New York: Academic Press; 1999. [Google Scholar]
- 18.Kankaanpää P, et al. BioImageXD: open general purpose and high-throughput image processing platform. Nat. Methods. doi: 10.1038/nmeth.2047. (In Press) [DOI] [PubMed] [Google Scholar]
- 19.de Chaumont F, et al. Icy: an open bioimage informatics platform for extended reproducible research. Nat. Methods. doi: 10.1038/nmeth.2075. (In Press) [DOI] [PubMed] [Google Scholar]
- 20.Schindelin J, et al. Fiji - an Open Source platform for biological image analysis. Nat. Methods. doi: 10.1038/nmeth.2019. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng H, Ruan Z, Long F, Simpson JH, Myers EW. V3D enables real-time 3D visualization and quantitative analysis of large-scale biological image data sets. Nat. Biotechnol. 2010;28:348–353. doi: 10.1038/nbt.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carpenter AE, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pieper S, Lorensen B, Schroeder W, Kikinis R. Proceedings of the 3rd IEEE International Symposium on Biomedical Imaging: From Nano to Macro. 2006;Vol. 1:698–701. [Google Scholar]
- 24.Fiala JC. Reconstruct: a free editor for serial section microscopy. J. Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- 25.Wan Y, Otsuna H, Chien C-B, Hansen C. Proceedings of Pacific Vis 2012. 2012:201–208. doi: 10.1109/pacificvis.2012.6183592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng D, et al. Stepping into the third dimension. J. Neurosci. 2007;27:12757–12760. doi: 10.1523/JNEUROSCI.2846-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosset A, Spadola L, Ratib O. OsiriX: An open-source software for navigating in multidimensional DICOM images. J. Digit. Imaging. 2004;17:205–216. doi: 10.1007/s10278-004-1014-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 29.Collins TJ. ImageJ for microscopy. Biotechniques. 2007;43:25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- 30.Abramoff M, Magalhaes P, Ram S. Image processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- 31.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. doi: 10.1038/nmeth.2089. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamentsky L, et al. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics. 2011;27:1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Preibisch S, Saalfeld S, Schindelin J, Tomancak P. Software for bead-based registration of selective plane illumination microscopy data. Nat. Methods. 2010;7:418–419. doi: 10.1038/nmeth0610-418. [DOI] [PubMed] [Google Scholar]
- 34.Tsai CL, et al. Robust, globally consistent and fully automatic multi-image registration and montage synthesis for 3-D multi-channel images. J. Microsc. 2011;243:154–171. doi: 10.1111/j.1365-2818.2011.03489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Preibisch S, Saalfeld S, Tomančák P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25:1463–1465. doi: 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saalfeld S. Elastic Volume Reconstruction from series of ultra-thin microscopy sections. Nat. Methods. doi: 10.1038/nmeth.2072. (In Press) [DOI] [PubMed] [Google Scholar]
- 37.Walter T, et al. Visualization of image data from cells to organisms. Nat. Methods. 2010;7:S26–S41. doi: 10.1038/nmeth.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saalfeld S, Cardona A, Hartenstein V, Tomančák P. CATMAID: collaborative annotation toolkit for massive amounts of image data. Bioinformatics. 2009;25:1984–1986. doi: 10.1093/bioinformatics/btp266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qu L, et al. Simultaneous recognition and segmentation of cells: application in C.elegans. Bioinformatics. 2011;27:2895–2902. doi: 10.1093/bioinformatics/btr480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Long F, Peng H, Liu X, Kim SK, Myers E. A 3D digital atlas of C. elegans and its application to single-cell analyses. Nat. Methods. 2009;6:667–672. doi: 10.1038/nmeth.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pau G, Fuchs F, Sklyar O, Boutros M, Huber W. EBImage--an R package for image processing with applications to cellular phenotypes. Bioinformatics. 2010;26:979–981. doi: 10.1093/bioinformatics/btq046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shamir L, Delaney JD, Orlov N, Eckley DM, Goldberg IG. Pattern recognition software and techniques for biological image analysis. PLoS Comput. Biol. 2010;6:e1000974. doi: 10.1371/journal.pcbi.1000974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy RF. An active role for machine learning in drug development. Nat. Chem. Biol. 2011;7:327–330. doi: 10.1038/nchembio.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nattkemper TW, Twellmann T, Ritter H, Schubert W. Human vs machine: evaluation of fluorescence micrographs. Comput. Biol. Med. 2003;33:31–43. doi: 10.1016/s0010-4825(02)00060-4. [DOI] [PubMed] [Google Scholar]
- 45.Johnston J, Iser WB, Chow DK, Goldberg IG, Wolkow CA. Quantitative image analysis reveals distinct structural transitions during aging in Caenorhabditis elegans tissues. PLoS One. 2008;3:e2821. doi: 10.1371/journal.pone.0002821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang K, Murphy RF. From quantitative microscopy to automated image understanding. J. Biomed. Opt. 2004;9:893–912. doi: 10.1117/1.1779233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shamir L, et al. Wndchrm - an open source utility for biological image analysis. Source Code Biol. Med. 2008;3:13. doi: 10.1186/1751-0473-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loo LH, Wu LF, Altschuler SJ. Image-based multivariate profiling of drug responses from single cells. Nat. Methods. 2007;4:445–453. doi: 10.1038/nmeth1032. [DOI] [PubMed] [Google Scholar]
- 49.Jackson C, Glory-Afshar E, Murphy RF, Kovacevic J. Model building and intelligent acquisition with application to protein subcellular location classification. Bioinformatics. 2011;27:1854–1859. doi: 10.1093/bioinformatics/btr286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perlman ZE, et al. Multidimensional drug profiling by automated microscopy. Science. 2004;306:1194–1198. doi: 10.1126/science.1100709. [DOI] [PubMed] [Google Scholar]
- 51.Chen X, Murphy RF. Objective clustering of proteins based on subcellular location patterns. J. Biomed. Biotechnol. 2005;2005:87–95. doi: 10.1155/JBB.2005.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones TR, et al. Scoring diverse cellular morphologies in image-based screens with iterative feedback and machine learning. Proc. Natl. Acad. Sci. USA. 2009;106:1826–1831. doi: 10.1073/pnas.0808843106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carpenter AE, Kamentsky L, Eliceiri KW. A call for bioimaging software usability. Nat. Methods. doi: 10.1038/nmeth.2073. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tomančák P, Cardona A. Open source as a winning strategy for bioimage informatics. Nat. Methods. doi: 10.1038/nmeth.2082. (In press) [DOI] [PubMed] [Google Scholar]
- 55.Nielsen M. Reinventing Discovery: The New Era of Networked Science. Princeton University Press; 2011. [Google Scholar]
- 56.Swedlow JR. Finding an image in a haystack: the case for public image repositories. Nat. Cell. Biol. 2011;13:183. doi: 10.1038/ncb0311-183. [DOI] [PubMed] [Google Scholar]
- 57.Linkert M, et al. Metadata matters: access to image data in the real world. J. Cell. Biol. 2010;189:777–782. doi: 10.1083/jcb.201004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Larson SD, Martone ME. Ontologies for Neuroscience: What are they and what are they good for? Front. Neurosci. 2009;3:60–67. doi: 10.3389/neuro.01.007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Plant AL, Elliott JT, Bhat TN. New concepts for building vocabulary for cell image ontologies. BMC Bioinformatics. 2011;12:487. doi: 10.1186/1471-2105-12-487. [DOI] [PMC free article] [PubMed] [Google Scholar]