

Abstract
Once released into the environment, engineered nanomaterials may be transformed by microbially mediated redox processes altering their toxicity and fate. Little information currently exists on engineered nanomaterial transformation under environmentally relevant conditions. Here, we report the development of an in vitro biomimetic assay for investigation of nanomaterial transformation under simulated oxidative environmental conditions. The assay is based on the extracellular hydroquinone-driven Fenton’s reaction used by lignolytic fungi. We demonstrate the utility of the assay using CdSecore/ZnSshell quantum dots (QDs) functionalized with poly(ethylene glycol). QD transformation was assessed by UV-Visible spectroscopy, inductively-coupled plasma-optical emission spectroscopy, dynamic light scattering, transmission electron microscopy (TEM), and energy dispersive x-ray spectroscopy (EDX). QDs were readily degraded under simulated oxidative environmental conditions: the ZnS shell eroded and cadmium was released from the QD core. TEM, electron diffraction analysis and EDX of transformed QDs revealed formation of amorphous Se aggregates. The biomimetic hydroquinone-driven Fenton’s reaction degraded QDs to a larger extent than did H2O2 and classical Fenton’s reagent (H2O2 + Fe2+). This assay provides a new method to characterize transformations of nanoscale materials expected to occur under oxidative environmental conditions.
Introduction
As nanomaterial production and use rise, introduction of engineered nanoparticles into the environment becomes inevitable. Inadvertent release of these materials into the environment may occur at numerous points during a product’s life cycle including production, transport, manufacturing, consumer use, recycling, and disposal (1,2). Release to the environment during recycling and disposal is of particular concern for nanoparticles incorporated into limited use and/or disposable products.
Environmental redox processes may induce breakdown of organic compounds and can also result in dissolution or precipitation of inorganic species. Microorganisms mediate many environmental redox reactions either directly through synthesis of enzymes or indirectly via biogenic oxidant/reductant production (3). Understanding possible transformations of engineered nanomaterials is essential for evaluating their potential environmental impact. Few systematic studies on the environmental fate of nanomaterials have been published. The stability of some nanomaterials in laboratory settings have been reported (4-8), but conditions employed were not representative of natural environments.
Understanding environmental transformations and fate of engineered nanomaterials will enable design and development of environmentally benign nanomaterials, as well as their use as environmental tracers, in environmental sensing, and in contaminant remediation. For example, functionalized Fe3O4 magnetic nanoparticles have been proposed as sorbents for removing toxic metals from natural waters and biological fluids (9). Their chemical stability was assessed by monitoring Fe release into various matrices after 2-h exposure (9). Mechanisms responsible for Fe release, possible changes in nanoparticle surface chemistry, and particle dissolution over longer exposure periods were not discussed. Thus, transformation of such nanoparticles in the environment and concomitant impacts on metal sorption remain unclear. Assessment of the potential environmental transformation of engineered nanomaterials would be facilitated by the development of test systems designed to represent specific environmental conditions. Such test systems for engineered nanomaterials are currently lacking.
The objective of this study was to develop a simple, yet robust, assay for investigating potential transformation of engineered nanomaterials under environmentally relevant oxidative conditions. To accomplish this, we developed an assay mimicking the extracellular chemistry of lignolytic fungi. To demonstrate the utility of the assay, we employed functionalized CdSecore/ZnSshell semiconductor nanocrystals or quantum dots (QDs) as prototypical nanoparticles and examined their transformation. We selected QDs because their optical properties provided a convenient means to qualitatively and quantitatively assess changes induced by the biomimetic oxidative assay. QDs cores are often composed of toxic heavy metal chalcogenides encapsulated in a shell composed of a wider band-gap material. Passivation by such shells improves optical emission (10), and is reported to protect the QD core from degradation (11). Extensive study of CdSe QDs in laboratory settings has yielded data on alterations to physical structure induced by severe oxidative treatments (viz. photo-oxidation, exposure to high H2O2 concentrations), and strongly acidic environments. These data can aid in understanding the extent of transformation induced by the biomimetic assay (4-7,10).
Methods
Quantum Dot Preparation
Synthesis and functionalization of CdSecore/ZnSshell QDs are described in the Supporting Information (SI).
Biomimetic Assay Design
The biomimetic oxidative assay was modeled after the extracellular chemistry generated by the lignolytic brown rot fungus Gloeophyllum trabeum. Brown rot fungi produce reactive oxygen species in soil environments to initiate decay of woody plant debris (12,13). One demonstrated system for extracellular ROS production by lignolytic fungi is a dimethoxyhydroquinone-driven Fenton’s reaction (12-15). In this system, fungi produce H2O2 and organic reductants, and use iron in the environment to sustain a Fenton’s reaction, producing hydroxyl radicals (12-15). In Fenton’s reaction, Fe2+ promotes H2O2 decomposition to produce hydroxyl radicals and Fe3+ (i.e., H2O2 + Fe2+ → •OH + OH− + Fe3+). Brown rot fungi produce organic reductants (e.g., 2,5-dimethoxyhydroquinone, 4,5-dimethyoxycatechol) to reduce Fe3+ to Fe2+, thus sustaining the reaction (13,14,16). Fenton’s reaction proceeds optimally at low pH (17) consistent with proton activities typical of extracellular environments of lignolytic fungi (viz. pH 3-5) (16). Laboratory G. trabeum cultures exhibit extracellular pH values between 4.3 and 4.7 (15) and extracellular H2O2 concentrations between 50 and 300 μM (13), while iron and 2,5-dimethoxyhydroquinone concentrations each range between 10 and 50 μM (12, 13, 15).
To mimic these conditions, solutions consisting of 200 μM H2O2, 20 μM Fe2+, 20 μM of an organic reductant, and 2 μM QDs in 10 mM buffer were prepared. Concentrations of assay components were environmentally relevant, and approach optimal H2O2:Fe molar ratios used in advanced oxidation processes based on Fenton’s chemistry (17,18). Although G. trabeum produces 2,5-dimethoxyhydroquinone as a reductant, this compound is not currently commercially available and its use would require synthesis and purification (13). We therefore tested two commercially available reductants to sustain the Fenton reaction: methoxyhydroquinone (MHQ) and l-ascorbic acid (AA). In principle, any reductant capable of rapidly reducing Fe3+ to Fe2+ could be used. To achieve a proton activity similar to that present in the vicinity of lignolytic fungal hyphae, we buffered reaction mixtures to pH 4.1 with 10 mM acetate. To provide a point of reference, we also exposed QDs to classical Fenton’s reagent using the same H2O2 and Fe2+ concentrations. As controls, we exposed QDs to each single assay constituent (viz. H2O2, MHQ, AA, Fe+2; Figure S6), as well as to every binary combination of constituents (e.g., H2O2 + MHQ, Fe+2 + AA; Figures S7 and S8) to verify that observed changes resulted from catalytically produced oxidants and not other assay components. All experiments and controls were conducted in triplicate and each experiment was repeated at least three times. The SI contains additional details on assay protocols.
Reactant and Product Characterization
UV-Visible spectrophotometry, X-ray photoelectron spectroscopy (XPS), attenuated total reflectance-Fourier transform infrared (ATR-FTIR), transmission electron microscopy (TEM), energy dispersive X-ray spectroscopy (EDX), dynamic light scattering (DLS), fluorescence emission spectroscopy, and inductively coupled plasma-optical emission spectroscopy (ICP-OES) were used to characterize QDs and changes induced by exposure to the assay (see the SI for details).
Results and Discussion
Characterization of as-synthesized QDs
The QDs investigated were poly(ethylene glycol) (PEG)-functionalized CdSecore/ZnSshell QDs (Figure 1a). The presence of the ZnS shell was verified by XPS (Figure S1). Shell thickness did not exceed 1 nm. Quantum dots were functionalized with methoxy-terminated PEG-thiol (either PEG350 or PEG5000) via interaction of the terminal thiol with Zn using a ligand exchange procedure (19). PEG renders QDs soluble and stable in aqueous solutions (19) and is biologically inert (20), making it a useful surface coating for biological applications of QDs (11) and facilitating toxicity assessment of parent and transformed species. Furthermore, PEG-thiols can serve as a model for the stability of other thiol ligand-exchanged QDs that are often further functionalized to produce bio-conjugates (11).
Figure 1.
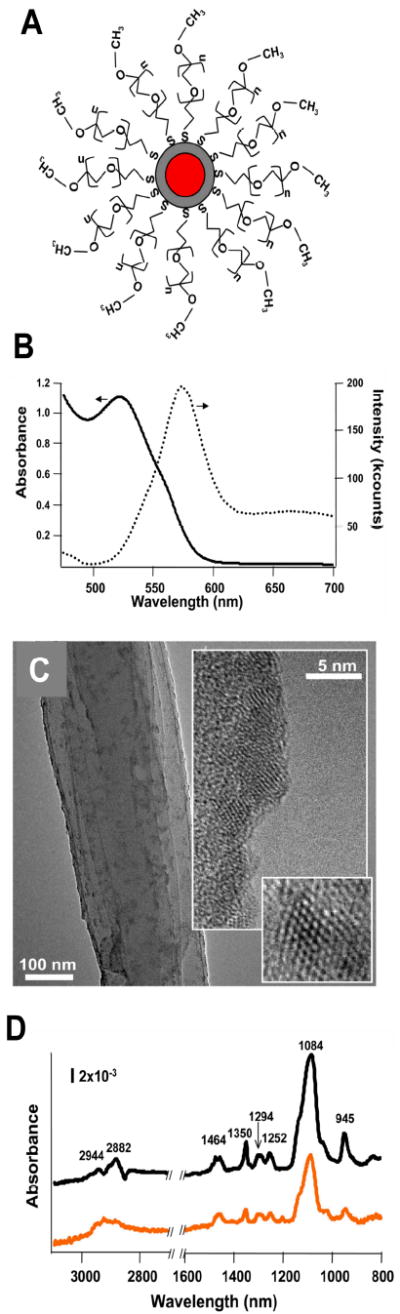
(a) Illustration of PEGylated CdSecore/ZnSshell quantum dot (n = 6 or 100 in this study). (b) UV-Visible absorption and fluorescence emission spectra of water-stable PEG5000-QD in unbuffered ddH2O (pH 8). (c) High resolution transmission electron micrographs of CdSecore/ZnSshell QDs on a lacey carbon-coated Cu grid, showing lattice-resolved images in the inserts (the smallest insert is a 5 nm × 5 nm box). Atomic spacing was 3.6 Å, sonsistent with wurtzite CdSe structures.(d) FTIR spectra of QDs ligand exchanged with PEG5000 (top, black) and PEG350 (bottom, orange). No evidence of coordinating ligands used in QD synthesis was observed by FTIR following ligand exchange with PEG.
Figure 1b shows typical UV-Visible and fluorescence emission spectra for the water-stable PEG5000-QDs conjugate stock suspension (unbuffered ddH2O, pH 8). The UV-Visible spectrum exhibits a steady increase in absorbance as wavelength decreases from 600 nm to a strong absorbance peak centered at 525 nm, caused by the first exciton of the CdSe core; absorbance increases nearly linearly as wavelength decreases further. Similar spectra were obtained from aqueous suspensions of PEG350-QD conjugates (data not shown). QD core diameter and number concentration can be determined from first exciton peak position and absorbance in the UV-Visible spectrum (21). QDs used in this study had first exciton peaks centered between 525 and 530 nm, corresponding to core diameters of 2.6 ± 0.1 nm (21). While the accuracy of UV-Visible spectroscopy methods for determining absolute measures of core diameter and QD concentration has recently received criticism (22), UV-Visible spectra retain utility for comparative analyses (e.g., examining changes induced by exposure to oxidative conditions). We employed TEM to independently verify QD size and observed crystalline nanoparticles with diameters of 3-4 nm (Figure 1c), in good agreement with the size of the core-shell QDs estimated from UV-Vis and XPS data (viz. 3.5-4.5 nm). FTIR spectra of QD-PEG conjugates in water exhibit at least eight clearly identifiable peaks (Figure 1d), all attributable to PEG (see the SI for peak assignments) Using an empirical relationship between effective diameter and molecular mass of free PEG molecules (23), we estimated the length of attached PEG350 and PEG5000 molecules to be 1 nm and 4.5 nm. Theoretical diameters of individual PEGylated QDs (based on estimated contributions of the CdSe core, ZnS shell and extended PEG ligands) were ~6.5 nm for PEG350-QDs and ~14 nm for PEG5000-QDs. Dynamic light scattering measurements, using number averaging techniques, revealed that in ddH2O PEG350- and PEG5000-QDs had hydrodynamic diameters of 26 ± 3 nm and 23 ± 1 nm, suggesting PEG5000-QDs formed very small aggregates, while PEG350-QDs aggregated to a larger extent. In 10 mM Na acetate, hydrodynamic diameters of PEG350- and PEG5000-QDs (29 ± 4 nm and 24 ± 1 nm) were statistically indistinguishable from those in ddH2O.
Transformation of PEG5000-QDs
Transformations of PEG5000-QDs conjugates induced by exposure to oxidizing solutions were monitored by UV-Visible absorption spectroscopy, fluorescence emission spectroscopy, DLS, TEM, EDX, and ICP-OES. Exposure of PEG5000-QDs to MHQ-Fenton’s reaction at pH 6, to examine the effect of raising assay pH on QD integrity, is reported in the SI.
Optical Characterization of Transformed QDs
UV-Visible spectra and fluorescence emission spectra (see the SI) were collected from exposed QD suspensions. UV-Visible absorption spectroscopy allows simultaneous monitoring of changes in the physical size of QD cores and changes in QD number concentration. Fluorescence emission spectra of QDs also respond to changes in the physical size of the core, but are also sensitive to many other factors (e.g., trapped states, shell condition, surface functionalization, solution chemistry). This complicates interpretation of changes in QD fluorescence emission spectra.
UV-Visible absorbance spectra of QDs in acetate buffer exhibited first exciton peaks centered near 530 nm, comparable to stock solutions in ddH2O (Figure 2a). Solution spectra of H2O2-exposed QDs exhibited similar shapes as those of buffer controls, but first exciton peak position was blue-shifted to ~515 nm (Figure 2a), indicating a ~0.2-nm reduction in core diameter, and peak height declined by ~8%. After exposure to the methoxyhydroquinone-driven Fenton’s (MHQ-Fenton’s) reaction, PEG5000-QD UV-Visible spectra increased monotonically as wavelength declined; the characteristic first exciton peak was absent from thesespectra. Compared to buffer and H2O2 controls, MHQ-Fenton’s spectra showed higher absorbance at λ < 525 nm. Methoxyhydroquinone in its reduced or oxidized states does not absorb strongly over the wavelength ranged shown in Figure 2a and therefore did not contribute significantly to the observed monotonically increasing absorbance (see the SI).
Figure 2.
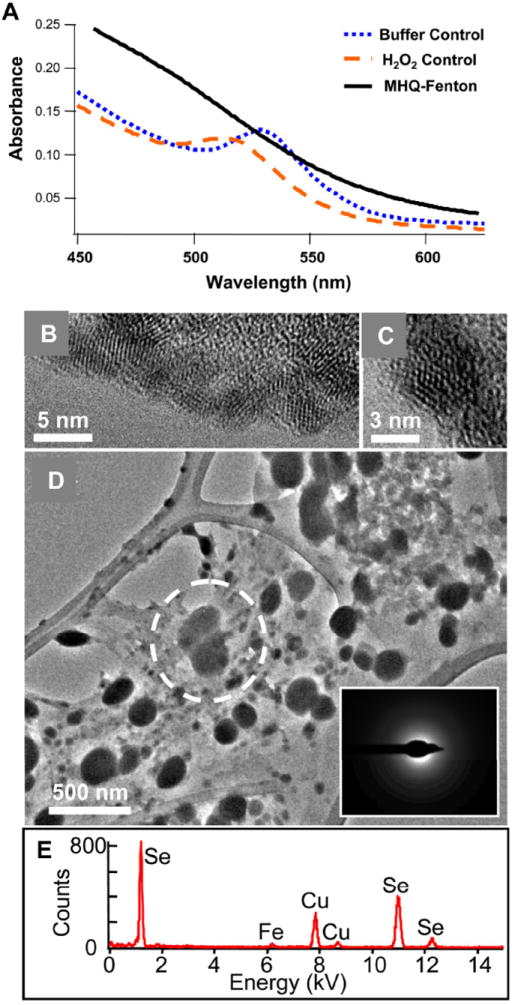
(a) UV-Visible absorption spectra of PEG5000-QDs exposed to methoxyhydroquinone-driven Fenton reaction. All reactions were conducted in 10 mM acetate buffer (pH 4.1). (b,c) Transmission electron micrographs of PEG5000-CdSecore/ZnSshell QDs after overnight suspension of in 10 mM acetate (pH 4.1) and deposition on an amorphous carbon background. (d) Nanoparticle aggregates obtained after overnight exposure of PEG5000-CdSecore/ZnSshell QDs to the MHQ-driven Fenton reaction. The dotted circle in (d) represents the area used in analysis by selected area electron diffraction (inset) and energy dispersive x-ray spectroscopy (e). The copper signal in (e) is due to the TEM grid.
In most experiments, differences in position and amplitude of the first exciton peak of QDs exposed to H2O2 or classical Fenton’s reaction (H2O2 + Fe2+) barely exceeded measurement uncertainty (± 1 nm wavelength, ± 0.01 absorbance; see the SI). The similarities in first exciton peak positions and absorbances for QDs exposed to H2O2 alone and to classical Fenton’s reagent indicated the amounts of •OH produced by classical Fenton’s reaction were insufficient to degrade PEG5000-QDs to a larger extent than did H2O2 by the end of the 16- to 18-h exposure period. Differences in UV-Visible spectra for QDs exposed to classical Fenton’s conditions and to ascorbic acid-driven Fenton’s reaction (AA-Fenton) also fell within experimental error, indicating AA failed to enhance •OH production to an extent that affected PEG5000-QD size or concentration. This was unexpected as AA was previously used to mimic the extracellular chemistry of brown rot fungi to study phenethyl polyacrylate oxidation (12). In contrast, lack of the first exciton peak in spectra of QDs exposed to MHQ-Fenton’s reaction indicates substantial alteration to QD cores and that methoxyhydroquinone enhances •OH production in a manner strongly impacting PEG5000-QD integrity.
Exposure of CdSecore/ZnSshell QDs to H2O2 induced a blue shift in first exciton peak position similar to that observed upon ~26 h exposure to high intensity UV light, while exposure to the MHQ-Fenton’s reaction produced changes analogous to sustained photo-oxidation on CdSe QDs (9). These data differ from the photo-oxidation experiments in that we employed a ZnS shell on our QDs. Zinc sulfide shells reportedly act as protective barriers to oxidation of the core, preventing release of Cd and Se(11). Contrary to these reports, the ZnS layer of our QDs did not prevent oxidation of the CdSe core by H2O2 or •OH produced by Fenton’s reagent.
Microscopic Investigation of Transformed QDs
To further examine changes to PEGylated QDs induced by exposure to H2O2 and (reductant-assisted) Fenton’s reaction, we separated particles with diameters ≳ 3 nm from solution with a 10-kDa nominal MWCO centrifugal concentrator. Given the particle sizes described above, unless disaggregated and severely degraded, QDs were not expected to pass the filter.
In one experiment, nanoparticles retained by the centrifugal filter were examined by TEM and EDX. Unaltered individual QDs from aqueous suspensions (10 mM acetate) had diameters < 5 nm (estimated estimated average diameters = 3-4 nm; Figure 2b) and had atomic spacing consistent with wurtzite structures. After exposing CdSecore/ZnSshell PEG5000-QDs to the MHQ-Fenton’s reaction, nanoparticles ranging in size from tens to hundreds of nanometers were apparent (Figure 2d). Selected-area electron diffraction demonstrated these particles were amorphous in nature (Figure 2d, inset). EDX revealed Se was the major component in these amorphous particles; trace amounts of Fe remained from the assay solution (Figure 2e). Copper signals were due to the Cu TEM grid; no cadmium signal was evident following PEG5000-QD exposure to the MHQ-Fenton’s reaction.
Metals Released from Transformed QDs
In independent experiments, we passed reaction mixtures through 10-kDa centrifugal filters at the conclusion of exposures, and determined Cd and Zn concentrations in filtrates and retentates by ICP-OES. Figure 3a shows recovered Cd mass normalized to a sample that was neither exposed to oxidants nor centrifuged, but diluted identically to exposed QDs. This sample provides a measure of expected total Cd content of the QDs. Cadmium in the retentate (corresponding to particles ≳ 3 nm) declined as oxidant strength increased (H2O2 vs. •OH from classical Fenton’s reagent) and as hydroxyl radical production accelerated upon addition of a reductant (Figure 3a; data for AA-driven Fenton’s reactions not shown). These data are consonant with enhanced QD degradation by the MHQ-Fenton’s reaction indicated by the UV-Visible absorbance data. For QDs suspended in 10 mM acetate (pH 4.1), nearly all Cd was retained by the centrifugal filter (79 ± 4%); little Cd was present in the filtrate (8.9 ± 0.4%). The MHQ-Fenton’s retentate contained only 19 ± 1%; most Cd passed the centrifugal filter (70 ± 3%). The presence of Cd in the retentate of QDs exposed to the MHQ-Fenton’s reaction contrasts with the UV-Visible spectroscopy data suggesting complete destruction. This discrepancy is only apparent and likely arises from the comparatively high detection limit for QD measurement by UV-Visible absorbance (~30 nM for as-synthesized QDs) coupled with enhanced background absorbance from degraded CdSe cores (e.g., amorphous Se-containing aggregates, Figure 3c). In addition, filter geometry prevented the filter from spinning dry leading to a small bias in the concentration of the retained solution (vide infra).
Figure 3.
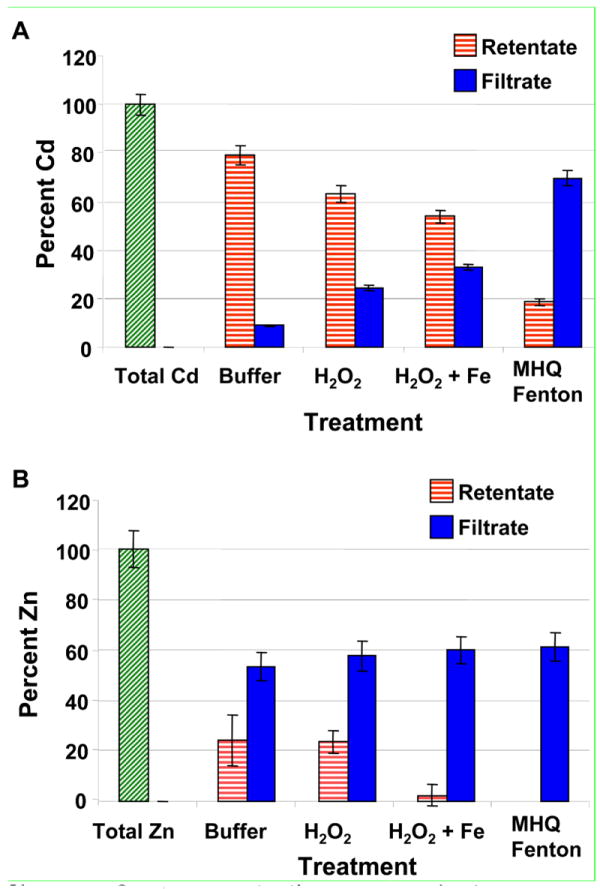
Concentrations of (a) Cd and (b) Zn from PEG5000-QDs exposed to H2O2 and (methoxyhydroquinone-driven) Fenton’s reagent, then filtered through a 10 kDa centrifugal concentrator (retaining particles with diameters > 3 nm). The total metals sample represents a QD suspension that was neither exposed nor filtered. Bars represent means of triplicate reactions, error bars correspond to one standard deviation. Data were normalized to the mass of the metals control (i.e., Total Cd and Zn bars).
Total cadmium recovery (retentate plus filtrate) ranged from 86% to 88%. Centrifugal filtration of CdCl2 solutions demonstrated ~30% of the Cd2+(aq) was lost to the regenerated cellulose filters, regardless of concentration or solution pH. Including Cd2+ losses of this magnitude in our calculations allowed closure of mass balances.
Unlike Cd, substantial amounts of the initial Zn were present in filtrates for all treatments (≥ 54 ± 6%), including buffer controls. While ZnS shells are generally assumed to protect the CdSe core from oxidation at near neutral pH values (11), here the shell dissolved under acidic conditions in the absence of oxidants. Zinc concentrations in filtrates rose as the oxidative potency of solutions increased (pairwise t-tests, p < 0.05). The trend in the amount of Zn in retentates paralleled that observed for Cd: the amount of zinc retained by centrifugal filters generally declined as solution oxidative potency increased. Thus, the Zn and Cd data are consistent and indicate QDs degradation increases as solution oxidative potency rises. Like Cd, zinc mass balances were closed after accounting for Zn2+(aq) losses to filters (~30% in control studies).
Fenton’s reaction is based on decomposition of H2O2 by Fe2+ to generate •OH and anions. Once hydroxyl radicals are produced, however, radical propagation makes the solution chemistry complicated. For example, •OH interaction with H2O2 or Fe+3 produces hydroperoxyl (HO2•) or superoxide anion (O2•−) radicals. These two species are in equilibrium with pKa = 4.8 (24). These radical oxygen species can react with organic molecules to produce a variety of reactive organic radicals. While •OH production by Fenton’s reaction is somewhat slow (reported rate constants range from 41.7 to 120 M-1s-1 at pH 4 (17,25,26)), formation of O2•−/ HO2• from •OH is rapid (rate constant of 3.3 × 107 M-1s-1 at pH 4 (17).
While we do not determine the radical species responsible for QD degradation here, the standard reduction potentials (EH0) for H2O2 (EH0 = +1.77 V, H2O2 + 2H+ + 2e− ↔ 2 H2O), (24) hydroxyl radical (EH0 = +1.90 V, ·OH + e− ↔ OH−) and superoxide/hydroperoxyl radical (EH0 = +0.75, ·HO2 + e− ↔ HO2−) (27) substantially exceed those for bulk ZnS (EH0 = −1.44 V, ZnS(s) + 2e− ↔ Zn0(s) + S2−) and CdSe (EH0 = −1.38 V, CdSe(s) + 2e− ↔ Cd0(s) + Se2−).(28) Using the Nernst equation, at pH 4 in water to mimic the condition used in the assay ({H+} = 10−4 M, {OH−} = 10−10 M, all other reaction components at unit activity), EH0(W, pH 4) is +1.53 V for H2O2 and +2.49 V for ·OH. The reduction potentials for O2−·/HO2 do not vary with pH. Therefore, at the low pH value used, Fenton’s reaction generates an oxidant (hydroxyl radical) with a reduction potential substantially higher than that of H2O2. Adding a reductant that regenerates Fe2+(aq) from Fe3+, sustains Fenton’s reaction and accelerates •OH production. Standard reduction potentials for ZnO and CdS decline as the quantum confinement regime is reached (29), indicating metal oxide and chalcogenide oxidation occurs more readily at the nanoscale. Comparable reports for CdSe are apparently lacking; however, similar size-dependent changes in EH0 are expected.
The ZnS shell is expected to deteriorate in the presence of Fe3+, as ferric iron oxidizes S2− to S0, releasing Zn2+ (Fe3+ + e− → Fe2+, EH0 = 0.771 V; S0 + 2e−→ S2−, EH0 = −0.447 V (28,30,31)). Production of Fe+3 in Fenton’s reactions would likely promote dissolution of the ZnS shell. This process would not have been operative in acetate buffer controls. Under acidic conditions, the ZnS shell could undergo dissolution through the reaction ZnS(s) + H+ ↔ Zn2+ + HS−. The solubility products (*Ks) for bulk wurtzite and sphalerite are 10−8.95 and 10−10.93(32), suggesting that the ZnS shell could dissolve under acidic conditions if the PEG-thiol coating provided insufficient protection from dissolution. Thiol-ligands desorb from CdS QD surfaces at QD concentrations ≤ 10 μM (7), implying they would not act as effective protective barriers under the conditions used here (2 μM CdSecore/ZnSshell QD concentration). Similarly, thiol-attached, self-assembled monolayers on gold are dynamic (i.e., attached ligands continually exchange with those in solution) (33). Gold has been detected in solutions used for functionalization indicating attachment/detachment dynamics can result in removal of surface atoms (33). If similar behavior occurs on ZnS, ligands may not only fail to form a weak protective barrier, but may also contribute to dissolution of the ZnS shell. Regardless of the mechanism, our data indicate better protective barriers are needed, possibly in addition to the ZnS shell, to maintain CdSe core integrity under acidic conditions.
Effect of PEG Chain-length on QD Transformation by MHQ-Fenton’s Reaction
Many biological labeling applications of QDs use short-length thiol-containing ligands as a platform for forming biocompatible conjugates (11,34). However, the photo-oxidation rate monothiol-terminated aliphatic ligands increases as chain length decreases (5). To determine whether the same trend held for oxidation by MHQ-Fenton’s reaction, we used a PEG hexamer (PEG350) to represent shorter chain-length ligands used in biological studies and to allow direct comparison with the PEG5000-QD data.
Initial experiments with PEG350-thiol ligands appeared to suggest shorter chain-length ligands improved stability against oxidation relative to PEG5000. Exposure of PEG350-QDs to MHQ-Fenton’s reaction yielded dots producing an absorption spectrum with a shape and first exciton peak energy indistinguishable from those for H2O2-exposed QDs (Figure 4a). We hypothesized that excess ligand in solution protected PEG350-QDs from oxidation by scavenging hydroxyl radicals, preventing them from attacking the QDs.
Figure 4.
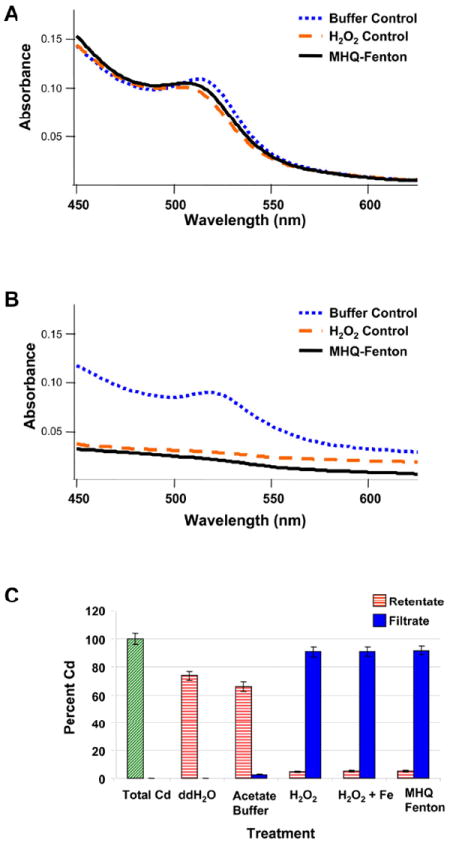
(a) UV-Visible absorption spectra of unfiltered PEG350-QDs exposed to methoxyhydroquinone (MHQ)-driven Fenton’s reagent at pH 4.1 (10 mM acetate). Excess ligand in solution prevented degradation of the PEG350-QDs by hydroxyl radicals generated from the MHQ-driven Fenton’s reaction. (b) UV-Visible absorption spectra of filtered PEG350-QDs exposed to methoxyhydroquinone (MHQ)-driven Fenton’s reagent at pH 4.1 (10 mM acetate). Quantum dot suspensions were filtered prior to exposure to H2O2 or MHQ-Fenton’s reaction to remove excess PEG350 ligand in solution. (c) Concentration of Cd from filtered PEG350-QDs exposed to H2O2 and (methoxyhydroquinone (MHQ)-driven) Fenton’s reagent, then separated through a centrifugal concentrator. The total Cd sample represents a QD suspension that was not exposed or filtered. Zn concentration data are presented in the SI. Bars represent means of triplicate reactions, error bars correspond to one standard deviation.
In subsequent experiments, prior to exposure to MHQ-Fenton’s reaction, PEG350-QD stock suspensions were filtered through centrifugal concentrators to separate QDs from much of the excess ligand. (See the SI for a full discussion) UV-Visible spectra of filtered PEG350-QD in buffer exhibited a first exciton peak centered at ~515 nm (Figure 4b). Upon exposure to H2O2, the UV-Visible spectrum became nearly flat, with absorbance increasing gradually from long to short wavelengths and lacking the characteristic first exciton peak (Figure 4b). Exposure to MHQ-Fenton’s reagent produced a UV-Visible spectrum with a small slope and lacking a first exciton peak (Figure 4b). These results indicate substantial dissolution of PEG350-QD by H2O2 and MHQ-Fenton’s reagent.
To confirm PEG350-QD shell and core degradation, we separated QDs from solution by centrifugal filtration and measured Cd and Zn concentrations in the filtrate and retentate by ICP-OES as described above. As in Figure 3, an unexposed, an unfiltered sample was used to determine the total metals content in QDs, and all recovered masses were normalized to this value. We also examined QDs suspended in ddH2O and otherwise treated like exposed QDs as a control to examine ZnS shell stability in the buffer solution. In pH 4.1 acetate solution, a small amount of Cd was observed in the filtrate (2.4 ± 0.1%); the majority remained in the retentate (66 ± 4%) (Figure 4c). Upon addition of H2O2, classical Fenton’s reagent or MHQ-Fenton’s reagent, the bulk (≥ 91%) of the Cd was present in the filtrate (Figure 4c). Retentate cadmium concentrations did not differ among treatments, nor did those of filtrates (p ≥ 0.05). These results corroborate the UV-Visible data indicating complete dissolution upon exposure. As noted above, discrepancies in Cd mass balances can be resolved by accounting for losses to filters, and the presence of Cd in retentates can be explained given detection limit of the UV-Visible spectrometer and retention to due filter geometry. Data on Zn release are reported in Figure S12.
Environmental Implications
Excess ligand in solution appears to protect QDs from oxidation by H2O2 and •OH. While excess synthetic ligands are unlikely to accompany nanoparticles after release into the environment, association with natural organic matter (NOM) may protect nanomaterials from biogenic oxidants (35). Lignolytic fungi replenish the reagents required to sustain Fenton’s chemistry in their extracellular environment, resulting in continuous •OH production. The extent of QD degradation in soils is expected to reflect a balance between continual •OH production and scavenging by NOM. The MHQ-Fenton assay is designed to provide an initial indication of the environmental stability of nanomaterials under such oxidative environments. The effect of NOM in protecting nanoparticles from oxidative transformation warrants investigation.
Our results suggest PEGylated CdSecore/ZnSshell QDs may be degraded in some aerobic soil environments. This may limit their utility as nanoparticle tracers in environmental fate and transport studies. Surface modifications such as silica shells (36) or amphiphilic polymer wrappings (22) may be needed to prevent oxidative decomposition.
The utility of the MHQ-Fenton assay was illustrated using CdSecore/ZnSshell QDs; however, this assay is applicable to many nanomaterials (application to some nanomaterials may require modification to the assay). Because of the environmental relevance of the conditions employed, this assay could be used to screen new nanomaterials for environmental stability and compatibility. We envision the MHQ-Fenton assay as contributing to a battery of tests that can be used to rapidly obtain initial indications of the propensity of engineered nanomaterials to be transformed in the environment. By examining the stability of new materials during development, researchers gain the opportunity to optimize nanomaterial design and deployment to minimize release into the environment.
Supplementary Material
Acknowledgments
This work was supported by NSF award DMR-0425880. We thank Kenneth Hammel and Chris Hunt for helpful discussions, Paige Wiecinski for laboratory assistance, Jackie Bastyr-Cooper for ICP-OES analysis, and Judith Burstyn and Michael Santiago Cintrón for assistance with fluorescence spectroscopy.
Footnotes
Supporting Information Available. QD synthesis and functionalization methods; FTIR peak assignments for poly(ethylene glycol); XPS spectra; controls for assay development ; DLS results, fluorescence emission spectra, effect of assay at pH 6. This material is available free of charge via the Internet at http://pubs.acs.org.
Literature Cited
- 1.Mueller NC, Nowack B. Exposure modeling of engineered nanoparticles in the environment. Environ Sci Technol. 2008;42:4447–4453. doi: 10.1021/es7029637. [DOI] [PubMed] [Google Scholar]
- 2.Köhler AR, Som C, Helland A, Gottschalk F. Studying the potential release of carbon nanotubes throughout the application life cycle. J Clean Prod. 2008;16:927–937. [Google Scholar]
- 3.Wiesner MR, Lowry GV, Alvarez P, Dionysiou D, Biswas P. Assessing the risks of manufactured nanomaterials. Environ Sci Technol. 2006;40:4336–4345. doi: 10.1021/es062726m. [DOI] [PubMed] [Google Scholar]
- 4.Aldana J, Lavelle N, Wang YJ, Peng XG. Size-dependent dissociation pH of thiolate ligands from cadmium chalcogenide nanocrystals. J Am Chem Soc. 2005;127:2496–2504. doi: 10.1021/ja047000+. [DOI] [PubMed] [Google Scholar]
- 5.Aldana J, Wang YA, Peng XG. Photochemical instability of CdSe nanocrystals coated by hydrophilic thiols. J Am Chem Soc. 2001;123:8844–8850. doi: 10.1021/ja016424q. [DOI] [PubMed] [Google Scholar]
- 6.Smith AM, Duan HW, Rhyner MN, Ruan G, Nie SM. A systematic examination of surface coatings on the optical and chemical properties of semiconductor quantum dots. Phys Chem Chem Phys. 2006;8:3895–3903. doi: 10.1039/b606572b. [DOI] [PubMed] [Google Scholar]
- 7.Dollefeld H, Hoppe K, Kolny J, Schilling K, Weller H, Eychmuller A. Investigations on the stability of thiol stabilized semiconductor nanoparticles. Phys Chem Chem Phys. 2002;4:4747–4753. [Google Scholar]
- 8.Mahendra S, Zhu H, Colvin VL, Alvarez PJ. Quantum Dot Weathering Results in Microbial Toxicity. Environ Sci Technol. 2008;42:9424–9430. doi: 10.1021/es8023385. [DOI] [PubMed] [Google Scholar]
- 9.Yantasee W, Warner CL, Sangvanich T, Addleman RS, Carter TG, Wiacek RJ, Fryxell GE, Timchalk C, Warner MG. Removal of heavy metals from aqueous systems with thiol functionalized superparamagnetic nanoparticles. Environ Sci Technol. 2007;41:5114–5119. doi: 10.1021/es0705238. [DOI] [PubMed] [Google Scholar]
- 10.Dabbousi BO, RodriguezViejo J, Mikulec FV, Heine JR, Mattoussi H, Ober R, Jensen KF, Bawendi MG. (CdSe)ZnS core-shell quantum dots: Synthesis and characterization of a size series of highly luminescent nanocrystallites. J Phys Chem B. 1997;101:9463–9475. [Google Scholar]
- 11.Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Quantum dot bioconjugates for imaging, labeling and sensing. Nat Mat. 2005;4:435–446. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- 12.Cohen R, Jensen KA, Houtman CJ, Hammel KE. Significant levels of extracellular reactive oxygen species produced by brown rot basidiomycetes on cellulose. FEBS Lett. 2002;531:483–488. doi: 10.1016/s0014-5793(02)03589-5. [DOI] [PubMed] [Google Scholar]
- 13.Kerem Z, Jensen KA, Hammel KE. Biodegradative mechanism of the brown rot basidiomycete Gloeophyllum trabeum: evidence for an extracellular hydroquinone-driven Fenton reaction. FEBS Lett. 1999;446:49–54. doi: 10.1016/s0014-5793(99)00180-5. [DOI] [PubMed] [Google Scholar]
- 14.Jensen KA, Houtman CJ, Ryan ZC, Hammel KE. Pathways for extracellular Fenton chemistry in the brown rot basidiomycete Gloeophyllum trabeum. Appl Environ Microb. 2001;67:2705–2711. doi: 10.1128/AEM.67.6.2705-2711.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki MR, Hunt CG, Houtman CJ, Dalebroux ZD, Hammel KE. Fungal hydroquinones contribute to brown rot of wood. Environ Microbiol. 2006;8:2214–2223. doi: 10.1111/j.1462-2920.2006.01160.x. [DOI] [PubMed] [Google Scholar]
- 16.Wood PM. Pathways of production of Fenton reagent by wood-rotting fungi. FEMS Microbiol Rev. 1994;13:313–320. [Google Scholar]
- 17.Duesterberg CK, Waite TD. Process optimization of Fenton oxidation using kinetic modeling. Environ Sci Technol. 2006;40:4189–4195. doi: 10.1021/es060311v. [DOI] [PubMed] [Google Scholar]
- 18.Tang WZ, Huang CP. Stochiometry of Fenton’s reagent in the oxidation of chlorinated aliphatic organic pollutants. Environ Technol. 1997;18:13–23. [Google Scholar]
- 19.Kang EC, Ogura A, Kataoka K, Nagasaki Y. Preparation of water-soluble PEGylated semiconductor nanocrystals. Chem Lett. 2004;33:840–841. [Google Scholar]
- 20.Harris JM, Martin NE, Modi M. PEGylation - A novel process for modifying pharmacokinetics. Clin Pharmacokinet. 2001;40:539–551. doi: 10.2165/00003088-200140070-00005. [DOI] [PubMed] [Google Scholar]
- 21.Yu WW, Qu LH, Guo WZ, Peng XG. Experimental determination of the extinction coefficient of CdTe, CdSe, and CdS nanocrystals. Chem Mat. 2003;15:2854–2860. [Google Scholar]
- 22.Yu WW, Chang E, Falkner JC, Zhang JY, Al-Somali AM, Sayes CM, Johns J, Drezek R, Colvin VL. Forming biocompatible and nonaggregated nanocrystals in water using amphiphilic polymers. JACS. 2007;129:2871–2879. doi: 10.1021/ja067184n. [DOI] [PubMed] [Google Scholar]
- 23.Sperling RA, Liedl T, Duhr S, Kudera S, Zanella M, Lin CAJ, Chang WH, Braun D, Parak WJ. Size determination of (bio)conjugated water-soluble colloidal nanoparticles: A comparison of different techniques. J Phys Chem C. 2007;111:11552–11559. [Google Scholar]
- 24.Hoffmann M, Hotze EM, Wiesner MR. Reactive oxygen species generation on nanoparticulate material. In: Wiesner MR, Bottero J-Y, editors. Environmental Nanotechnology. McGraw-Hill; New York: 2007. pp. 155–203. [Google Scholar]
- 25.Kwan WP, Voelker BM. Decomposition of hydrogen peroxide and organic compounds in the presence of dissolved iron and ferrihydrite. Environ Sci Technol. 2002;36:1467–1476. doi: 10.1021/es011109p. [DOI] [PubMed] [Google Scholar]
- 26.Cheves W. Fenton’s reagent revisited. Acc Chem Res. 1975;8:125–131. [Google Scholar]
- 27.Schwarzenbach RP, Gschwend PM, Imboden DM. Environmental Organic Chemistry. 2. John Wiley & Sons; Hoboken, New Jersey: 2003. [Google Scholar]
- 28.Bard AJ, Parsons R, Jordan J. Standard Potentials in Aqueous Solution. Marcel Dekker; New York: 1985. [Google Scholar]
- 29.Hoyer P, Weller H. Size-dependent redox potentials of quantized zinc-oxide measured with an optically transparent thin-layer electrode. Chem Phys Lett. 1994;221:379–384. [Google Scholar]
- 30.Fowler TA, Crundwell FK. Leaching of zinc sulfide by Thiobacillus ferrooxidans: Bacterial oxidation of the sulfur product layer increases the rate of zinc sulfide dissolution at high concentrations of ferrous ions. Appl Environ Microbiol. 1999;65:5285–5292. doi: 10.1128/aem.65.12.5285-5292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crundwell FK. Kinetics and mechanism of the oxidative dissolution of a zinc-sulfide concentrate in ferric sulfate-solutions. Hydrometallurgy. 1987;19:227–242. [Google Scholar]
- 32.Stumm W, Morgan JJ. Aquatic Chemistry. 3. John Wiley & Sons; New York: 1996. [Google Scholar]
- 33.Ulman A. Formation and structure of self-assembled monolayers. Chem Rev. 1996;96:1533–1554. doi: 10.1021/cr9502357. [DOI] [PubMed] [Google Scholar]
- 34.Chan WCW, Nie SM. Quantum dot bioconjugates for ultrasensitive nonisotopic detection. Science. 1998;281:2016–2018. doi: 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- 35.Hyung H, Fortner JD, Hughes JB, Kim JH. Natural organic matter stabilizes carbon nanotubes in the aqueous phase. Environ Sci Technol. 2007;41:179–184. doi: 10.1021/es061817g. [DOI] [PubMed] [Google Scholar]
- 36.Gerion D, Pinaud F, Williams SC, Parak WJ, Zanchet D, Weiss S, Alivisatos AP. Synthesis and properties of biocompatible water-soluble silica-coated CdSe/ZnS semiconductor quantum dots. J Phys Chem B. 2001;105:8861–8871. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.