

Abstract
Hedgehog Interacting Protein (HHIP) was implicated in chronic obstructive pulmonary disease (COPD) by genome-wide association studies (GWAS). However, it remains unclear how HHIP contributes to COPD pathogenesis. To identify genes regulated by HHIP, we performed gene expression microarray analysis in a human bronchial epithelial cell line (Beas-2B) stably infected with HHIP shRNAs. HHIP silencing led to differential expression of 296 genes; enrichment for variants nominally associated with COPD was found. Eighteen of the differentially expressed genes were validated by real-time PCR in Beas-2B cells. Seven of 11 validated genes tested in human COPD and control lung tissues demonstrated significant gene expression differences. Functional annotation indicated enrichment for extracellular matrix and cell growth genes. Network modeling demonstrated that the extracellular matrix and cell proliferation genes influenced by HHIP tended to be interconnected. Thus, we identified potential HHIP targets in human bronchial epithelial cells that may contribute to COPD pathogenesis.
Keywords: Hedgehog interacting protein (HHIP), Gene expression profiling, COPD (Chronic obstructive pulmonary disease), extracellular matrix (ECM), network modeling
1. Introduction
Chronic obstructive pulmonary disease (COPD), the third leading cause of death in the U.S. [1], is a complex disease strongly influenced by cigarette smoking and genetic predisposition [2, 3] COPD is characterized by emphysematous destruction of the alveoli and thickening of the airway walls in response to chronic exposure to cigarette smoke (CS). While the emphysematous destruction of the lung parenchyma probably results from the cytotoxic and pro-inflammatory activities of CS, the pathogenic mechanisms of the disease remain to be fully defined [4]. It has been shown that smoke-induced cell apoptosis and autophagy are likely to be important causes for lung injury during COPD development; imbalance of proteinases and proteinase inhibitors as well as abnormal tissue repair are other mechanisms likely involved in the pathogenesis of COPD [5].
Genome-wide association studies (GWAS) of COPD have provided compelling evidence for a disease susceptibility locus on chromosome 4q31 [6, 7]. This locus has been significantly associated with lung function in the general population [8, 9] as well as with COPD [10, 11]. Our previous work has identified the gene encoding hedgehog interacting protein (HHIP) as the causative gene for this highly replicated genetic association with COPD susceptibility [12]. Specifically, we have shown that SNPs upstream of the HHIP gene modulate expression of HHIP, implicating reduced HHIP gene expression in the pathogenesis of COPD [12]. However, the biological mechanisms by which HHIP influences COPD susceptibility have not been determined.
Hhip is a cellular membrane protein that directly binds to Sonic Hedgehog (Shh), Indian Hedgehog (Ihh) and Desert Hedgehog (Dhh), which trigger the hedgehog signaling pathway by binding to their membrane receptor, Ptch1. By competing with Ptch1 for binding of hedgehog ligands, Hhip negatively regulates the hedgehog pathway, a crucial pathway for the development of the lungs and other organs [13–15].
To study the biological pathways connecting HHIP to COPD pathogenesis, we determined the effect of HHIP knock-down through lentivirus-based shRNAs on gene expression in the bronchial epithelial cell line Beas-2B, which has detectable expression of HHIP at a relatively higher level than primary human airway epithelial cells (data not shown). Gene expression differences detected by expression array were validated by RT-PCR in Beas-2B and normal human bronchial epithelial cells (NHBE). In these systems, we evaluated the activity of the canonical Hedgehog pathway as well as other novel biological pathways identified by gene expression array analysis.
2. Results
2.1. Expression profiling in HHIP depleted Beas-2B cells
To identify the full range of cellular targets of HHIP in human bronchial epithelial cells, we performed expression microarray analysis in Beas-2B cells with and without knock-down of HHIP by stable infection with lentivirus-based shRNAs targeting HHIP. To limit possible off-target effects of the shRNAs, four different HHIP shRNAs were used, with up to four replicates for each shRNA (Figure 1B). Efficiency of HHIP knockdown was shown by at least 70% reduction in HHIP mRNA in Beas-2B cells after infection with HHIP shRNAs compared to cells infected with non-targeting control shRNA (NT) (data not shown).
Figure 1.
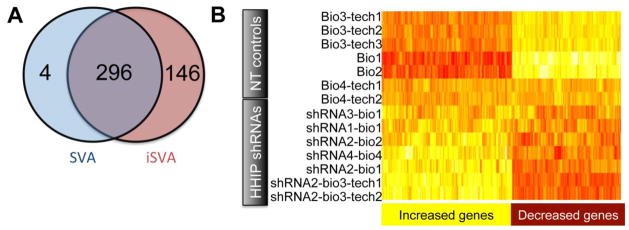
Gene expression profiling analysis in Beas-2B cells stably infected with HHIP shRNAs. A. Venn diagram showing the number of differentially expressed genes based on iSVA or SVA. B. Expression heat map of 296 differentially expressed genes (columns) (p adjusted <0.05). Each row represents a separate sample (array). Bio: biological replicate; tech: technical replicate; NT shRNA: non-targeting shRNA control; shRNA: HHIP-targeting shRNA
Since there is no consensus in the literature regarding the optimal analytical approach for batch effect analysis of gene expression array data, two methods were used. Surrogate variable analysis [16] and its modification, independent surrogate variable analysis (iSVA) [17] coupled with the linear regression models with an empirical Bayes shrinkage method (limma), found 317 gene probes (representing 300 unique genes) and 469 gene probes (442 genes) respectively, to be differentially expressed (p adjusted <0.05) in HHIP shRNA-infected Beas-2B cells compared to non-targeted shRNA-infected control cells. Of note, 296 genes were identified as showing differential gene expression by both analysis methods (Figure 1A). Figure 1B depicts the differential gene expression pattern for different biological and technical replicates with and without HHIP silencing with four individual HHIP-targeted shRNAs, showing that the effect of HHIP knock-down on gene expression is consistently greater than technical batch effects. The top 20 differentially expressed genes identified by iSVA analysis are shown in Supplemental Table 3. As expected, HHIP ranks first on this list. Protein level reductions of Hhip caused by shRNAs were confirmed by Western Blot (Supplemental Figure 1A).
2.2. Genetic Association Analysis of Differentially Expressed Genes
To assess the biological importance of these differential gene expression signals, we evaluated the affected genes for common variant associations with COPD based on our previously published genome-wide association study of four cohorts: The National Emphysema Treatment Trial/Normative Aging Study, GenKOLS, ECLIPSE, and COPDGene (first 1000 subjects) [18].
Among the 296 differentially expressed genes after HHIP silencing (including the HHIP genic region but not the COPD GWAS region upstream from the HHIP gene), 271 were located on autosomes and harbored high-quality imputed or genotyped SNPs within a window of 5kb upstream and 1kb downstream from the transcribed gene boundaries. One of the 271 genes, RAB4B is located within the chromosome 19 GWAS region shown to be strongly associated with COPD. While no other SNPs located within the 271 genes showed an association with COPD that reached genome-wide significance after a conservative Bonferroni correction within this subset, 115 genes contained at least one SNP showing association with COPD with a nominal P value of < 0.05. Based on the method by Lipman and colleagues [19], these results are consistent with a significant enrichment in low P-values (P = 0.02). A histogram plot of LD-pruned p-values used for the Lipman analysis demonstrated the enrichment of lower p values (Supplemental Figure 2), supporting a role of these differentially expressed genes in COPD pathogenesis.
2.3. Extracellular matrix genes are differentially expressed by HHIP silencing
Next, we evaluated enrichment of the 296 genes differentially expressed after HHIP silencing in Gene Ontology (GO) biological processes, using DAVID [20] functional annotation analysis (http://david.abcc.ncifcrf.gov/home.jsp) and GeneMANIA (http://www.genemania.org/) [21]. Both found significant enrichment (P < 0.01, using DAVID or GeneMANIA) of genes involved in extracellular matrix remodeling (ECM, Table 1). Based on these pathway analysis results, we used SYBR-green based RT-PCR analysis to attempt to validate differential expression of 18 ECM-related genes in Beas-2B cells infected with three HHIP shRNAs or one control shRNA (Table 2).
Table 1.
DAVID and GeneMANIA functional annotation clusters for genes regulated by HHIP
| DAVID | |||||
|---|---|---|---|---|---|
| Clusters | Enrichment score | Top GO term | Top term | Gene count | |
| Fold enrichment | P value | ||||
| 1 | 2.12 | GO:0031012~extracellular matrix | 2.93 | <0.001 | 14 |
| 2 | 1.75 | GO:0047115~trans-1,2- dihydrobenzene-1,2-diol dehydrogenase activity | 50.45 | 0.0013 | 4 |
| 3 | 1.74 | GO:0042612~MHC class I protein complex | 12.89 | <0.001 | 5 |
| GeneMANIA | |||||
| Clusters | GO annotation | FDR | coverage query genes | ||
| 1 | extracellular matrix | 1.82E-04 | 15/178 | ||
| 2 | proteinaceous extracellular matrix | 1.82E-04 | 12/105 | ||
| 3 | DNA conformation change | 4.86E-04 | 11/97 | ||
Table 2.
Effect of HHIP knock-down on expression of selected ECM genes, as measured by microarray and SYBR-green based RT-PCR.
| Gene symbol | Gene name | Microarray | RT-PCR | |||
|---|---|---|---|---|---|---|
| Statistics* | adjusted p-value | Mean§ | SEM§ | p- value† | ||
| OLFML2 | Olfactomedin-like 2A | 4.63 | 0.02 | 1.22 | 0.11 | 0.213 |
| TIMP1 | TIMP metallopeptidase inhibitor 1 | 4.21 | 0.04 | 1.01 | 0.05 | 0.96 |
| EFEMP2 | EGF-containing fibulin-like extracellular matrix protein 2 | 4.94 | 0.017 | 1.16 | 0.11 | 0.45 |
| SPARC | Secreted protein, acidic, cycsteine-rich (osteonectin) | 4.39 | 0.03 | 0.92 | 0.11 | 0.44 |
| BGN | biglycan | 4.63 | 0.02 | 1.31 | 0.24 | 0.21 |
| NTN4 | Netrin 4 | 4.01 | 0.049 | 0.58 | 0.08 | 0.0016 |
| COL6A2 | Collagen type VI, alpha2 | 5.25 | .0.013 | 1.13 | 0.06 | 0.19 |
| GSN | Gelsolin | 5.95 | 0.006 | 1.37 | 0.14 | 0.1 |
| ECM1 | Extracellular matrix protein 1 | 4.68 | 0.02 | 1.39 | 0.14 | 0.095 |
| FBLN7 | Fibulin 7 | 5.78 | 0.007 | 2.07 | 0.21 | <0.001 |
| SERPING1 | serpin peptidase inhibitor, clade G (C1 inhibitor), member 1 | 5.60 | 0.009 | 4.35 | 1.88 | 0.0039 |
| PI3 | peptidase inhibitor 3, skin-derived | 4.19 | 0.039 | 3.50 | 1.18 | 0.077 |
| RECK | reversion-inducing-cysteine-rich protein with kazal motifs | 6.12 | 0.005 | 2.01 | 0.17 | <0.001 |
| TULP3 | Tubby like protein 3 | 6.56 | 0.004 | 1.47 | 0.07 | <0.001 |
| SERPINB7 | serine (or cysteine) proteinase inhibitor, clade B (ovalbumin), member 7 | −5.33 | 0.012 | 0.20 | 0.03 | <0.001 |
| WNT5B | Wingless-type MMTV integration site family, member 5B | −5.18 | 0.014 | 0.34 | 0.07 | <0.001 |
| COL13A1 | Collagen, typeXIII, alpha 1 | −5.14 | 0.014 | 0.59 | 0.09 | 0.0249 |
| SDC1 | syndecan 1 | −4.56 | 0.026 | 0.50 | 0.11 | <0.001 |
Moderated t-test statistic testing for the hypothesis that a gene probe is differentially expressed between HHIP hairpins arrays and control hairpin arrays adjusting for the 2 estimated independent surrogate variables (iSVA).
mean and standard error of mean (sem) of log2 fold change of hairpins versus controls.
p-value for testing if a gene probe is differentially expressed between HHIP hairpins and NT controls based on linear mixed effects models. Genes with p<0.01 were regarded as significantly differentially expressed genes validated by RT-PCR, which were highlighted in bold.
Using a linear mixed effects (LME) model (see details in Supplemental Materials and Methods section), we found that SYBR-green RT-PCR expression analysis confirmed the microarray results for seven genes, demonstrating significantly (P<0.01) increased (FBLN7, SERPING1, RECK, TULP3) or decreased (SERPINB7, WNT5B, SDC1) mRNA levels following HHIP silencing in RT-PCR (Table 2). For one gene, NTN4 (Netrin 4), SYBR-green RT-PCR results contradicted the microarray results. For the remaining ten genes, expression changes measured by SYBR-green RT-PCR did not reach statistical significance, although a trend was noted for PI3 (P=0.077). Furthermore, TaqMan-based RT-PCR confirmed the significant or near-significant SYBR-green based RT-PCR results, except for NTN4, which showed no statistically significant expression changes (Figure 2). Similar results were obtained when TBP was used as an additional reference gene (data not shown).
Figure 2.

Significantly increased (A) and decreased (B) expression (**, p<0.01) of HHIP target genes associated with extracellular matrix (ECM) after HHIP silencing in Beas-2B cells, as measured by TaqMan assay-based RT-PCR. Three different HHIP-targeted shRNAs were used for knock-down. Relative expression values were calculated as described in Materials and Methods and represent means ± standard errors from two independent shRNA infections.
2.4. Hedgehog pathway activity lacks significant changes in HHIP silenced Beas-2B cells
Hhip has been shown to inhibit the Hedgehog pathway by competitively binding to three Hedgehog morphogens (Shh, Ihh and Dhh) during development [13]. Surprisingly, we did not observe a significant enrichment of canonical Hedgehog pathway members other than HHIP among genes that were found to be differentially expressed by microarray analysis after HHIP knockdown. To further explore the Hedgehog pathway activity in Beas-2B cells with HHIP silencing, we used TaqMan based qPCR to measure the expression of five genes coding for critical components of the canonical Hedgehog pathway (SHH, PTCH1, GLI1, GLI2, GLI3, IHH, and DHH [22]) in Beas-2B cells stably infected with two of the HHIP shRNAs that were used in the microarray analysis.
No statistically significant expression changes were detected for PTCH1 or GLI3, and SHH expression was under the detection limit in Beas-2B cells (Figure 1B); this low expression level of SHH in airway cells was confirmed in primary small airway epithelial cells (data not shown). GLI1 demonstrated increased expression with HHIP shRNAs but was still expressed at a relatively low level. In addition, GLI2, previously reported to regulate cell proliferation during lung development [23], demonstrated a statistically significant reduction of 20–30% in expression after HHIP knock-down with any of the three HHIP shRNAs (Supplemental Figure 1B). Two other Hedgehog pathway ligands, IHH and DHH, were also barely detected in Beas-2B cells by qPCR at baseline and after HHIP silencing (data not shown).
These qPCR data indicate that, HHIP silencing by shRNAs had a limited effect on canonical Hedgehog pathway members in our current experimental settings. Hence, these minimal changes of the canonical Hedgehog pathway may not explain all of the other gene expression changes that we observed. These results raised the possibility that other biological pathways may be influenced by silencing HHIP.
2.5. Cell proliferation-related genes have reduced gene expression upon HHIP silencing
Ten differentially expressed genes, including two additional genes identified by the iSVA analysis method [BDNF (brain-derived neurotrophic factor) and BTG2 (B-cell translocation gene 2)], associated with cell growth were chosen for further validation by TaqMan based RT-PCR in Beas-2B cells (Figure 3A). Two cell-cycle related genes, CCND1, encoding Cyclin D1, and G0S2 (G0/G1 switch regulatory protein 2) demonstrated more than 50% reduced expression by HHIP shRNAs in Beas-2B cells.
Figure 3.

Genes associated with cell growth that demonstrated significant differential expression (p<0.05) after HHIP-knock-down in Beas-2B (A) or normal human bronchial epithelial cells (NHBE) (B), as measured by TaqMan assay-based RT-PCR. Three different HHIP-targeted shRNAs were used for knock-down. Relative expression values were calculated as described in Materials and Methods and represent means ± standard errors from two (A) or three (B) replicates for each hairpin. P values in figure 3B are calculated based on two biological replicates and one is represented here. C. Expression of CCND1, TGM2 and BIRC5 in Beas-2B cells infected with HHIP-targeted or non-target (NT) control shRNA, as detected by western blot. α-actin was used as loading control. Band density was quantified using Image J and then normalized to α-actin control.
Other significantly decreased genes include FGF2 (fibroblast growth factor 2), BIRC5 (baculoviral IAP repeat containing 5), TGM2 (transglutaminase 2), CREB3L2 (cAMP responsive element binding protein 3-like 2), and ATF5 (activating transcription factor 5), which are all known to promote cell growth and/or survival ([24], [25], [26], [27]), as well as LETM1 (leucine zipper-EF-hand containing transmembrane protein 1) and BDNF.
In contrast, BTG2, the only anti-proliferation gene included here [28], demonstrated significantly increased expression up to ~5 fold in HHIP shRNA-infected cells. For BIRC5, BDNF, FGF2 and BTG2, the effect of HHIP knock-down on expression levels was validated in primary human bronchial epithelial cell lines (NHBE, Lonza) (Figure 3B). We attempted to validate protein level changes for three of our differentially expressed genes, CCND1, TGM2 and BIRC5; we detected protein level reductions in Beas-2B cells infected with HHIP shRNAs for all three of these genes (Figure 3C).
2.6. HHIP target genes are also differentially expressed in human COPD lung tissues
We have previously shown that HHIP expression is reduced in human COPD compared to control lung tissues [12]. To provide additional evidence that HHIP target genes associated with cell proliferation or ECM are relevant to COPD pathogenesis, we examined expression of 11 selected HHIP target genes in human COPD and control lung tissues from the Lung Tissue Research Consortium (LTRC) by RT-PCR to determine whether COPD is associated with expression patterns similar to those seen following knock-down of HHIP in vitro.
Among-ECM related genes, PI3 showed a trend of increased expression in human COPD lung tissues compared to control tissues from ex-smokers (p=0.08); while WNT5B and SDC1 demonstrated significantly decreased expression in human COPD lung tissues (Figure 4A, p<0.01, unpaired t test), recapitulating the pattern seen after HHIP knock-down in cell culture. Among genes associated with cell growth, CREB3L2, CCND1, ATF5 and LETM1 showed significantly decreased expression (p<0.05) and TGM2 showed a trend for decreased expression (p=0.052) in human COPD lung tissues compared with controls, again recapitulating the effect of HHIP knock-down in cell culture.
Figure 4.
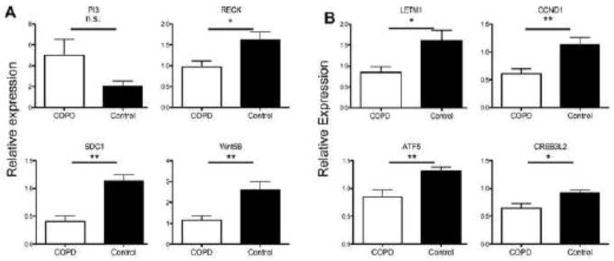
Expression of HHIP target genes associated with ECM (A) or cell growth (B) were measured in human COPD and control lung tissues by RT-PCR. Relative expression values were calculated as described in Materials and Methods and represent means± standard errors for COPD (n=15) and Controls (n=18). **p<0.01 and *p<0.05. n.s., non-significant.
However, for RECK (Figure 4A, which was statistically significant in the direction opposite from our expectation based on RNAi studies), G0S2 and BTG2 (data not shown), the expression pattern in our set of COPD and control lung tissues did not confirm the statistically significant changes observed after HHIP knock-down. Overall, these data suggest that most of the novel Hhip targets identified in this study may participate in human COPD pathogenesis.
Furthermore, we measured gene expression of Sonic Hedgehog pathway members including SHH, PTCH1, GLI1, GLI2 and GLI3 in human lung tissue samples that contain mixed cell populations. There are relatively low expression levels of GLI1 and SHH in human lung tissues, consistent with previous reports [29]. Compared to samples from control smokers, we detected significantly decreased expression of SHH, PTCH1, GLI1, GLI2, and GLI3 (Supplemental Figure 3) in lung tissue samples from COPD subjects. Hence, the canonical Hedgehog pathway might participate in COPD pathogenesis in other cell types (or in epithelial cells after cellular injury) [29].
2.7. Network building based on Beas-2B cell microarray analysis
To understand the relationships between the genes found to be regulated by HHIP, we performed data mining and curation using the Predictive Networks web application [30] with default parameters. Top differentially expressed genes (n=442) with p-adjusted <0.05 found using iSVA after HHIP silencing were used as seeds for network analysis. The resulting network includes 7489 connections among 1958 genes, including 87 genes that were differentially expressed by HHIP shRNAs (highlighted in red circles in Supplemental Figure 4). In this network, gene clusters centered on some validated target genes such as CCND1, SERPING1, and RECK were observed (supplemental Figure 4).
Next, we asked whether the network showed interconnections between HHIP target genes associated with cell growth and extracellular matrix. Within this network analysis model, we found several primary and secondary connections between the 18 validated cell proliferation and extracellular matrix genes (Figure 5); in addition, 26 additional differentially expressed genes showed direct interactions with the 18 validated genes, and 220 differentially expressed genes are within two degrees of separation from the validated genes. The large number of connections observed between differentially expressed genes suggests that the genes which have differential expression after HHIP knock-down may have shared biological functions.
Figure 5.
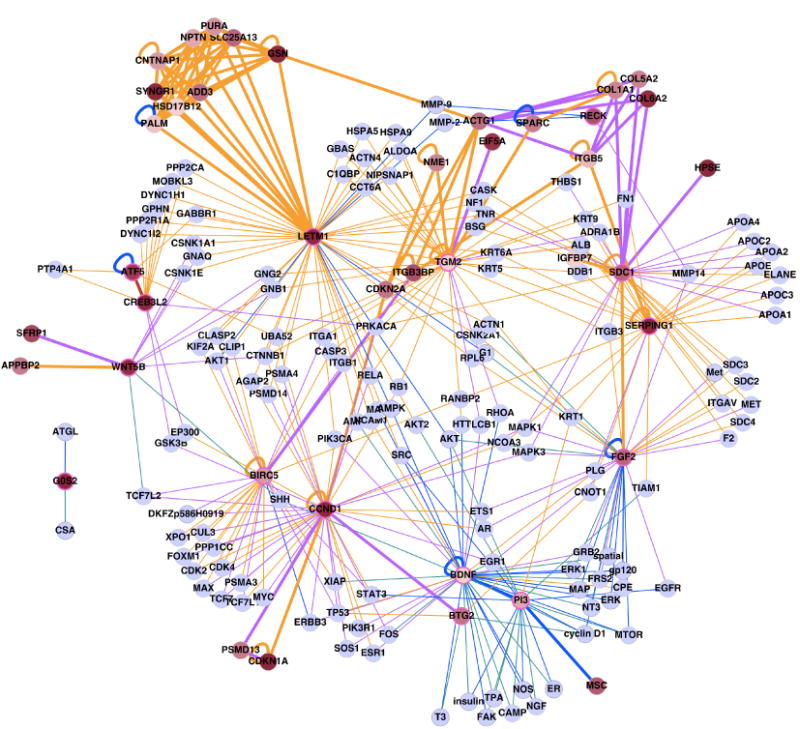
Network analysis of validated HHIP targets related to ECM and cell proliferation (highlighted by pink circles). Edge colors represent sources of the interaction (brown: Pathway Commons, purple: functional interaction, blue: PubMed abstract, teal: Medline abstract, red: manual curation based on recently published literature) and node colors represent the level of differential expression (red: highly differential, gray: no expression change.) Thickened edges connect pairs of differentially expressed genes.
To estimate the statistical significance of the number of connections between ECM and proliferation genes seen in our network, we generated 10,000 gene sets of the same size as in our validated gene set, drawing randomly from the genome; for each, we generated literature-based networks using the network reconstruction protocol described previously. While the number of connections between ECM genes in our validated gene set is not significant (p=0.4, Supplemental Figure 5A left panel), the number of connections between cell proliferation genes is significantly greater than expected by chance (p<0.01, Supplemental Figure 5A middle panel).
Given the representation of ECM and proliferation genes in the validated gene set, the enrichment of connections between these genes may not be surprising. To test the statistical significance of the observed associations, we constructed 10,000 random networks consisting of only ECM genes (N=7) and proliferation genes (N=11). Based on this analysis, the connections within the ECM gene subset, and within the cell proliferation gene subset, compared to the random networks are not significant (p=0.67 and 0.09, respectively). However, the number of observed connections between ECM genes and cell proliferation genes is significantly greater than we would expect by chance (p=0.0035, Supplemental Figure 5B), suggesting that we may have discovered important new connections between the ECM and proliferative processes in the cell. Using GO annotations, 12 of the 18 validated genes are connected to cell proliferation genes, and 9 are connected to at least one extracellular matrix modification gene. For example LETM1 (a cell proliferation gene) has connections with MMP2 and MMP9, which are inhibited by RECK (extracellular matrix genes).
3. Discussion
Genetic variants near HHIP have been consistently associated with COPD susceptibility, and a functional variant influencing HHIP expression has been previously reported by our group [12]. Based on these genetic association results, we hypothesized that HHIP would influence COPD susceptibility through the Hedgehog pathway. The Hedgehog (HH) signaling pathway is critical for organogenesis and carcinogenesis [22].
Vertebrate organisms have three types of Hedgehog ligands: Sonic Hedgehog (SHH), Indian Hedgehog (IHH), and Desert Hedgehog (DHH). Binding of the Hedgehog ligand to its membrane receptor, protein patched homolog 1 (PTCH1), triggers a series of cytoplasmic signaling cascades and eventually activates transcription factors GLI1, GLI2, and GLI3 [22]. HHIP, through its extracellular domain, binds to all three types of HH ligands and inhibits HH signaling [13, 31, 32].
In adults, detectable expression of HHIP was previously found in both whole lung [12] and primary bronchial epithelial cells (unpublished data). However, in our study, SHH was barely detectable in both Beas-2B and primary small airway epithelial cells. Moreover, induced expression of PTCH1 and GLI1 are usually regarded as hallmarks for the activation of the canonical Hedgehog pathway. Nonetheless, no significant expression changes of PTCH1 or GLI3 were observed after HHIP knockdown, and GLI1 demonstrated increased but still relatively low expression in Beas-2B cells with HHIP silencing.
Only a subset of the HHIP targets identified here, such as TULP3 [33] and CCND1 [23, 33], are known targets of the Hedgehog pathway. Based on these observations, it is unlikely that the canonical Hedgehog pathway can explain all of the gene expression changes that we observed in the Beas-2B cells treated with HHIP shRNAs. However, given the complexity of the Hedgehog pathway, which includes both ligand-dependent and ligand-independent activation [34, 35] mechanisms, more comprehensive evaluations are needed to address whether Hedgehog signaling regulates expression of these novel HHIP targets in human bronchial epithelial cells through non-canonical pathways. Interestingly, when we assessed the canonical Hedgehog pathway in human COPD lung tissues by qPCR targeting the major pathway components: SHH, PTCH1, GLI1, GLI2 and GLI3 (Supplemental Figure 3), we detected overall reduced pathway activity in COPD subjects compared to controls. This may indicate a plausible role of the Hedgehog pathway in human COPD. Based on the gene expression levels that we observed in lung epithelial cell lines, we speculate that the involvement of the Hedgehog pathway in COPD could be mediated in other cell types or in lung epithelial cells only after cellular injury.
We found 296 genes with significant alterations in their expression levels after HHIP silencing in Beas-2B cells. The occurrence of the RAB4B gene (within the chromosome 19 COPD GWAS region) and the observed enrichment of other COPD genetic association signals which do not reach conventional levels of genome-wide significance suggest that SNPs within other members of this set of differentially expressed genes could influence COPD susceptibility. Further work to clarify which of these genes are associated with COPD in larger sample sizes and other study designs (e.g., eQTL analysis) could lead to the identification of novel genes for COPD susceptibility that interact with HHIP.
Our study provides some helpful insights regarding the mechanisms through which HHIP may affect COPD susceptibility. In particular, the results shown here suggest that HHIP may influence an individual’s susceptibility to smoke-induced COPD by regulating extracellular matrix (ECM) remodeling and cell growth pathways. The expression levels of multiple extracellular matrix-related genes were significantly changed by HHIP shRNAs based on gene enrichment analysis.
Additionally, a subset of these ECM-related genes also demonstrated significant differential expression in human COPD lung samples. For example, SDC1 expression was significantly decreased in both Beas-2B cells with HHIP silencing and in human COPD lungs. SDC1 encodes a heparan sulfate proteoglycans transmembrane protein that is an important component of ECM as well as regulating matrix assembly [36]. It also protects lung epithelial cells from injury by removing C-X-C chemokines during inflammation [37].
Another differentially expressed gene, PI3/Elafin, is an inhibitor for neutrophil elastase, while RECK (a membrane-bound protein) can exert inhibitory effects on the transcription, synthesis, activation, and activity of MMPs, including MMP2, MMP7, and MMP9 [38–41]. It is not entirely surprising to detect significant expression changes of RECK in human COPD lungs given its crucial roles in regulating the activities of MMPs, and the importance of imbalance between proteinases and anti-proteinases in human lungs contributing to the pathogenesis of COPD/emphysema.
However, how HHIP affects expression of ECM-related genes requires further mechanistic investigations. It is possible that HHIP, as a cellular membrane protein [13], directly cross-talks with other cellular transmembrane proteins such as Syndecan-1 to facilitate its anti-inflammatory and extracellular matrix functions.
Among the most significant differentially expressed genes after HHIP silencing found by microarray analysis, we used RT-PCR to confirm reductions in expression of several cell cycle and proliferation-related genes, G0S2, CCND1, FGF2, BIRC5 and TGM2, as well as increased expression of an anti-proliferation gene, BTG2. This apparent regulation by HHIP of cell growth genes in Beas-2B cells (Figure 3) is consistent with previous findings in HHIP conditional transgenic mice that over-expression of HHIP lacking the transmembrane domain led to hyper-proliferation of small intestine epithelium [42].
Moreover, GLI2 has been reported to regulate lung epithelial growth by modulating Cyclin expression including Cyclin D1 [23], consistent with our findings of decreased expression of CCND1 and GLI2 (Supplemental Figure 1B) with HHIP shRNAs. Given that the HHIP gene locus was also associated with height in multiple populations [43], it is conceivable that HHIP may regulate cell growth and proliferation.
In addition, apoptosis has been implicated in COPD pathogenesis [44]. HHIP target genes FGF-2 [45] and ATF5 [27] positively regulate expression of Bcl-2 anti-apoptotic protein, which showed decreased expression accompanied by increased pro-apoptotic protein Bax expression in cigarette smoke (CS) induced cell death in vitro [46] and in vivo [47]. Additionally, BIRC5, a potent anti-apoptosis protein, interferes with cell death through caspase-dependent and independent mechanisms [48]. Hence, gene expression changes including decreased expression of FGF2, ATF5 and BIRC5 in HHIP-depleted cells may contribute to smoke-induced lung injury due to enhanced cell apoptosis.
When we used a recently developed network modeling approach to analyze our microarray results, we found not only that most of our validated hits are clustered in this “predictive network” (Supplemental Figure 4) but also that there are direct or indirect connections (0–1 connectors) between our validated cell proliferation and ECM-related genes (Figure 5), suggesting that the genes in these two pathways that are perturbed by HHIP knock-down may have shared biological functions. This network analysis included a variety of genes which have been previously implicated in COPD pathogenesis, but which were not directly identified by our gene expression analysis after HHIP silencing. For example, EGR1 was identified as a key differentially expressed gene in a microarray gene expression analysis of human COPD and normal lung tissues [49]. Multiple members of the integrin family also appear in this HHIP network, including ITGAV, which has been implicated in TGF-β activation [50]. Thus, the network identified after HHIP depletion could relate directly to previously reported components of COPD pathobiology.
Nonetheless, there are some limitations related to our study. First, the gene expression changes induced by HHIP shRNAs may result from both primary effects of genetic perturbation of HHIP expression or the downstream signaling events related to these primary effects. Second, only a subset of the differentially expressed genes identified by microarray analysis were validated by RT-PCR; further work will be required to determine whether these inconsistencies represent false-positive microarray results or false-negative validation attempts. Finally, it is possible that the mixed cell population in whole lung homogenates from COPD subjects, phenotypic heterogeneity within COPD subjects, and the moderate sample size of our COPD case and control lung tissue population accounts for our ability to confirm some, but not all, of the gene expression differences that we observed in cell-based gene expression analysis.
4. Conclusion
In summary, we have applied transcriptome profiling in human bronchial epithelial cell lines that were silenced with HHIP shRNAs to identify novel targets of the HHIP gene that may participate in the pathogenesis of human COPD. Our whole-transcriptome analysis suggests novel pathways by which HHIP may affect COPD pathogenesis. Further functional validation of the roles of these intriguing targets of HHIP in smoke-induced murine COPD models is warranted.
5. Materials and methods
5.1. Cell culture and human lung tissues
Human Beas-2B bronchial epithelial cells (#CRL-9609) were purchased from ATCC and cultured in complete DMEM medium supplemented with 10% fetal bovine serum, penicillin (50 units/ml), streptomycin (50 μg/ml) and gentamicin (10 μg/ml). Normal Human Bronchial Epithelial cells (NHBE) from healthy non-smoker donors and medium for culturing NHBE were purchased from Lifeline, Inc.
Human lung tissue samples from 15 COPD patients (FEV1<80%) and 18 control subjects with normal lung function were obtained from the NHLBI Lung Tissue Research Consortium (LTRC), a national biorepository of lung tissues.
5.2. Gene silencing of HHIP by RNA interference in Beas-2B cells
Four individual HHIP-targeting shRNAs and one non-targeting scrambled shRNA (Open Biosystems, Thermo Scientific) were transfected into 293T cells for packaging into lentivirus particles. Beas-2B cells were infected with lentivirus-based shRNAs using standard methods [49]. Approximately 24 hours post-infection, puromycin was added at 1.5 μg/ml for 3–4 days to select for successfully infected cells before RNA and protein extraction.
5.3. Detection of differential gene expression
Total RNA from Beas-2B cells was extracted using the RNeasy Mini Kit (Qiagen, Germantown, MD). Differential gene expression detected by gene expression microarrays was validated by real-time PCR with gene-specific primers on an ABI 7900 HT instrument (Applied Biosystems, Foster City, CA), using SYBR Green for detection. If statistically significant expression changes were confirmed, RT-PCR was repeated using TaqMan probes for detection. Relative gene expression was calculated based on the standard 2−ΔΔCT method, using GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) and control cells as references. TBP (TATA box binding protein) was also used as a reference gene in all TaqMan based RT-PCR validations. Primer and probe sequences are listed in the Supplemental Table 1 and 2.
5.4. Network analysis
Unbiased, automated literature mining was performed on the differentially expressed genes (adjusted p-value < 0.05) using the Predictive Networks application [30]. We gathered gene pairs representing genetic interactions, regulatory functions, and pathways collected from PubMed and Medline abstracts and other published sources. Networks were visualized by Cystoscape (http://www.cytoscape.org). A sub-network centered around the RT-PCR validated genes was produced to illustrate connections between the differentially expressed genes and cell proliferation/survival and extracellular matrix functions. First, only validated genes and their interactions were selected. Then, all non-differentially expressed genes with one interacting partner were removed, retaining only genes that connect to differentially expressed genes.
To estimate the statistical significance of the number of connections to ECM and proliferation genes seen in our network, we generated 10,000 random gene sets of the same size (18) as in our validated gene sets and used them to generate literature-based networks using the network reconstruction protocol described previously. We then counted the total number of connections in that network, the number of connections between ECM genes (defined by GO:0005576 and GO:0005615 classes), the number of connections between cell proliferation genes (defined by GO:0008283), and the number of connections between ECM and cell proliferation genes (excluding internal connections among ECM genes and connections among proliferation genes) for each of the random networks. The empirical p-values for enrichment are calculated by taking the fraction of random networks having a greater number of connections than the observed values. This was repeated with random gene sets generated by using the same numbers of ECM genes (7) and proliferation genes (11) as in the validated gene set.
5.5. Additional details are provided in the online supplemental material and methods
Supplementary Material
Highlights.
Gene expression analysis was performed after silencing HHIP, a COPD GWAS gene.
SNPs near differentially expressed genes were nominally associated with COPD.
Extracellular matrix and cell growth genes were enriched and tend to be interconnected.
Seven differentially expressed genes showed significant changes in human COPD lungs.
Novel targets of HHIP that may contribute to COPD pathogenesis are identified.
Acknowledgments
This work was supported by U.S. National Institutes of Health (NIH) grants R01 HL075478, R01 HL084323, P01 HL083069, P01 HL105339, U01 HL089856 (Silverman), U01 HL089897, and P01 H105339. The National Emphysema Treatment Trial was supported by the National Heart, Lung, and Blood Institute, the Centers for Medicare and Medicaid Services and the Agency for Healthcare Research and Quality. The Normative Aging Study is supported by the Cooperative Studies Program/ERIC of the US Department of Veterans Affairs and is a component of the Massachusetts Veterans Epidemiology Research and Information Center (MAVERIC). The Norway GenKOLS study (Genetics of Chronic Obstructive Lung Disease, GSK code RES11080), the ECLIPSE study (clinicaltrials.gov identifier NCT00292552; GSK code SCO104960) and the ICGN study are funded by GlaxoSmithKline. The COPDGene project is also supported by the COPD Foundation through contributions made by an Industry Advisory Board comprised of AstraZeneca, Boehringer Ingelheim, Novartis, Pfizer and Sunovion. This study used biological specimens and data provided by the Lung Tissue Research Consortium (LTRC) supported by the National Heart, Lung, and Blood Institute (NHLBI).
The members of the COPDGene® study group include: Ann Arbor VA: Jeffrey Curtis, MD (PI), Ella Kazerooni, MD (RAD). Baylor College of Medicine, Houston, TX: Nicola Hanania, MD, MS (PI), Philip Alapat, MD, Venkata Bandi, MD, Kalpalatha Guntupalli, MD, Elizabeth Guy, MD, Antara Mallampalli, MD, Charles Trinh, MD (RAD), Mustafa Atik, MD. Brigham and Women’s Hospital, Boston, MA: Dawn DeMeo, MD, MPH (Co-PI), Craig Hersh, MD, MPH (Co-PI), George Washko, MD, Francine Jacobson, MD, MPH (RAD). Columbia University, New York, NY: R. Graham Barr, MD, DrPH (PI), Byron Thomashow, MD, John Austin, MD (RAD).
Duke University Medical Center, Durham, NC: Neil MacIntyre, Jr, MD (PI), Lacey Washington, MD (RAD), H. Page McAdams, MD (RAD). Fallon Clinic, Worcester, MA: Richard Rosiello, MD (PI), Timothy Bresnahan, MD (RAD). Health Partners Research Foundation, Minneapolis, MN: Charlene McEvoy, MD, MPH (PI), Joseph Tashjian, MD (RAD). Johns Hopkins University, Baltimore, MD: Robert Wise, MD (PI), Nadia Hansel, MD, MPH, Robert Brown, MD (RAD), Gregory Diette, MD. Los Angeles Biomedical Research Institute at Harbor UCLA Medical Center, Los Angeles, CA: Richard Casaburi, MD (PI), Janos Porszasz, MD, PhD, Hans Fischer, MD, PhD (RAD), Matt Budoff, MD.
Michael E. DeBakey VAMC, Houston, TX: Amir Sharafkhaneh, MD (PI), Charles Trinh, MD (RAD), Hirani Kamal, MD, Roham Darvishi, MD. Minneapolis VA: Dennis Niewoehner, MD (PI), Tadashi Allen, MD (RAD), Quentin Anderson, MD (RAD), Kathryn Rice, MD. Morehouse School of Medicine, Atlanta, GA: Marilyn Foreman, MD, MS (PI), Gloria Westney, MD, MS, Eugene Berkowitz, MD, PhD (RAD). National Jewish Health, Denver, CO: Russell Bowler, MD, PhD (PI), Adam Friedlander, MD, David Lynch, MB (RAD), Joyce Schroeder, MD (RAD), John Newell, Jr., MD (RAD).
Temple University, Philadelphia, PA: Gerard Criner, MD (PI), Victor Kim, MD, Nathaniel Marchetti, DO, Aditi Satti, MD, A. James Mamary, MD, Robert Steiner, MD (RAD), Chandra Dass, MD (RAD). University of Alabama, Birmingham, AL: William Bailey, MD (PI), Mark Dransfield, MD (Co-PI), Hrudaya Nath, MD (RAD). University of California, San Diego, CA: Joe Ramsdell, MD (PI), Paul Friedman, MD (RAD).
University of Iowa, Iowa City, IA: Geoffrey McLennan, MD, PhD (PI), Edwin JR van Beek, MD, PhD (RAD), Brad Thompson, MD (RAD), Dwight Look, MD. University of Michigan, Ann Arbor, MI: Fernando Martinez, MD (PI), MeiLan Han, MD, Ella Kazerooni, MD (RAD). University of Minnesota, Minneapolis, MN: Christine Wendt, MD (PI), Tadashi Allen, MD (RAD). University of Pittsburgh, Pittsburgh, PA: Frank Sciurba, MD (PI), Joel Weissfeld, MD, MPH, Carl Fuhrman, MD (RAD), Jessica Bon, MD. University of Texas Health Science Center at San Antonio, San Antonio, TX: Antonio Anzueto, MD (PI), Sandra Adams, MD, Carlos Orozco, MD, Mario Ruiz, MD (RAD).
Administrative Core: James Crapo, MD (PI), Edwin Silverman, MD, PhD (PI), Barry Make, MD, Elizabeth Regan, MD, Sarah Moyle, MS, Douglas Stinson.
Genetic Analysis Core: Terri Beaty, PhD, Barbara Klanderman, PhD, Nan Laird, PhD, Christoph Lange, PhD, Michael Cho, MD, Stephanie Santorico, PhD, John Hokanson, MPH, PhD, Dawn DeMeo, MD, MPH, Nadia Hansel, MD, MPH, Craig Hersh, MD, MPH, Jacqueline Hetmanski, MS, Tanda Murray.
Imaging Core: David Lynch, MB, Joyce Schroeder, MD, John Newell, Jr, MD, John Reilly, MD, Harvey Coxson, PhD, Philip Judy, PhD, Eric Hoffman, PhD, George Washko, MD, Raul San Jose Estepar, PhD, James Ross, MSc, Rebecca Leek, Jordan Zach, Alex Kluiber, Jered Sieren, Heather Baumhauer, Verity McArthur, Dzimitry Kazlouski, Andrew Allen, Tanya Mann, Anastasia Rodionova.
PFT QA Core, LDS Hospital, Salt Lake City, UT: Robert Jensen, PhD.
Biological Repository, Johns Hopkins University, Baltimore, MD: Homayoon Farzadegan, PhD, Stacey Meyerer, Shivam Chandan, Samantha Bragan.
Data Coordinating Center and Biostatistics, National Jewish Health, Denver, CO: James Murphy, PhD (deceased), Douglas Everett, PhD, Carla Wilson, MS, Ruthie Knowles, Amber Powell, Joe Piccoli, Maura Robinson, Margaret Forbes, Martina Wamboldt.
Epidemiology Core, University of Colorado School of Public Health, Denver, CO: John Hokanson, MPH, PhD, Marci Sontag, PhD, Jennifer Black-Shinn, MPH, Gregory Kinney, MPH.
Principal investigators and centers participating in ECLIPSE include: Bulgaria: Y. Ivanov, Pleven; K. Kostov, Sofia. Canada: J. Bourbeau, Montreal; M. Fitzgerald, Vancouver; P. Hernández, Halifax; K. Killian, Hamilton; R. Levy, Vancouver; F. Maltais, Montreal; D. O’Donnell, Kingston. Czech Republic: J. Krepelka, Praha.
Denmark: J. Vestbo, Hvidovre. The Netherlands: E. Wouters, Horn. New Zealand: D. Quinn, Wellington. Norway: P. Bakke, Bergen, Slovenia: M. Kosnik, Golnik. Spain: A. Agusti, Jaume Sauleda, Palma de Mallorca. Ukraine: Y. Feschenko, Kiev; V. Gavrisyuk, Kiev; L. Yashina, Kiev. UK: L. Yashina, W. MacNee, Edinburgh; D. Singh, Manchester; J. Wedzicha, London. USA: A. Anzueto, San Antonio, TX; S. Braman, Providence. RI; R. Casaburi, Torrance CA; B. Celli, Boston, MA; G. Giessel, Richmond, VA; M. Gotfried, Phoenix, AZ; G. Greenwald, Rancho Mirage, CA; N. Hanania, Houston, TX; D. Mahler, Lebanon, NH; B. Make, Denver, CO; S. Rennard, Omaha, NE; C. Rochester, New Haven, CT; P. Scanlon, Rochester, MN; D. Schuller, Omaha, NE; F. Sciurba, Pittsburgh, PA; A. Sharafkhaneh, Houston, TX; T. Siler, St Charles, MO; E. Silverman, Boston, MA; A. Wanner, Miami, FL; R. Wise, Baltimore, MD; R. ZuWallack, Hartford, CT.
Steering Committee: H. Coxson (Canada), C. Crim (GlaxoSmithKline, USA), L. Edwards (GlaxoSmithKline, USA), D. Lomas (UK), W. MacNee (UK), E. Silverman (USA), R. Tal Singer (Co-chair, GlaxoSmithKline, USA), J. Vestbo (Co-chair, Denmark), J. Yates (GlaxoSmithKline, USA).
Scientific Committee: A. Agusti (Spain), P. Calverley (UK), B. Celli (USA), C. Crim (GlaxoSmithKline, USA), B. Miller (GlaxoSmithKline, USA), W. MacNee (Chair, UK), S. Rennard (USA), R. Tal-Singer (GlaxoSmithKline, USA), E. Wouters (The Netherlands), J. Yates (GlaxoSmithKline, USA).
The National Emphysema Treatment Trial was supported by the National Heart, Lung, and Blood Institute contracts N01HR76101, N01HR76102, N01HR76103, N01HR76104, N01HR76105, N01HR76106, N01HR76107, N01HR76108, N01HR76109, N01HR76110, N01HR76111, N01HR76112, N01HR76113, N01HR76114, N01HR76115, N01HR76116, N01HR76118 and N01HR76119.
Co-investigators in the NETT Genetics Ancillary Study also include J. Benditt, G. Criner, M. DeCamp, P. Diaz, M. Ginsburg, L. Kaiser, M. Katz, M. Krasna, N. MacIntyre, R. McKenna, F. Martinez, Z. Mosenifar, J. Reilly, A. Ries, P. Scanlon, F. Sciurba and J. Utz
Footnotes
Competing Interests Statement:
E.K.S. has received grant support and consulting and speaker’s fees from GlaxoSmithKline, consulting and speaker’s fees from Astra-Zeneca, and consulting fees from Merck. X.Z., W.Q., M.H.C., J.F.S., J.D.M., T.L., D.M.T, G.L., P.S.B., A.G., D.A.L., T.H.B., C.P.H., C.A., U.G., B.A.R., S.I.R., M.A.P., A.M.K.C. and J.Q. do not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arialdi M, Miniño MJXMD, Kenneth D, Kochanek MA. Division of Vital Statistics: Deaths: Preliminary Data for 2008. National Vital Statistics Reports. (2010/12/09) 2010;59:1–72. [PubMed] [Google Scholar]
- 2.McCloskey SC, Patel BD, Hinchliffe SJ, Reid ED, Wareham NJ, Lomas DA. Siblings of patients with severe chronic obstructive pulmonary disease have a significant risk of airflow obstruction. Am J Respir Crit Care Med. 2001;164:1419–1424. doi: 10.1164/ajrccm.164.8.2105002. [DOI] [PubMed] [Google Scholar]
- 3.Silverman EK, Chapman HA, Drazen JM, Weiss ST, Rosner B, Campbell EJ, O’Donnell WJ, Reilly JJ, Ginns L, Mentzer S, et al. Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease. Risk to relatives for airflow obstruction and chronic bronchitis. Am J Respir Crit Care Med. 1998;157:1770–1778. doi: 10.1164/ajrccm.157.6.9706014. [DOI] [PubMed] [Google Scholar]
- 4.Salazar LM, Herrera AM. Fibrotic response of tissue remodeling in COPD. Lung. 2011;189:101–109. doi: 10.1007/s00408-011-9279-2. [DOI] [PubMed] [Google Scholar]
- 5.Gill SE, Parks WC. Metalloproteinases and their inhibitors: regulators of wound healing. The international journal of biochemistry & cell biology. 2008;40:1334–1347. doi: 10.1016/j.biocel.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pillai SG, Ge D, Zhu G, Kong X, Shianna KV, Need AC, Feng S, Hersh CP, Bakke P, Gulsvik A, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sovio U, Bennett AJ, Millwood IY, Molitor J, O’Reilly PF, Timpson NJ, Kaakinen M, Laitinen J, Haukka J, Pillas D, et al. Genetic determinants of height growth assessed longitudinally from infancy to adulthood in the northern Finland birth cohort 1966. PLoS Genet. 2009;5:e1000409. doi: 10.1371/journal.pgen.1000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hancock DB, Eijgelsheim M, Wilk JB, Gharib SA, Loehr LR, Marciante KD, Franceschini N, van Durme YM, Chen TH, Barr RG, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 42:45–52. doi: 10.1038/ng.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Repapi E, Sayers I, Wain LV, Burton PR, Johnson T, Obeidat M, Zhao JH, Ramasamy A, Zhai G, Vitart V, et al. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 42:36–44. doi: 10.1038/ng.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Durme YM, Eijgelsheim M, Joos GF, Hofman A, Uitterlinden AG, Brusselle GG, Stricker BH. Hedgehog-interacting protein is a COPD susceptibility gene: the Rotterdam Study. Eur Respir J. 2010;36:89–95. doi: 10.1183/09031936.00129509. [DOI] [PubMed] [Google Scholar]
- 11.Young RP, Whittington CF, Hopkins RJ, Hay BA, Epton MJ, Black PN, Gamble GD. Chromosome 4q31 locus in COPD is also associated with lung cancer. Eur Respir J. 2010;36:1375–1382. doi: 10.1183/09031936.00033310. [DOI] [PubMed] [Google Scholar]
- 12.Zhou X, Baron RM, Hardin M, Cho MH, Zielinski J, Hawrylkiewicz I, Sliwinski P, Hersh CP, Mancini JD, Lu K, et al. Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Human molecular genetics. 2012;21:1325–1335. doi: 10.1093/hmg/ddr569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chuang PT, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature. 1999;397:617–621. doi: 10.1038/17611. [DOI] [PubMed] [Google Scholar]
- 14.Kawahira H, Ma NH, Tzanakakis ES, McMahon AP, Chuang PT, Hebrok M. Combined activities of hedgehog signaling inhibitors regulate pancreas development. Development. 2003;130:4871–4879. doi: 10.1242/dev.00653. [DOI] [PubMed] [Google Scholar]
- 15.Chuang PT, Kawcak T, McMahon AP. Feedback control of mammalian Hedgehog signaling by the Hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev. 2003;17:342–347. doi: 10.1101/gad.1026303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS genetics. 2007;3:1724–1735. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teschendorff AE, Zhuang J, Widschwendter M. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics. 2011;27:1496–1505. doi: 10.1093/bioinformatics/btr171. [DOI] [PubMed] [Google Scholar]
- 18.Cho MH, Castaldi PJ, Wan ES, Siedlinski M, Hersh CP, Demeo DL, Himes BE, Sylvia JS, Klanderman BJ, Ziniti JP, et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Human molecular genetics. 2012;21:947–957. doi: 10.1093/hmg/ddr524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lipman PJ, Cho MH, Bakke P, Gulsvik A, Kong X, Lomas DA, Anderson W, Silverman EK, Lange C. On the follow-up of genome-wide association studies: an overall test for the most promising SNPs. Genetic epidemiology. 2011;35:303–309. doi: 10.1002/gepi.20578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome biology. 2008;9 (Suppl 1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nature reviews Genetics. 2011;12:393–406. doi: 10.1038/nrg2984. [DOI] [PubMed] [Google Scholar]
- 23.Rutter M, Wang J, Huang Z, Kuliszewski M, Post M. Gli2 influences proliferation in the developing lung through regulation of cyclin expression. American journal of respiratory cell and molecular biology. 2010;42:615–625. doi: 10.1165/rcmb.2008-0390OC. [DOI] [PubMed] [Google Scholar]
- 24.Ono S, Hane M, Kitajima K, Sato C. Novel regulation of fibroblast growth factor 2 (FGF2)-mediated cell growth by polysialic acid. The Journal of biological chemistry. 2012;287:3710–3722. doi: 10.1074/jbc.M111.276618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKenzie JA, Grossman D. Role of the apoptotic and mitotic regulator survivin in melanoma. Anticancer research. 2012;32:397–404. [PubMed] [Google Scholar]
- 26.Sheng Z, Li L, Zhu LJ, Smith TW, Demers A, Ross AH, Moser RP, Green MR. A genome-wide RNA interference screen reveals an essential CREB3L2-ATF5-MCL1 survival pathway in malignant glioma with therapeutic implications. Nature medicine. 2010;16:671–677. doi: 10.1038/nm.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dluzen D, Li G, Tacelosky D, Moreau M, Liu DX. BCL-2 is a downstream target of ATF5 that mediates the prosurvival function of ATF5 in a cell type-dependent manner. The Journal of biological chemistry. 2011;286:7705–7713. doi: 10.1074/jbc.M110.207639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nature reviews Cancer. 2011;11:558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 29.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313–317. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 30.Haibe-Kains B, Olsen C, Djebbari A, Bontempi G, Correll M, Bouton C, Quackenbush J. Predictive networks: a flexible, open source, web application for integration and analysis of human gene networks. Nucleic acids research. 2012;40:D866–875. doi: 10.1093/nar/gkr1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bosanac I, Maun HR, Scales SJ, Wen X, Lingel A, Bazan JF, de Sauvage FJ, Hymowitz SG, Lazarus RA. The structure of SHH in complex with HHIP reveals a recognition role for the Shh pseudo active site in signaling. Nature structural & molecular biology. 2009;16:691–697. doi: 10.1038/nsmb.1632. [DOI] [PubMed] [Google Scholar]
- 32.Chuang PT, Kawcak T, McMahon AP. Feedback control of mammalian Hedgehog signaling by the Hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes & development. 2003;17:342–347. doi: 10.1101/gad.1026303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patterson VL, Damrau C, Paudyal A, Reeve B, Grimes DT, Stewart ME, Williams DJ, Siggers P, Greenfield A, Murdoch JN. Mouse hitchhiker mutants have spina bifida, dorso-ventral patterning defects and polydactyly: identification of Tulp3 as a novel negative regulator of the Sonic hedgehog pathway. Human molecular genetics. 2009;18:1719–1739. doi: 10.1093/hmg/ddp075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Onishi H, Kai M, Odate S, Iwasaki H, Morifuji Y, Ogino T, Morisaki T, Nakashima Y, Katano M. Hypoxia activates the hedgehog signaling pathway in a ligand-independent manner by upregulation of Smo transcription in pancreatic cancer. Cancer science. 2011;102:1144–1150. doi: 10.1111/j.1349-7006.2011.01912.x. [DOI] [PubMed] [Google Scholar]
- 35.Polizio AH, Chinchilla P, Chen X, Manning DR, Riobo NA. Sonic Hedgehog activates the GTPases Rac1 and RhoA in a Gli-independent manner through coupling of smoothened to Gi proteins. Science signaling. 2011;4:pt7. doi: 10.1126/scisignal.2002396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi Y, Chung H, Jung H, Couchman JR, Oh ES. Syndecans as cell surface receptors: Unique structure equates with functional diversity. Matrix biology : journal of the International Society for Matrix Biology. 2011;30:93–99. doi: 10.1016/j.matbio.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Hayashida K, Parks WC, Park PW. Syndecan-1 shedding facilitates the resolution of neutrophilic inflammation by removing sequestered CXC chemokines. Blood. 2009;114:3033–3043. doi: 10.1182/blood-2009-02-204966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, Nishimura S, Imamura Y, Kitayama H, Alexander DB, et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107:789–800. doi: 10.1016/s0092-8674(01)00597-9. [DOI] [PubMed] [Google Scholar]
- 39.Liu LT, Chang HC, Chiang LC, Hung WC. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer research. 2003;63:3069–3072. [PubMed] [Google Scholar]
- 40.Sasahara RM, Brochado SM, Takahashi C, Oh J, Maria-Engler SS, Granjeiro JM, Noda M, Sogayar MC. Transcriptional control of the RECK metastasis/angiogenesis suppressor gene. Cancer detection and prevention. 2002;26:435–443. doi: 10.1016/s0361-090x(02)00123-x. [DOI] [PubMed] [Google Scholar]
- 41.Takagi S, Simizu S, Osada H. RECK negatively regulates matrix metalloproteinase-9 transcription. Cancer research. 2009;69:1502–1508. doi: 10.1158/0008-5472.CAN-08-2635. [DOI] [PubMed] [Google Scholar]
- 42.Madison BB, Braunstein K, Kuizon E, Portman K, Qiao XT, Gumucio DL. Epithelial hedgehog signals pattern the intestinal crypt-villus axis. Development. 2005;132:279–289. doi: 10.1242/dev.01576. [DOI] [PubMed] [Google Scholar]
- 43.Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, Mangino M, Freathy RM, Perry JR, Stevens S, Hall AS, et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nature genetics. 2008;40:575–583. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. American journal of respiratory cell and molecular biology. 2003;28:555–562. doi: 10.1165/rcmb.2002-0090OC. [DOI] [PubMed] [Google Scholar]
- 45.Sgadari C, Barillari G, Palladino C, Bellino S, Taddeo B, Toschi E, Ensoli B. Fibroblast Growth Factor-2 and the HIV-1 Tat Protein Synergize in Promoting Bcl-2 Expression and Preventing Endothelial Cell Apoptosis: Implications for the Pathogenesis of AIDS-Associated Kaposi’s Sarcoma. International journal of vascular medicine. 2011;2011:452729. doi: 10.1155/2011/452729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu W, Xie J, Zhao J, Xu Y, Yang S, Ni W. Involvement of Bcl-2 family in apoptosis and signal pathways induced by cigarette smoke extract in the human airway smooth muscle cells. DNA and cell biology. 2009;28:13–22. doi: 10.1089/dna.2008.0782. [DOI] [PubMed] [Google Scholar]
- 47.Wu CH, Lin HH, Yan FP, Wang CJ. Immunohistochemical detection of apoptotic proteins, p53/Bax and JNK/FasL cascade, in the lung of rats exposed to cigarette smoke. Archives of toxicology. 2006;80:328–336. doi: 10.1007/s00204-005-0050-4. [DOI] [PubMed] [Google Scholar]
- 48.Cheung CH, Cheng L, Chang KY, Chen HH, Chang JY. Investigations of survivin: the past, present and future. Frontiers in bioscience : a journal and virtual library. 2011;16:952–961. doi: 10.2741/3728. [DOI] [PubMed] [Google Scholar]
- 49.Baldwin A, Li W, Grace M, Pearlberg J, Harlow E, Munger K, Grueneberg DA. Kinase requirements in human cells: II. Genetic interaction screens identify kinase requirements following HPV16 E7 expression in cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16478–16483. doi: 10.1073/pnas.0806195105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.