

Abstract
Congenital hyperinsulinism (CHI) is the result of unregulated insulin secretion from the pancreatic β-cells leading to severe hypoglycaemia. In these patients it is important to make an accurate diagnosis and initiate the appropriate management so as to avoid hypoglycemic episodes and prevent the potentially associated complications like epilepsy, neurological impairment and cerebral palsy. At a genetic level abnormalities in eight different genes (ABCC8, KCNJ11, GLUD1, GCK, HADH, SLC16A1, HNF4A and UCP2) have been reported with CHI. Loss of function mutations in ABCC8/KCNJ11 lead to the most severe forms of CHI which are usually medically unresponsive. At a histological level there are two major subgroups, diffuse and focal, each with a different genetic etiology. The focal form is sporadic in inheritance and is localized to a small region of the pancreas whereas the diffuse form is inherited in an autosomal recessive (or dominant) manner. Imaging using a specialized positron emission tomography scan with the isotope fluroine-18 L-3, 4-dihydroxyphenyalanine (18F-DOPA-PET-CT) is used to accurately locate the focal lesion pre-operatively and if removed can cure the patient from hypoglycemia. Understanding the molecular mechanisms, the histological basis, improvements in imaging modalities and surgical techniques have all improved the management of patients with CHI.
Keywords: Diazoxide, hypoglycemia, hyperinsulinism
INTRODUCTION
Congenital hyperinsulinism (CHI) is an important cause of hyperinsulinaemic hypoglycaemia (HH) in the neonatal, infancy and childhood periods. It occurs due to the unregulated secretion of insulin form pancreatic β-cells leading to severe and persistent hypoglycemia. The early recognition, diagnosis and immediate management are important as delay in the diagnosis and inappropriate management can lead to hypoglycemic brain injury.[1] CHI is associated with an increased risk of brain injury secondary to the metabolic actions of insulin. Unregulated insulin secretion leads to suppression of glucose production by inhibition of glycogenolysis and gluconeogenesis.[2] In the absence of glucose as energy source, brain tends to utilise the ketone bodies as an alternative substrate. However, in CHI, brain is deprived of both glucose and ketones as insulin inhibits lipolysis and ketogenesis.
The other forms of hyperinsulinaemic hypoglycaemia (HH) include transient hyperinsulinism due to risk factors such as birth asphyxia, intra-uterine growth retardation (IUGR) and maternal diabetes mellitus (gestational or insulin dependent), several developmental syndromes (such as Beckwith-Wiedemann) and rare metabolic conditions such as CDG syndromes.[3]
CHI is extremely heterogeneous in terms of clinical presentation, histological subgroups and underlying molecular biology. The incidence of CHI can vary from 1 in 35,000-40,000 in the general population[4] to 1 in 2500 in some communities with high rates of consanguinity.[5] The molecular basis of CHI involves genetic defects in eight different genes (ABCC8, KCNJ11, GLUD1, GCK, HADH, SLC16A1, HNF4A and UCP2) that are involved in regulating insulin secretion from β-cells.[6]
At a histological level two major subtypes of CHI (diffuse and focal) have been described.[7] The diffuse form is inherited in an autosomal recessive (or dominant) manner whereas the focal form is sporadic in inheritance. The differentiation of these two histological subtypes is important from the management point of view. The diffuse from may require a near total pancreatectomy (with the risk of diabetes mellitus and pancreatic exocrine insufficiency) whereas the focal form will only require a limited focal lesionectomy. Recently a specialized positron emission tomography scan using the isotope Fluorine 18 L-3, 4-dihydroxyphenyalanine (18F-DOPA-PET) has become the gold standard for the localisation of focal lesions within the pancreas.[8]
The aim of this review is to provide an overview of pancreatic β-cell physiology and insulin secretion, to review the different molecular mechanisms of CHI, to outline the clinical presentation and describe the recent advances in imaging and management.
PANCREATIC β-CELL PHYSIOLOGY, GLUCOSE METABOLISM AND INSULIN SECRETION
Pancreatic β-cells play a key role in regulating insulin production and secretion. Insulin secretion is tightly linked to glucose metabolism by pancreatic β-cell ATP-sensitive K+ channels. These ATP-sensitive K+ channel (KATP channel) plays a fundamental role in glucose homeostasis by linking glucose metabolism to electrical excitability of the β-cell and insulin secretion.[9] The β-cell KATP channel is a hetero-octameric complex (arranged in a 4:4 stoichiometry) composed of two types of subunits: Four inward-rectifying potassium channel pore-forming (Kir6.2) subunits and four high-affinity sulfonylurea receptor 1 (SUR 1) subunits.[10] The Kir6.2 forms the core of the channel whilst the SUR1 (an ATP binding cassette transporter) acts as a regulatory subunit. KATP channels can only function if they are assembled and correctly transported to the cell membrane surface (trafficking). The assembly and trafficking of KATP channels are intricately linked processes. Only octameric KATP channel complexes are capable of expressing on the cell membrane surface. For example both Kir6.2 and SUR1 possess an endoplasmic reticulum (ER) retention signal (RKR) that prevents the trafficking of each subunit to the plasma membrane in the absence of the other subunit.[11] Co expression of two subunits masks these retention signals, allowing them to traffic to the plasma membrane. The retention signal is present in the C terminal region of Kir6.2 and in an intracellular loop between TM11 and NBF-1 in SUR1. Truncation of the C-terminus of Kir6.2 deletes its retention signal, allowing functional expression of Kir6.2 in the absence of SUR1 subunit.[12]
KATP channels are regulated by adenine nucleotides to convert changes in cellular metabolic levels into membrane excitability.[13] The Kir6.2 subunit determines the biophysical properties of the channel complex including K+ selectivity, rectification, and inhibition by ATP and activation by acyl-CoAs.[12] The sulfonylurea receptors endow KATP channels with sensitivity to the stimulatory actions of Mg-nucleotides and KATP channel openers like diazoxide and the inhibitory effects of sulfonylureas.[14]
Glucose metabolism in the β-cells regulates insulin secretion. Metabolism raises the intracytosolic ATP/ADP ratio which inhibits the plasma membrane sulfonylurea receptor 1 (SUR 1). This results in the closure of the KATP channel which in turn leads to cell membrane depolarisation and Ca2+ influx into the cell via voltage gated calcium channels. The increase in the intracellular concentration of calcium triggers the release of insulin. When glucose levels are low, KATP channels are open and potassium diffusing via these channels maintains the resting membrane potential at a hyperpolarized level.
Glucose stimulates insulin secretion by triggering (KATP channel dependent) and amplifying (augmentation pathway) signals in beta-cells. The triggering pathway is well characterized involving the pancreatic beta-cells. The augmentation pathway is less well defined but involves an increase in the efficacy of Ca2+ on the exocytosis of insulin granules. The amplification pathway serves to optimize the secretory response not only to glucose but also to non-glucose stimuli.
AETIOLOGY OF CONGENITAL HYPERINSULINISM [FIGURE 1]
Figure 1.
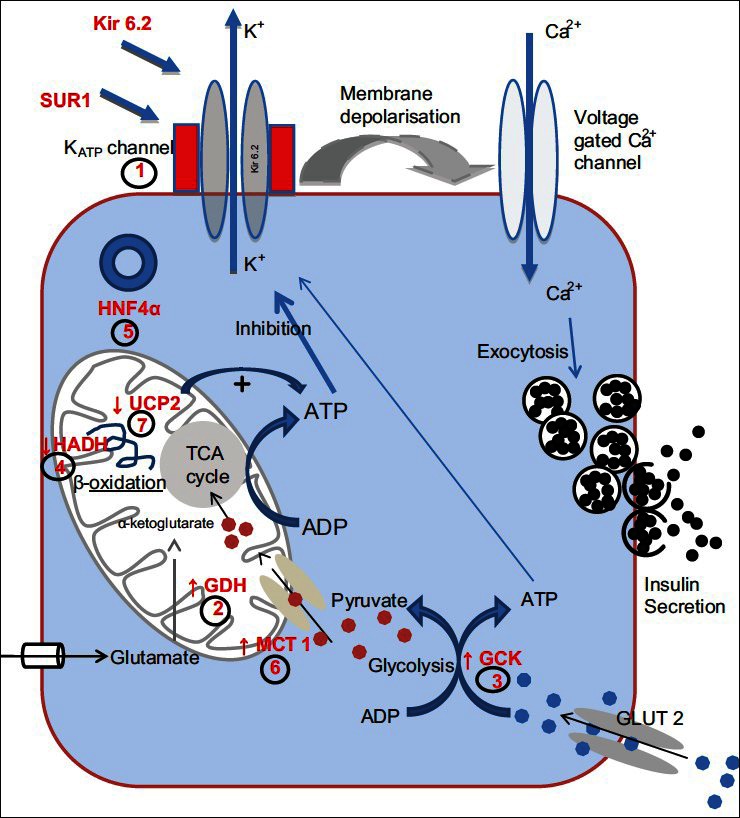
Common mutations associated with CHI. (1) ATP gated K+ channel (KATP) encoded by ABCC8 and KCNJ11; (2) Glutamate Dehydrogenase (GDH) encoded by GLUD1; (3) Glucokinase (GCK) encoded by GCK gene; (4) L-3-hyroxyacyl-coenzyme A dehydrogenase (HADH) encoded by HADH; (5) Hepatocyte Nuclear Factor 4α (HNF4α) encoded by HNF4A gene; (6) The moncarboxylate transporter (MCT1) encoded by SLC16A1; (7) Uncoupling Protein 2 (UCP2)
So far at a genetic level, abnormalities in eight different genes have been described which lead to unregulated insulin secretion. The most common forms are due to defects in the genes which encode the components of the pancreatic KATP channel. Below is a brief description of the different forms of CHI.
Pancreatic β-cell KATP channel defects
The Kir6.2 and SUR1 subunits are encoded by the genes KCNJ11 and ABCC8 (localized to chromosome 11p15.1), respectively. Recessive mutations in ABCC8 and KCNJ11 are the most common causes of CHI.[15,16] The inactivating mutations in ABCC8/KCNJ11 reduce or completely abolish the activity of the KATP channel, leading to unregulated insulin release despite severe hypoglycemia.[15] Germline mutations in ABCC8/KCNJ11 are found in approximately 50% of CHI patients.[16] To date about 150 homozygous, compound heterozygous and heterozygous inactivating mutations in ABCC8 and around 24 KCNJ11 mutations have been reported.[16]
The recessive inactivating mutations in ABCC8 and KCNJ11 usually cause severe CHI which is unresponsive to medical treatment with diazoxide. The molecular basis of recessive inactivating ABCC8 and KCNJ11 mutations involves defects in KATP channel biogenesis and turnover,[17] channel trafficking from the ER and Golgi apparatus[18,19] to the plasma membrane and alterations of channels in response to nucleotide regulation and open state frequency.[20] Dominant inactivating mutations in ABCC8 and KCNJ11 usually cause CHI with a milder phenotype[21,22] although medically unresponsive forms have recently been reported.[23]
Hyperinsulinism-hyperammonaemia syndrome
Hyperinsulinism-hyperammonemia syndrome (HI/HA), the second most common form of (CHI) is associated with activating missense mutations in the GLUD1 gene, which encodes the mitochondrial matrix enzyme, glutamate dehydrogenase (GDH). Patients present with recurrent symptomatic postprandial hypoglycemia following protein-rich meals (leucine-sensitive hypoglycemia) as well as fasting hypoglycemia accompanied by asymptomatic elevations of plasma ammonia. GDH is expressed in liver, kidney, brain, and pancreatic β-cells. Mutations in GLUD1 lead to a gain of enzyme function by reducing its sensitivity to allosteric inhibition by the high-energy phosphates such as GTP and ATP and allowing activation by the amino acid leucine.[24]
GDH catalyses the reversible oxidative deamination of glutamate to alpha-ketoglutarate and ammonia using NAD or NADP as co-factors. The increased GDH activity leads to inappropriate insulin secretion in pancreatic β-cells, as well as to excessive ammonia production and reduced urea synthesis in the liver. Recent animal studies have suggested the role of renal ammoniagenesis due to activation of GDH as a source of hyperammonemia in these patients.[25] The phenotype is characterized by recurrent postprandial hypoglycemia following protein-rich meals as well as fasting hypoglycemia accompanied by asymptomatic hyperammonemia.[26]
Though hyperammonemia has remained the most consistent feature of HI/HA, there is a rare group of patients with GLUD1 mutations who demonstrate leucine hypersensitivity but have normal serum ammonia level.[27] These patients may be mosaic for GDH enzyme activity (with normal GDH activity in the liver but elevated activity in the pancreas), however, this remains to be proven.
The phenotype of HI/HA is reported to be milder in contrast to other forms of CHI, thus escaping recognition for the first few months of life.[27] Patients with HI/HA syndrome have more neurological complications such as epilepsy and learning disabilities.[28] However, in contrast to hyperammonemia patients due to urea cycle disorders, patients with HI/HA syndrome do not experience lethargy, headaches, or manifest CNS symptoms that might be expected for their degree of hyperammonemia, and are resistant to ammonia scavenging agents or protein restriction. Routine measurement of plasma ammonia concentrations in all patients with hypoglycemia is an essential screening test for the disorder.
HADH and hyperinsulinism
HADH encodes for the enzyme 3-hydroxyacyl-coenzyme A dehydrogenase (HADH) and catalyses the penultimate reaction in the beta-oxidation of fatty acids the NAD+-dependent conversion of L-3-hydroxyacyl-CoA to 3-ketoacyl-CoA. Mutations in the mitochondrial HADH gene (encoding the enzyme L-3-hydroxyacyl-Coenzyme A dehydrogenase, HADH), are a rare cause of CHI.[29] This enzyme catalyses the conversion of L3-hydroxyacyl CoAs of variable chain length to their corresponding 3-ketoacyl CoAs and exerts highest activity to 3-hydroxybutyryl-CoA. HADH gene has been mapped to chromosome 4q22-26[30] and is expressed in most tissues, although the enzyme activity is high in the pancreas, particularly in the islets of Langerhans.[31] HADH expression is regulated by transcription factors such as Foxa2, which are essential for β-cell differentiation and mice which have Foxa2 knocked out, showed a 3-fold down regulation of HADH mRNA and severe HH.[32]
HADH gene mutations can lead either to severe neonatal HH or to mild late onset HH.[32,33] All patients reported so far have responded to diazoxide and some had abnormal acylcarnitine metabolites (raised plasma hydroxybutyrylcarnitine and urinary 3-hydroxyglutarate levels). Protein sensitivity has been demonstrated in patients with HADH mutations[34] and this has been confirmed in the HADH knockout mouse.[35] However, the precise mechanism of dysregulated insulin secretion in patients with a HADH deficiency is not understood but might involve an interaction between GDH and HADH.[35] Genetic analysis for HADH gene is recommended in patients with diazoxide responsive HH from consanguineous families, who are negative for mutations in the KATP channels.[36]
HNF4A and hyperinsulinism
HNF4A gene encodes for the transcription factor hepatocyte nuclear factor 4α (HNF-4α), which belongs to the nuclear hormone receptor superfamily and has been shown to control the expression of genes involved in glucose stimulated insulin secretion.[37] Heterozygous mutations in the HNF4A gene causes maturity-onset diabetes of the young type 1 (MODY1), which is characterized by progressive β-cell dysfunction and failure of glucose induced insulin secretion.[38]
Recently heterozygous mutations in the HNF4A gene were also reported to result in transient[39] or persistent HH.[40] The phenotype of these patients is characterized by macrosomia and neonatal HH.[41,42] The severity of HH in these patients varies from diet-controlled neonatal hypoglycemia to persistent HH requiring diazoxide treatment. In a recent series of 11 patients with HNF4A mutations, the HH was noted to range from three months to eight years with on-going need for diazoxide therapy.[43] Interestingly, only few of the parents of children in this series had diabetes, suggesting that the absence of a history of diabetes in the parents should not preclude sequencing of the HNF4A gene. HNF4A mutation has been noted to have variable penetrance with only a minority of HNF4A mutation carriers developing HH. The precise mechanism by which HNF4A mutations cause HH is not clear but might involve a reduction in expression of the potassium channel subunit Kir6.2[37] or reduction in the levels of PPARα.[44] PPARα is a transcription factor that is known to control the expression of genes encoding enzymes of the beta oxidation pathway of fatty acids. Low levels of PPARα are reported in HNF-4α deficient β-cells.[37] It can be postulated that HNF-4α deficiency causes lower levels of PPARα and a decrease in beta-oxidation of fatty acids resulting in the accumulation of lipids (such as malonyl-CoA) in the cytoplasm. Increased malonyl-CoA is thought to inhibit the enzyme carnitine-palmitoyltransferase1 thereby increasing cytosolic long-chain acyl-CoA levels, which signals insulin release.[45] In support of this hypothesis PPARα null mice develop fasting HH suggesting that PPARα is important for regulated insulin secretion during fasting.[44]
Exercise-induced hyperinsulinism (SLC16A1)
Exercise-induced hyperinsulinism (EIHI) is a dominantly inherited and characterized by the inappropriate insulin secretion during anaerobic exercise or on pyruvate load.[46,47,48] To date, 13 patients have been reported, 12 from two Finnish pedigrees and one unrelated patient.[49] Affected patients become hypoglycemic typically 30-45 minutes after a period of intensive anaerobic exercise.[47]
The transport of lactate and pyruvate into the β-cells are mediated by monocarboxylate transporter 1 (MCT1) which is encoded by the SLC16A1 (solute carrier family 16, member 1) gene. Under normal physiological conditions lactate and pyruvate concentrations are low in β-cells and do not stimulate insulin secretion.[50] However, promoter-activating mutations in SLC16A1 induce the expression of MCT1 in β-cells (where this gene is not usually transcribed) permitting pyruvate uptake and pyruvate-stimulated insulin release despite ensuing hypoglycemia.[49] This novel disease mechanism results from the failure of cell-specific transcriptional silencing of a gene (SLC16A1) that is highly expressed in other tissues. During strenuous anaerobic exercise there is accumulation of lactate and pyruvate which then act as insulin secretagogues.
Glucokinase induced hyperinsulinism
Glucokinase is a key glycolytic enzyme that plays a pivotal role as a glucose sensor in the pancreatic β-cell and appears to have a similar role in entero-endocrine cells, hepatocytes, and hypothalamic neurons.[51] In β-cells, glucokinase a rate-limiting enzyme for glucose metabolism governs glucose-stimulated insulin secretion. Heterozygous activating mutations of glucokinase lead to CHI.[52] These glucokinase mutations result in increased affinity of the enzyme for glucose, resulting in an increase in the ATP: ADP ratio in the pancreatic β-cell, closure of KATP channel, and inappropriate insulin secretion.
The activating glucokinase mutations are inherited in an autosomal dominant manner with the severity of symptoms varying markedly within and between families.[53] The age of presentation of glucokinase induced hyperinsulinism can range widely from infancy to adulthood.[54] Affected children have fasting hypoglycemia and may manifest variable responsiveness to medical treatment. Some appear to respond well to pharmacologic intervention with diazoxide, whilst others require more intensive medical management including octreotide and even surgery.[55]
Mutations in the UCP2 gene and HH
Uncoupling protein 2 (UCP2) is expressed in several tissues, and acts in the protection against oxidative stress, in fatty acid metabolism and in the negative regulation of insulin secretion by beta cells.[56] UCP2 knockout mice exhibit HH, suggesting an involvement of UCP2 in insulin secretion. A few patients have been described with mild HH due to loss of function mutations in the UCP2 gene.[57] Parental-inherited heterozygous UCP2 variants encoding amino-acid changes were found in two unrelated children with CHI.[57] Functional assays in yeast and in insulin-secreting cells revealed an impaired activity of UCP2 mutants.[57]
HISTOLOGICAL SUBTYPES OF CONGENITAL HYPERINSULINISM
There are two major histological subtypes of CHI; diffuse and focal.[58] The diffuse form consists of hyper-functioning pancreatic β-cells and affects the whole of pancreas. The most common causes of diffuse CHI are the recessive and dominant mutations in ABCC8 and KCNJ11. Patients with diffuse disease due to recessive mutations in ABCC8 and KCNJ11 do not usually respond to diazoxide.
The second histological subtype of CHI is the focal disease, which involves a small localized region of pancreas (2-10 mm in diameter). It is characterized by nodular hyperplasia of islet-like cell clusters, including ductuloinsular complexes and giant β-cell nuclei surrounded by a histologically and functionally normal pancreatic tissue.[59] The focal lesions can sometimes be deeply embedded within the pancreatic tissue.
The focal form has a distinctive genetic etiology from that of the diffuse disease and involves two independent events, the first of which is the inheritance of a paternal mutation in ABCC8 or KCNJ11.[60] The second event is the somatic loss of the maternal 11p allele (11p15.1 to 11p15.5) involving the ABCC8 and KCNJ11 region within the focal lesion.[61] This paternal uniparental disomy unmasks the paternally inherited KATP channel mutation, which leads to altered expression of a number of imprinted genes, including the maternally expressed tumour suppressor genes H19 and CDKN1C, and the paternally expressed growth factor IGF2.[61] These events eventually give rise to the increase in proliferation of β-cells evolving into a focal adenomatous hyperplasia. The focal disease is always sporadic in origin.
DIFFERENTIAL DIAGNOSIS
Transient HH
Transient HH generally refers to the group of patients in whom HH resolves spontaneously within few days after birth. Transient HH is associated with certain risk factors such as maternal diabetes mellitus (gestational or insulin dependent), intra-uterine growth retardation, perinatal asphyxia, erythroblastosis fetalis, maternal use of drugs like sulphonylureas, and intravenous maternal glucose infusions during labor. Occasionally, some patients with intra-uterine growth retardation and perinatal asphyxia have a protracted form of HH which resolves over several months and may require treatment with diazoxide.[62] The mechanism of transient HH is not clear.
Metabolic conditions associated with HH
Congenital disorders of glycosylation
Congenital disorders of glycosylation (CDG) are a rapidly evolving family of inherited multisystem disorders resulting from defects in the synthesis of the glycan moiety of glycoconjugates (mainly glycoproteins or glycolipids) or in the attachment of glycans to macromolecules.[63] CDG type Ia is the most common and is caused by mutations in the phosphomannomutase 2 gene (PMM2) gene. The clinical spectrum of CDG type Ia has expanded since its initial description to now include rare features such as HH congenital nephrotic syndrome and obstructive cardiomyopathy.[64] A milder disease with single organ involvement, presenting as isolated HH has been reported in a female patient.[65]
HH as a leading symptom has been described predominantly in CDG type Ib (phosphomannose-isomerase deficiency).[64] The phenotype is characterized by protein-losing enteropathy, congenital hepatic fibrosis, and coagulopathy without overt neurologic manifestations that are commonly seen in other CDGs. Early diagnosis is essential, because patients can be successfully treated with oral mannose.[66] HH has also been described in patients with CDG Id.[67]
The precise mechanism for insulin dysregulation is unknown, but the rapid resolution of HH in CDG type Ib patients with oral mannose supplementation suggests a role of glycosylation in maintenance of normoglycemia, perhaps at the level of the sulfonylurea receptor. CDGs should be considered in patients with HH of undiagnosed etiology.
Postprandial forms of HH
Postprandial hyperinsulinemic hypoglycemia (PPHH) refers to the development of hypoglycemia within a few hours of meal ingestion. It is associated with inappropriate insulin secretion in response to the meal. The most common cause is due to the “dumping” syndrome in infants who have undergone gastro-oesophageal surgery.[68] It has also been observed in adult patients who have undergone gastric bypass surgery for morbid obesity.[69] It has been noted that children with PPHH after Nissen fundoplication have abnormally exaggerated secretion of glucagon like peptide-1 (GLP-1) which may contribute to the exaggerated insulin surge and resultant hypoglycemia.[70] PPHH is also observed in the insulin autoimmune syndrome which is characterized by the presence of insulin-binding autoantibodies in subjects who have not been previously exposed to exogenous insulin.[71]
A syndrome of autosomal dominant PPHH with onset in adolescence to adulthood and linked to a mutation (Arg1174Gln) in the insulin receptor kinase gene has been reported.[72] Impaired insulin clearance and insulin resistance due to mutations in insulin receptor have been hypothesized to affect insulin action differently in various tissues leading to hypoglycemia.
In adults, a syndrome of “non-insulinoma pancreatogenous” PPHH has been recognized.[73] These patients demonstrate neuroglycopenic episodes from hypoglycemia within 4hrs of meal ingestion and have negative 72 hrs controlled fasts. The exact mechanism of hypoglycemia is not clear.
Syndromes associated with HH
A large number of developmental syndromes may present in the newborn period with HH [Table 1]. The most common syndrome associated with HH is Beckwith-Wiedemann syndrome (BWS).[74] This syndrome is characterized by prenatal and/or postnatal overgrowth, macroglossia, anterior abdominal wall defects, organomegaly, hemihypertrophy, ear lobe creases, helical pits, and renal tract abnormalities. HH is observed in about 50% of patients with BWS and in the vast majority of patients with BWS, HH is usually transient and resolves spontaneously in a few days.[74] However, a small number of patients (5% of cases), have persistent HH requiring medical therapy or even sub-total pancreatectomy.
Table 1.
Summary of the syndromes associated with hyperinsulinaemic hypoglycaemia
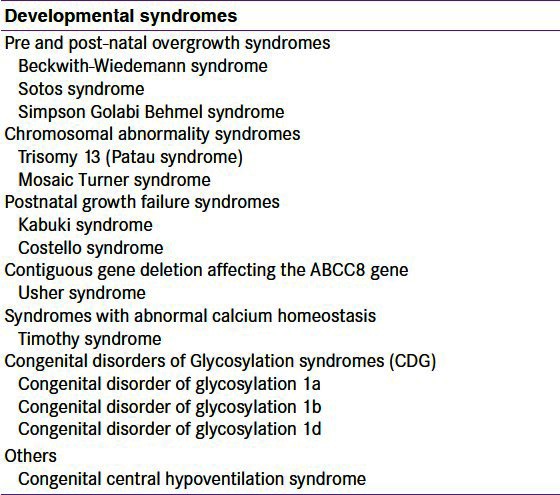
Other causes of HH
An insulinoma must be considered in older children or adolescents with HH.[75] Insulinoma may be a part of multiple endocrine neoplasia syndrome type 1 (MEN1) and hence a family history may provide a diagnostic clue in the familial cases. Munchausen by proxy can present as factitious HH due to administration of insulin or anti diabetic drugs such as sulphonylureas. In some cases, this has led to misdiagnosis and consequent pancreatectomy.[76]
CLINICAL PRESENTATION
CHI most commonly presents in the newborn but it can also present during infancy childhood and rarely in adults. The clinical presentation of CHI is most severe in the newborn and may be quite subtle in the infancy and childhood periods. The CHI due to recessive mutations in ABCC8/KCNJ11 genes is usually refractory to oral feeds and requires high concentrations of intravenous glucose to maintain normoglycemia.[1] However, the milder forms may be able to maintain normoglycemia on oral feeds. Hypoglycemic symptoms may vary from being non-specific (such as poor feeding, lethargy and irritability) to severe (such as apnea, seizures or coma).
As a result of the foetal hyperinsulinemia, newborns with CHI may be macrosomic, however; the absence of macrosomia does not exclude CHI. Hypertrophic cardiomyopathy and hepatomegaly is observed in some patients with CHI. The mechanism of cardiomyopathy and hepatomegaly in these patients is unclear but might be related to the effect of fetal hyperinsulinemia.[1] In some patients, hypoglycemia and associated symptoms manifest only following protein ingestion or exercise. In dumping syndrome, hypoglycemia occurs typically post feed. Hence it is important to ask these specific questions in the history to identify the aetiology.
DIAGNOSIS OF CONGENITAL HYPERINSULINISM
It is essential to make an early diagnosis of CHI so as to avoid hypoglycemic brain injury hence clinicians should have a low threshold for recognizing these patients. Any patient with recurrent or persistent hypoglycemia can potentially have CHI. CHI is the only condition, wherein hypoglycemia can persist despite continuous intravenous administration of glucose. An intravenous glucose infusion rate of >8 mg/kg/min (normal 4-6 mg/kg/min) is virtually diagnostic of CHI.[1] Factors like duration of fasting, protein sensitivity (hypoglycemia precipitated by meals) and hypoglycemia induced by exercise aid the diagnosis of milder forms of CHI.
The key feature of CHI is an inappropriate concentration of serum insulin (and/or c-peptide) for the level of blood glucose (spontaneous or provoked). Any detectable insulin during an episode of hypoglycemia is abnormal. However, some assays (Delphia) will detect insulin even when the blood glucose is low making a cut off more difficult. The other biochemical features are inappropriately low levels of serum ketone bodies and fatty acids during the hypoglycemic episode, which are secondary to the metabolic effects of in appropriate insulin levels. There is no correlation between the serum insulin concentration and the severity of the hypoglycemia.[77] The level of hypoglycaemia is not only dependent on augmented insulin secretion, but also on the lack of insulin switch off at low glucose levels. An elevated C peptide could be of additional diagnostic benefit. The diagnostic criteria for HH are summarized in Table 2.
Table 2.
Diagnostic criteria for patients with hyperinsulinaemic hypoglycaemia

A rise in the blood glucose concentration of >1.5 mmol/l within 30 minutes following an intramuscular/intravenous injection of glucagon at the time of hypoglycemia provides supportive evidence in some patients.[78] An improvement in hypoglycemia to a subcutaneous dose of octreotide may also aid diagnosis along with decreased serum levels of insulin growth factor binding protein 1 (IGFBP-1) as insulin suppresses the transcription of the IGFBP-1 gene.[79] HI/HA syndrome is a possibility when elevated serum ammonia concentration is noted in a patient with CHI.[25] Raised plasma hydroxybutyrylcarnitine and urinary 3-hydroxyglutarate might suggest HADH deficiency.[30]
Diagnosis of certain types of CHI needs provocation testing. Patients with HI/HA syndrome have fasting as well as protein induced hypoglycemia. A protein/leucine load test will be necessary to demonstrate hypoglycemia in HI/HA patients.[27] Similarly, patients with exercise induced CHI will require a formal exercise test and or a pyruvate load to demonstrate hypoglycemia.[48,49]
As CHI is a rare disorder, it is important to network with the local tertiary centers where the expertise is available to guide on appropriate diagnosis and management.
MANAGEMENT OF CONGENITAL HYPERINSULINISM
A prompt diagnosis and initiation of appropriate management will reduce the risk of brain injury in these patients. A higher threshold of blood glucose concentration is recommended to diagnose hypoglycemia and initiate treatment due to the metabolic effects of high insulin levels.[2] It is important to maintain the blood glucose concentrations within the normal range (3.5-6 mmol/l),[2] which may necessitate the use of high concentrations of glucose through a central venous catheter. A combination of intravenous fluids and oral feeds with a glucose polymer (such as Maxijul or Polycal) can be used to simulate orality and maintain normoglycaemia.
In situations where it is difficult to secure a venous access, intramuscular glucagon (0.5-1 mg) can be administered in order to temporarily improve blood glucose concentrations.[1] Glucagon acts immediately by releasing glycogen stores from the liver and also has actions on gluconeogenesis, ketogenesis and lipolysis. It is important to follow this with intravenous glucose infusion to prevent rebound hypoglycemia (as glucagon in high doses causes paradoxical insulin secretion). Glucagon can also be administered as continuous intravenous or subcutaneous infusion to stabilize blood glucose concentrations in the acute management of infants with CHI.
Figure 2 gives an overview of the management of patients with CHI.
Figure 2.

Outline of the suggested diagnostic and management cascade of patients presenting with CHI. The assessment of the response to diazoxide is critical in terms of planning further investigations
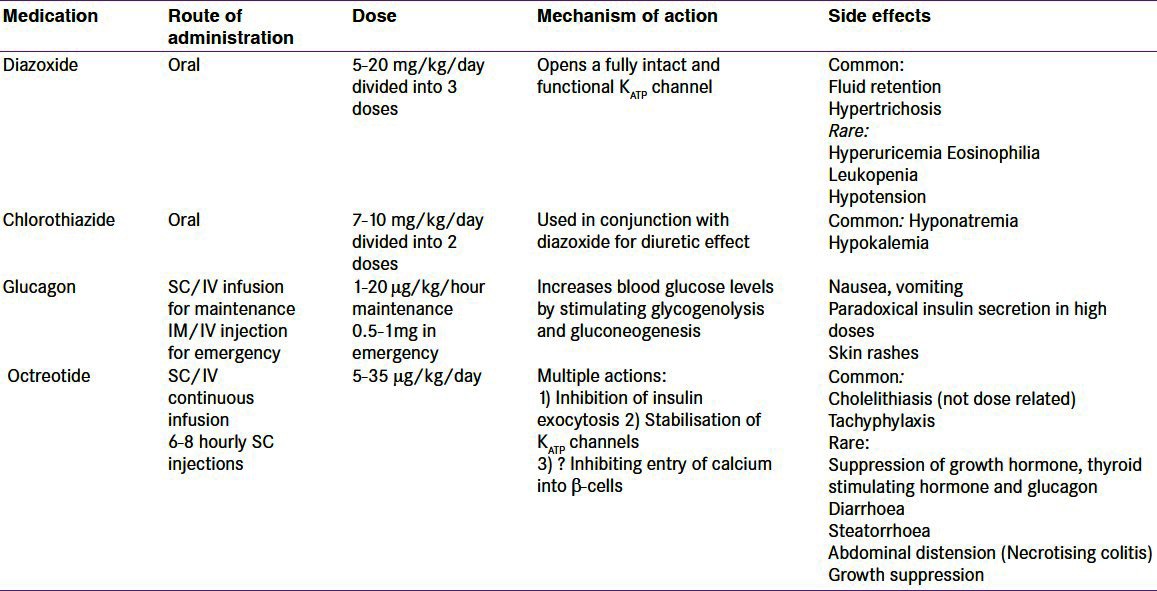
The mainstay of medical therapy is diazoxide, which is used as a first line drug.[1] Diazoxide binds and activates intact KATP channels, thereby reducing the insulin secretion. The side effects include fluid retention and hypertrichosis. The fluid retention is mostly observed in the neonatal period, and may cause cardiac failure. A thiazide diuretic is usually added to prevent fluid retention. However, routine use of thiazide diuretic is not necessary in older children when there is no clinical evidence of fluid retention. Diazoxide responsiveness is noted when (a) feeding with normal frequency and volume (b) able to fast appropriately for age and maintain normal blood glucose levels (c) serum insulin level low or undetectable at the end of the fast (d) appropriate increase in serum fatty acids and ketone bodies at the end of the fast. However, diazoxide is generally ineffective in diffuse CHI due to inactivating mutations in ABCC8 and KCNJ11 and in most patients with focal CHI.
Rapid genetic analysis for mutations in ABCC8 and KCNJ11 allows identification of the majority of patients with diffuse disease (homozygous or compound heterozygous mutations in ABCC8 and KCNJ11).[80] Patients with a paternal mutation in ABCC8 and KCNJ11 (or those with no mutations in these genes) potentially have focal disease. It is important to precisely locate the lesion in the pancreas preoperatively, as removal of this lesion can be curative in focal CHI. Fluorine-18-L dihydroxyphenylalanine positron emission tomography (18F-DOPA PET) offers precise pre-operative localization of the focal lesion, thus guiding the extent of surgical resection.[8] The uptake of the positron emitting tracer 18F-DOPA is increased in β-cells with a high rate of insulin synthesis and secretion compared to unaffected areas allowing visualization of the focal lesion. The sensitivity for detecting focal lesions varies between 88 and 94% with a specificity of 100%.[81]
The treatment options for the diazoxide unresponsive infants with confirmed (genetically/by 18F-DOPA-PET scanning) diffuse disease includes long term medical therapy with octreotide or surgery. Some of these patients respond to long term subcutaneous octreotide injections in combination with frequent feeding[82] as the hypoglycemia gradually gets milder over time. A gastrostomy may be necessary in these patients to allow delivery of daytime bolus and overnight continuous feeds. The use of a long acting octreotide formulation has now been described in two studies.[83,84] Tachyphylaxis has been observed on long term use of Octreotide.[85] Recently, necrotising enterocolitis (NEC) has been described in infants treated with octreotide especially in the presence of other risk factors.[86]
Table 3 summarizes the medical therapy for CHI.
Table 3.
Summary of the drugs used in the management of patients with hyperinsulinaemic hypoglycaemia
In the severe forms of diffuse CHI patients, near total pancreatectomy may be required when all medical therapy has failed.[87] Near-total pancreatectomy is associated with a high incidence of diabetes mellitus and pancreatic exocrine insufficiency. The surgery is traditionally carried out by open approach and is associated with peri and post-operative complications. The use of laparoscopy represents a new approach to the diagnosis and management of infants with both diffuse and focal CHI.[88]
SUMMARY
CHI is caused by the unregulated secretion of insulin from pancreatic β-cells. The molecular basis of CHI involves defects in key genes (ABCC8, KCNJ11, GLUD1, GCK, HADH, SLC16A1, HNF4A and UCP2) which regulate insulin secretion. The advent of rapid genetic analysis, imaging with 18F-DOPA-PET/CT and new surgical techniques have changed the clinical approach to these complex patients.
Footnotes
Source of Support: Nil
Conflict of Interest: No
REFERENCES
- 1.Aynsley-Green A, Hussain K, Hall J, Saudubray JM, Nihoul-Fékété C, De Lonlay-Debeney P, et al. Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed. 2000;82:F98–107. doi: 10.1136/fn.82.2.F98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hussain K, Blankenstein O, De Lonlay P, Christesen HT. Hyperinsulinaemic hypoglycaemia: Biochemical basis and the importance of maintaining normoglycaemia during management. Arch Dis Child. 2007;92:568–70. doi: 10.1136/adc.2006.115543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Senniappan S, Shanti B, James C, Hussain K. Hyperinsulinaemic hypoglycaemia: Genetic mechanisms, diagnosis and management. J Inherit Metab Dis. 2012;35:589–601. doi: 10.1007/s10545-011-9441-2. [DOI] [PubMed] [Google Scholar]
- 4.Bruining GJ. Recent advances in hyperinsulinism and the pathogenesis of diabetes mellitus. Curr Opinion Pediatr. 1990;2:758–65. [Google Scholar]
- 5.Mathew PM, Young JM, Abu-Osba YK, Mulhern BD, Hammoudi S, Hamdan JA, et al. Persistent neonatal hyperinsulinism. Clin Pediatr (Phila) 1988;27:148–51. doi: 10.1177/000992288802700307. [DOI] [PubMed] [Google Scholar]
- 6.James C, Kapoor RR, Ismail D, Hussain K. The genetic basis of congenital hyperinsulinism. J Med Genet. 2009;46:289–99. doi: 10.1136/jmg.2008.064337. [DOI] [PubMed] [Google Scholar]
- 7.Rahier J, Guiot Y, Sempoux C. Persistent hyperinsulinaemic hypoglycaemia of infancy: A heterogeneous syndrome unrelated to nesidioblastosis. Arch Dis Child Fetal Neonatal Ed. 2000;82:F108–12. doi: 10.1136/fn.82.2.F108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otonkoski T, Näntö-Salonen K, Seppänen M, Veijola R, Huopio H, Hussain K, et al. Noninvasive diagnosis of focal hyperinsulinism of infancy with [18F]-DOPA positron emission tomography. Diabetes. 2006;55:13–8. [PubMed] [Google Scholar]
- 9.Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature. 1984;312:446–8. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- 10.Inagaki N, Gonoi T, Clement JP, 4th, Namba N, Inazawa J, Gonzalez G, et al. Reconstitution of IKATP: An inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–70. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- 11.Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K (ATP) channels. Neuron. 1999;22:537–48. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
- 12.Tucker SJ, Gribble FM, Proks P, Trapp S, Ryder TJ, Haug T, et al. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 1998;17:3290–6. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook DL, Hales CN. Intracellular ATP directly blocks K + channels in pancreatic B-cells. Nature. 1984;311:271–3. doi: 10.1038/311271a0. [DOI] [PubMed] [Google Scholar]
- 14.Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, 4 th, Boyd AE, 3rd, González G, et al. Cloning of the beta cell high-affinity sulfonylurea receptor: A regulator of insulin secretion. Science. 1995;268:423–6. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- 15.Kane C, Shepherd RM, Squires PE, Johnson PR, James RF, Milla PJ, et al. Loss of functional KATP channels in pancreatic beta-cells causes persistent hyperinsulinemic hypoglycemia of infancy. Nat Med. 1996;2:1344–7. doi: 10.1038/nm1296-1344. [DOI] [PubMed] [Google Scholar]
- 16.Flanagan SE, Clauin S, Bellanné-Chantelot C, de Lonlay P, Harries LW, Gloyn AL, et al. Update of mutations in the genes encoding the pancreatic beta-cell K (ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum Mutat. 2009;30:170–80. doi: 10.1002/humu.20838. [DOI] [PubMed] [Google Scholar]
- 17.Crane A, Aguilar-Bryan L. Assembly, maturation, and turnover of K (ATP) channel subunits. J Biol Chem. 2004;279:9080–90. doi: 10.1074/jbc.M311079200. [DOI] [PubMed] [Google Scholar]
- 18.Cartier EA, Conti LR, Vandenberg CA, Shyng SL. Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc Natl Acad Sci USA. 2001;98:2882–7. doi: 10.1073/pnas.051499698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan FF, Lin YW, MacMullen C, Ganguly A, Stanley CA, Shyng SL. Congenital hyperinsulinism associated ABCC8 mutations that cause defective trafficking of ATP-sensitive K+channels: Identification and rescue. Diabetes. 2007;56:2339–48. doi: 10.2337/db07-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin YW, MacMullen C, Ganguly A, Stanley CA, Shyng SL. A novel KCNJ11 mutation associated with congenital hyperinsulinism reduces the intrinsic open probability of beta-cell ATP-sensitive potassium channels. J Biol Chem. 2006;281:3006–12. doi: 10.1074/jbc.M511875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huopio H, Reimann F, Ashfield R, Komulainen J, Lenko HL, Rahier J, et al. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J Clin Invest. 2000;106:897–906. doi: 10.1172/JCI9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, et al. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest. 2008;118:2877–86. doi: 10.1172/JCI35414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flanagan SE, Kapoor RR, Banerjee I, Hall C, Smith VV, Hussain K, et al. Dominantly acting ABCC8 mutations in patients with medically unresponsive hyperinsulinaemic hypoglycaemia. Clin Genet. 2011;79:582–7. doi: 10.1111/j.1399-0004.2010.01476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stanley CA, Lieu YK, Hsu BY, Burlina AB, Greenberg CR, Hopwood NJ, et al. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med. 1998;338:1352–7. doi: 10.1056/NEJM199805073381904. [DOI] [PubMed] [Google Scholar]
- 25.Treberg JR, Clow KA, Greene KA, Brosnan ME, Brosnan JT. Systemic activation of glutamate dehydrogenase increases renal ammoniagenesis: Implications for the hyperinsulinism/hyperammonemia syndrome. Am J Physiol Endocrinol Metab. 2010;298:E1219–25. doi: 10.1152/ajpendo.00028.2010. [DOI] [PubMed] [Google Scholar]
- 26.Hsu BY, Kelly A, Thornton PS, Greenberg CR, Dilling LA, Stanley CA. Protein-sensitive and fasting hypoglycemia in children with the hyperinsulinism/hyperammonemia syndrome. J Pediatr. 2001;138:383–9. doi: 10.1067/mpd.2001.111818. [DOI] [PubMed] [Google Scholar]
- 27.Kapoor RR, Flanagan SE, Fulton P, Chakrapani A, Chadefaux B, Ben-Omran T, et al. Hyperinsulinism-hyperammonaemia syndrome: Novel mutations in the GLUD1 gene and genotype-phenotype correlations. Eur J Endocrinol. 2009;161:731–5. doi: 10.1530/EJE-09-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bahi-Buisson N, Roze E, Dionisi C, Escande F, Valayannopoulos V, Feillet F, et al. Neurological aspects of hyperinsulinism-hyperammonaemia syndrome. Dev Med Child Neurol. 2008;50:945–9. doi: 10.1111/j.1469-8749.2008.03114.x. [DOI] [PubMed] [Google Scholar]
- 29.Clayton PT, Eaton S, Aynsley-Green A, Edginton M, Hussain K, Krywawych S, et al. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. J Clin Invest. 2001;108:457–65. doi: 10.1172/JCI11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vredendaal PJ, van den Berg IE, Malingré HE, Stroobants AK, Olde Weghuis DE, Berger R. Human short-chain L-3-hydroxyacyl-CoA dehydrogenase: Cloning and characterization of the coding sequence. Biochem Biophys Res Commun. 1996;223:718–23. doi: 10.1006/bbrc.1996.0961. [DOI] [PubMed] [Google Scholar]
- 31.Hardy OT, Hohmeier HE, Becker TC, Manduchi E, Doliba NM, Gupta RK, et al. Functional genomics of the beta-cell: Short-chain 3-hydroxyacyl-coenzyme A dehydrogenase regulates insulin secretion independent of K+currents. Mol Endocrinol. 2007;21:765–73. doi: 10.1210/me.2006-0411. [DOI] [PubMed] [Google Scholar]
- 32.Molven A, Matre GE, Duran M, Wanders RJ, Rishaug U, Njølstad PR, et al. Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes. 2004;53:221–7. doi: 10.2337/diabetes.53.1.221. [DOI] [PubMed] [Google Scholar]
- 33.Martins E, Cardoso ML, Rodrigues E, Barbot C, Ramos A, Bennett MJ, et al. Short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: The clinical relevance of an early diagnosis and report of four new cases. J Inherit Metab Dis. 2011;34:835–42. doi: 10.1007/s10545-011-9287-7. [DOI] [PubMed] [Google Scholar]
- 34.Kapoor RR, James C, Flanagan SE, Ellard S, Eaton S, Hussain K. 3-Hydroxyacyl-coenzyme A dehydrogenase deficiency and hyperinsulinemic hypoglycemia: Characterization of a novel mutation and severe dietary protein sensitivity. J Clin Endocrinol Metab. 2009;94:2221–5. doi: 10.1210/jc.2009-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li C, Chen P, Palladino A, Narayan S, Russell LK, Sayed S, et al. Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J Biol Chem. 2010;285:31806–18. doi: 10.1074/jbc.M110.123638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flanagan SE, Patch AM, Locke JM, Akcay T, Simsek E, Alaei M, et al. Genome-wide homozygosity analysis reveals HADH mutations as a common cause of diazoxide-responsive hyperinsulinemic-hypoglycemia in consanguineous pedigrees. J Clin Endocrinol Metab. 2011;96:E498–502. doi: 10.1210/jc.2010-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupta RK, Vatamaniuk MZ, Lee CS, Flaschen RC, Fulmer JT, Matschinsky FM, et al. The MODY1 gene HNF-4alpha regulates selected genes involved in insulin secretion. J Clin Invest. 2005;115:1006–15. doi: 10.1172/JCI200522365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, et al. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1) Nature. 1996;384:458–60. doi: 10.1038/384458a0. [DOI] [PubMed] [Google Scholar]
- 39.Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, et al. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. 2007;4:e118. doi: 10.1371/journal.pmed.0040118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapoor RR, Locke J, Colclough K, Wales J, Conn JJ, Hattersley AT, et al. Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young due to heterozygous HNF4A mutations. Diabetes. 2008;57:1659–63. doi: 10.2337/db07-1657. [DOI] [PubMed] [Google Scholar]
- 41.Fajans SS, Bell GI. Macrosomia and neonatal hypoglycaemia in RW pedigree subjects with a mutation (Q268X) in the gene encoding hepatocyte nuclear factor 4alpha (HNF4A) Diabetologia. 2007;50:2600–1. doi: 10.1007/s00125-007-0833-7. [DOI] [PubMed] [Google Scholar]
- 42.Pingul MM, Hughes N, Wu A, Stanley CA, Gruppuso PA. Hepatocyte nuclear factor 4α gene mutation associated with familial neonatal hyperinsulinism and maturity-onset diabetes of the young. J Pediatr. 2011;158:852–4. doi: 10.1016/j.jpeds.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Flanagan SE, Kapoor RR, Mali G, Cody D, Murphy N, Schwahn B, et al. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol. 2010;162:987–92. doi: 10.1530/EJE-09-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gremlich S, Nolan C, Roduit R, Burcelin R, Peyot ML, Delghingaro-Augusto V, et al. Pancreatic islet adaptation to fasting is dependent on peroxisome proliferator-activated receptor alpha transcriptional up-regulation of fatty acid oxidation. Endocrinology. 2005;146:375–82. doi: 10.1210/en.2004-0667. [DOI] [PubMed] [Google Scholar]
- 45.Prentki M, Joly E, El-Assaad W, Roduit R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: Role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes. 2002;51(Suppl 3):S405–13. doi: 10.2337/diabetes.51.2007.s405. [DOI] [PubMed] [Google Scholar]
- 46.Meissner T, Otonkoski T, Feneberg R, Beinbrech B, Apostolidou S, Sipilä I, et al. Exercise induced hypoglycaemic hyperinsulinism. Arch Dis Child. 2001;84:254–7. doi: 10.1136/adc.84.3.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meissner T, Friedmann B, Okun JG, Schwab MA, Otonkoski T, Bauer T, et al. Massive insulin secretion in response to anaerobic exercise in exercise-induced hyperinsulinism. Horm Metab Res. 2005;37:690–4. doi: 10.1055/s-2005-870583. [DOI] [PubMed] [Google Scholar]
- 48.Otonkoski T, Kaminen N, Ustinov J, Lapatto R, Meissner T, Mayatepek E, et al. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes. 2003;52:199–204. doi: 10.2337/diabetes.52.1.199. [DOI] [PubMed] [Google Scholar]
- 49.Otonkoski T, Jiao H, Kaminen-Ahola N, Tapia-Paez I, Ullah MS, Parton LE, et al. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic beta cells. Am J Hum Genet. 2007;81:467–74. doi: 10.1086/520960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao C, Wilson MC, Schuit F, Halestrap AP, Rutter GA. Expression and distribution of lactate/monocarboxylate transporter isoforms in pancreatic islets and the exocrine pancreas. Diabetes. 2001;50:361–6. doi: 10.2337/diabetes.50.2.361. [DOI] [PubMed] [Google Scholar]
- 51.Matschinsky FM. Regulation of pancreatic beta-cell glucokinase: From basics to therapeutics. Diabetes. 2002;51(Suppl 3):S394–404. doi: 10.2337/diabetes.51.2007.s394. [DOI] [PubMed] [Google Scholar]
- 52.Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, et al. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med. 1998;338:226–30. doi: 10.1056/NEJM199801223380404. [DOI] [PubMed] [Google Scholar]
- 53.Wabitsch M, Lahr G, Van de Bunt M, Marchant C, Lindner M, von Puttkamer J, et al. Heterogeneity in disease severity in a family with a novel G68V GCK activating mutation causing persistent hyperinsulinaemic hypoglycaemia of infancy. Diabet Med. 2007;24:1393–9. doi: 10.1111/j.1464-5491.2007.02285.x. [DOI] [PubMed] [Google Scholar]
- 54.Christesen HB, Jacobsen BB, Odili S, Buettger C, Cuesta-Munoz A, Hansen T, et al. The second activating glucokinase mutation (A456V): Implications for glucose homeostasis and diabetes therapy. Diabetes. 2002;51:1240–6. doi: 10.2337/diabetes.51.4.1240. [DOI] [PubMed] [Google Scholar]
- 55.Cuesta-Muñoz AL, Huopio H, Otonkoski T, Gomez-Zumaquero JM, Näntö-Salonen K, Rahier J, et al. Severe persistent hyperinsulinemic hypoglycemia due to a de novo glucokinase mutation. Diabetes. 2004;53:2164–8. doi: 10.2337/diabetes.53.8.2164. [DOI] [PubMed] [Google Scholar]
- 56.Chan CB, De Leo D, Joseph JW, McQuaid TS, Ha XF, Xu F, et al. Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: Mechanism of action. Diabetes. 2001;50:1302–10. doi: 10.2337/diabetes.50.6.1302. [DOI] [PubMed] [Google Scholar]
- 57.González-Barroso MM, Giurgea I, Bouillaud F, Anedda A, Bellanné-Chantelot C, Hubert L, et al. Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS One. 2008;3:e3850. doi: 10.1371/journal.pone.0003850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rahier J, Guiot Y, Sempoux C. Persistent hyperinsulinaemic hypoglycaemia of infancy: A heterogeneous syndrome unrelated to nesidioblastosis. Arch Dis Child Fetal Neonatal Ed. 2000;82:F108–12. doi: 10.1136/fn.82.2.F108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sempoux C, Guiot Y, Jaubert F, Rahier J. Focal and diffuse forms of congenital hyperinsulinism: The keys for differential diagnosis. Endocr Pathol. 2004;15:241–6. doi: 10.1385/ep:15:3:241. [DOI] [PubMed] [Google Scholar]
- 60.Verkarre V, Fournet JC, de Lonlay P, Gross-Morand MS, Devillers M, Rahier J, et al. Paternal mutation of the sulfonylurea receptor (SUR1) gene and maternal loss of 11p15 imprinted genes lead to persistent hyperinsulinism in focal adenomatous hyperplasia. J Clin Invest. 1998;102:1286–91. doi: 10.1172/JCI4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fournet JC, Mayaud C, de Lonlay P, Gross-Morand MS, Verkarre V, Castanet M, et al. Unbalanced expression of 11p15 imprinted genes in focal forms of congenital hyperinsulinism: Association with a reduction to homozygosity of a mutation in ABCC8 or KCNJ11. Am J Pathol. 2001;158:2177–84. doi: 10.1016/S0002-9440(10)64689-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fafoula O, Alkhayyat H, Hussain K. Prolonged hyperinsulinaemic hypoglycaemia in newborns with intrauterine growth retardation. Arch Dis Child Fetal Neonatal Ed. 2006;91:F467. doi: 10.1136/adc.2006.095919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jaeken J. Komrower Lecture. Congenital disorders of glycosylation (CDG): It's all in it! J Inherit Metab Dis. 2003;26:99–118. doi: 10.1023/a:1024431131208. [DOI] [PubMed] [Google Scholar]
- 64.Böhles H, Sewell AA, Gebhardt B, Reinecke-Lüthge A, Klöppel G, Marquardt T. Hyperinsulinaemic hypoglycaemia–leading symptom in a patient with congenital disorder of glycosylation Ia (phosphomannomutase deficiency) J Inherit Metab Dis. 2001;24:858–62. doi: 10.1023/a:1013944308881. [DOI] [PubMed] [Google Scholar]
- 65.Shanti B, Silink M, Bhattacharya K, Howard NJ, Carpenter K, Fietz M, et al. Congenital disorder of glycosylation type Ia: Heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis. 2009 doi: 10.1007/s10545-009-1180-2. [DOI] [PubMed] [Google Scholar]
- 66.de Lonlay P, Cuer M, Vuillaumier-Barrot S, Beaune G, Castelnau P, Kretz M, et al. Hyperinsulinemic hypoglycemia as a presenting sign in phosphomannose isomerase deficiency: A new manifestation of carbohydrate-deficient glycoprotein syndrome treatable with mannose. J Pediatr. 1999;135:379–83. doi: 10.1016/s0022-3476(99)70139-3. [DOI] [PubMed] [Google Scholar]
- 67.Sun L, Eklund EA, Chung WK, Wang C, Cohen J, Freeze HH. Congenital disorder of glycosylation id presenting with hyperinsulinemic hypoglycemia and islet cell hyperplasia. J Clin Endocrinol Metab. 2005;90:4371–5. doi: 10.1210/jc.2005-0250. [DOI] [PubMed] [Google Scholar]
- 68.Bufler P, Ehringhaus C, Koletzko S. Dumping syndrome: A common problem following Nissen fundoplication in young children. Pediatr Surg Int. 2001;17:351–5. doi: 10.1007/s003830000525. [DOI] [PubMed] [Google Scholar]
- 69.Foster-Schubert KE. Hypoglycemia complicating bariatric surgery: Incidence and mechanisms. Curr Opin Endocrinol Diabetes Obes. 2011;18:129–33. doi: 10.1097/MED.0b013e32834449b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Palladino AA, Sayed S, Levitt Katz LE, Gallagher PR, De León DD. Increased glucagon-like peptide-1 secretion and postprandial hypoglycemia in children after Nissen fundoplication. J Clin Endocrinol Metab. 2009;94:39–44. doi: 10.1210/jc.2008-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hirata Y. [Insulin autoimmune syndrome] Nihon Rinsho. 1973;31:2227–31. [PubMed] [Google Scholar]
- 72.Højlund K, Hansen T, Lajer M, Henriksen JE, Levin K, Lindholm J, et al. A novel syndrome of autosomal-dominant hyperinsulinemic hypoglycemia linked to a mutation in the human insulin receptor gene. Diabetes. 2004;53:1592–8. doi: 10.2337/diabetes.53.6.1592. [DOI] [PubMed] [Google Scholar]
- 73.Service FJ, Natt N, Thompson GB, Grant CS, van Heerden JA, Andrews JC, et al. Noninsulinoma pancreatogenous hypoglycemia: A novel syndrome of hyperinsulinemic hypoglycemia in adults independent of mutations in Kir6.2 and SUR1 genes. J Clin Endocrinol Metab. 1999;84:1582–9. doi: 10.1210/jcem.84.5.5645. [DOI] [PubMed] [Google Scholar]
- 74.Munns CF, Batch JA. Hyperinsulinism and Beckwith-Wiedemann syndrome. Arch Dis Child Fetal Neonatal Ed. 2001;84:F67–9. doi: 10.1136/fn.84.1.F67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shin JJ, Gorden P, Libutti SK. Insulinoma: Pathophysiology, localization and management. Future Oncol. 2010;6:229–37. doi: 10.2217/fon.09.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Giurgea I, Ulinski T, Touati G, Sempoux C, Mochel F, Brunelle F, et al. Factitious hyperinsulinism leading to pancreatectomy: Severe forms of Munchausen syndrome by proxy. Pediatrics. 2005;116:e145–8. doi: 10.1542/peds.2004-2331. [DOI] [PubMed] [Google Scholar]
- 77.Palladino AA, Bennett MJ, Stanley CA. Hyperinsulinism in infancy and childhood: When an insulin level is not always enough. Clin Chem. 2008;54:256–63. doi: 10.1373/clinchem.2007.098988. [DOI] [PubMed] [Google Scholar]
- 78.Finegold DN, Stanley CA, Baker L. Glycemic response to glucagon during fasting hypoglycemia: An aid in the diagnosis of hyperinsulinism. J Pediatr. 1980;96:257–9. doi: 10.1016/s0022-3476(80)80817-1. [DOI] [PubMed] [Google Scholar]
- 79.Levitt Katz LE, Satin-Smith MS, Collett-Solberg P, Thornton PS, Baker L, Stanley CA, et al. Insulin-like growth factor binding protein-1 levels in the diagnosis of hypoglycemia caused by hyperinsulinism. J Pediatr. 1997;131:193–9. doi: 10.1016/s0022-3476(97)70153-7. [DOI] [PubMed] [Google Scholar]
- 80.Christesen HB, Brusgaard K, Alm J, Sjöblad S, Hussain K, Fenger C, et al. Rapid genetic analysis in congenital hyperinsulinism. Horm Res. 2007;67:184–8. doi: 10.1159/000097063. [DOI] [PubMed] [Google Scholar]
- 81.Hardy OT, Hernandez-Pampaloni M, Saffer JR, Scheuermann JS, Ernst LM, Freifelder R, et al. Accuracy of [18F] fluorodopa positron emission tomography for diagnosing and localizing focal congenital hyperinsulinism. J Clin Endocrinol Metab. 2007;92:4706–11. doi: 10.1210/jc.2007-1637. [DOI] [PubMed] [Google Scholar]
- 82.Glaser B, Landau H, Smilovici A, Nesher R. Persistent hyperinsulinaemic hypoglycaemia of infancy: Long-term treatment with the somatostatin analogue Sandostatin. Clin Endocrinol (Oxf) 1989;31:71–80. doi: 10.1111/j.1365-2265.1989.tb00455.x. [DOI] [PubMed] [Google Scholar]
- 83.Modan-Moses D, Koren I, Mazor-Aronovitch K, Pinhas-Hamiel O, Landau H. Treatment of congenital hyperinsulinism with lanreotide acetate (Somatuline Autogel) J Clin Endocrinol Metab. 2011;96:2312–7. doi: 10.1210/jc.2011-0605. [DOI] [PubMed] [Google Scholar]
- 84.Le Quan Sang KH, Arnoux JB, Mamoune A, Saint-Martin C, Bellanné-Chantelot C, Valayannopoulos V, et al. Successful treatment of congenital hyperinsulinism with long-acting release octreotide. Eur J Endocrinol. 2012;166:333–9. doi: 10.1530/EJE-11-0874. [DOI] [PubMed] [Google Scholar]
- 85.Thornton PS, Alter CA, Katz LE, Baker L, Stanley CA. Short- and long-term use of octreotide in the treatment of congenital hyperinsulinism. J Pediatr. 1993;123:637–43. doi: 10.1016/s0022-3476(05)80969-2. [DOI] [PubMed] [Google Scholar]
- 86.Laje P, Halaby L, Adzick NS, Stanley CA. Necrotizing enterocolitis in neonates receiving octreotide for the management of congenital hyperinsulinism. Pediatr Diabetes. 2010;11:142–7. doi: 10.1111/j.1399-5448.2009.00547.x. [DOI] [PubMed] [Google Scholar]
- 87.Fékété CN, de Lonlay P, Jaubert F, Rahier J, Brunelle F, Saudubray JM. The surgical management of congenital hyperinsulinemic hypoglycemia in infancy. J Pediatr Surg. 2004;39:267–9. doi: 10.1016/j.jpedsurg.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 88.Bax KN, van der Zee DC. The laparoscopic approach toward hyperinsulinism in children. Semin Pediatr Surg. 2007;16:245–51. doi: 10.1053/j.sempedsurg.2007.06.006. [DOI] [PubMed] [Google Scholar]