

Abstract
The current management of thalassemia includes regular transfusion programs and chelation therapy. It is important that physicians be aware that endocrine abnormalities frequently develop mainly in those patients with significant iron overload due to poor compliance to treatment, particularly after the age of 10 years. Since the quality of life of thalassemia patients is a fundamental aim, it is vital to monitor carefully their growth and pubertal development in order to detect abnormalities and to initiate appropriate and early treatment. Abnormalities should be identified and treatment initiated in consultation with a pediatric or an adult endocrinologist and managed accordingly. Appropriate management shall put in consideration many factors such as age, severity of iron overload, presence of chronic liver disease, thrombophilia status, and the presence of psychological problems. All these issues must be discussed by the physician in charge of the patient's care, the endocrinologist and the patient himself. Because any progress in research in the field of early diagnosis and management of growth disorders and endocrine complications in thalassemia should be passed on to and applied adequately to all those suffering from the disease, on the 8 May 2009 in Ferrara, the International Network on Endocrine Complications in Thalassemia (I-CET) was founded in order to transmit the latest information on these disorders to the treating physicians. The I-CET position statement outlined in this document applies to patients with transfusion-dependent thalassemia major to help physicians to anticipate, diagnose, and manage these complications properly.
Keywords: Endocrine complications, growth, guidelines, iron overload, thalassemia, treatment
INTRODUCTION
The hemoglobinopathies (thalassemias and sickle-cell disease) are the most commonly inherited genetic disorders, with some 240 000 infants born annually with major hemoglobinopathies and at least 190 million carriers worldwide.[1,2]
The current management of thalassemia (TM) includes regular transfusion programs and chelation therapy. Current guidelines recommend a pretransfusion threshold not exceeding 9.5% g/dl, which seems to be associated with adequate marrow inhibition and a relatively low iron burden.[2–5] The aim is to transfuse 10-20 ml/kg body weight of packed filtered red cells over a period of 2-3 h every 2 to 5 weeks throughout life.[2–5]
The natural course of the disease is dramatically altered by transfusion side effects, which need to be monitored and treated. Iron overload resulting in end-organ damage and blood-borne infectious agents still represent the principal causes of morbidity and mortality.[1–9]
TRANSFUSION-RELATED IRON OVERLOAD AND IRON TOXICITY
The precise underlying mechanism of iron overload-induced organ dysfunction presently remains unclear. However, when iron levels in the body become too high, this leads to saturation of transferrin, and non-transferrin-bound iron (NTBI) species circulate in the plasma. Unbound iron within cells or in plasma is labile and can redox cycle between Fe2+ and Fe3+, thereby generating reactive oxygen species (ROS), leading to lipid peroxidation. Lipid peroxidation under conditions of iron overload leads to the generation of both unsaturated (malondialdehyde and hydroxynonenal) and saturated (hexanal) aldehydes. Both have been implicated in cellular dysfunction, cytotoxicity, and cell death.[10,11] Certain tissues are particularly susceptible to excess iron incorporation when NTBI is present.
Apart from iron overload, other factors responsible for organ damage have been previously pointed out, including chronic hypoxia due to anemia that may potentiate the toxicity of iron deposition in endocrine glands.[12] Also, viral infections as well as individual susceptibility have been implicated in causing endocrine dysfunction.[13]
IRON CHELATION THERAPY
Current practice is to begin iron chelation therapy when the serum ferritin levels reach 1000 ng/ml or when child reaches 3 years of age or has received about 10-20 transfusions.[2,3,13]
Iron chelators in current clinical use include monotherapy with subcutaneous or intravenous desferrioxamine (DFO), oral deferiprone or oral deferasirox, and combination therapy with SC DFO and deferiprone.[7]
DFO is most commonly self-administered as a subcutaneous infusion over 8-10 h, 3-7 nights weekly. Oral deferiprone and deferasirox are mainly used for patients in whom chelation with DFO has been inadequate or intolerable.[3,13–15] Recently, oral monotherapy with deferasirox has been used, as it is equally effective to DFO and deferiprone.[16]
Effective chelation reduces or prevents iron accumulation and iron-mediated organ damage, resulting in a consistent decrease of morbidity and mortality.[3–5,13–15]
PREMISE OF INTERNATIONAL NETWORK ON ENDOCRINE COMPLICATIONS IN THALASSEMIA ACTIVITIES
Optimal quality care for hemoglobin disorders is the basic prerequisite to achieve long survival and a good quality of life. To achieve this aim in dealing with a multi-organ pathology as encountered in TM, a coordinated multidisciplinary team of specialists is required. Such a team must include an endocrinologist, preferably experienced in the management of hormonal deficiencies caused early in life by transfusion-induced iron overload in TM patients.[17]
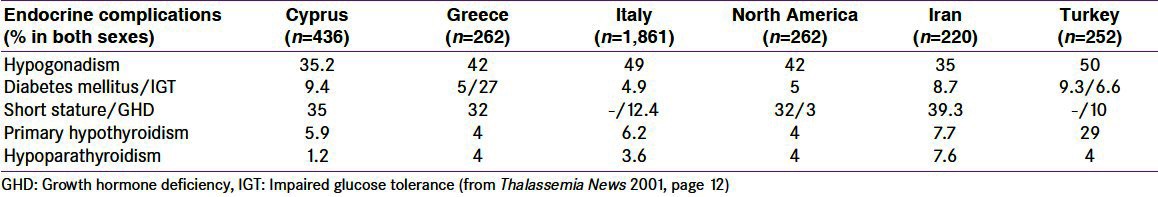
Endocrine complications are very common in multi-transfused TM patients (De Sanctis V, unpublished data, Berlin 2010) [Table 1]. Despite this, a recent survey conducted by the Thalassemia International Federation (TIF) on 316 regularly transfused patients older than 10 years across Europe for the Enerca Project (European Rare Anaemias) showed that only 177 (56%) are ever seen by an endocrinologist. Of these, 134 (42%) are seen at least annually, while the rest seem to be followed occasionally (every 2 years or less).[18] A similar survey was conducted by TIF in the Middle East (data unpublished, 2009) on 90 health professionals who look after TM patients from various countries in the region; 86.4% of them reported a lack of multi-disciplinary support. Of 96 multi-transfused patients from the same region, 62.4% stated that they have never visited an endocrinologist, 3.6% have been seen occasionally (every 2 years or less), and 34% have been seen yearly.[17]
Table 1.
Incidence/prevalence of endocrinopathies in iron overloaded ί-thalassemia major patients in different geographical areas
The major difficulties reported by hematologists or pediatric endocrinologists experienced in TM or TM syndromes[17] in following growth disorders and endocrine complications concern:
Facilities (27%)
“Familiarity” with medical treatment of endocrine complications (40%)
Presence of pediatric endocrinologist for consultation on growth disorders and endocrine complications (27%)
Interpretation of endocrine tests (30%)
Others (17%)
Cost (65%)
OBJECTIVES OF THE INTERNATIONAL NETWORK ON ENDOCRINE COMPLICATIONS IN THALASSEMIA EDUCATIONAL TRAINING PROGRAM
The International Network on Endocrine Complications in Thalassemia (I-CET) was founded on the 8 May 2009, in Ferrara, Italy, to transfer any progress in research in the field of growth and endocrine complications in TM into real-life applications for patients with these disorders.[17]
To accomplish this goal, the endocrine I-CET program aims to train pediatricians/physicians and hematologists to:
Acquire adequate and updated knowledge about growth and endocrine manifestations in patients with TM, TM syndromes, and sickle cell disease;
Improve their ability to select and interpret data originating from laboratory and radiological investigations to solve these problems;
Reach and follow an international acceptable standard on management of these growth and endocrine complications in TM;
Become competent to maintain optimal comprehensive care of children and adolescents suffering from endocrine disorders due to TM and
Encourage research in the field of these disorders in TM syndromes.
COURSE OUTLINE
The course defines and discusses the endocrine disorders in TM and TM syndromes and their relation to the transfusion regimen, iron overload, chelation therapy, and severity of the disease.
Doctors will learn in detail their clinical manifestations and laboratory and imaging tests that are most useful for diagnosis.
In-depth case studies will allow participants to put together and interpret all the clinical, hematological, biochemical, molecular and imaging data to arrive at a probable diagnostic conclusion.
Management plans will be discussed for each endocrine abnormality.
Contents of the I-CET course
Defining TM and explaining the molecular basis and pathophysiology of TM syndromes
Identifying the clinical manifestations of growth, pubertal and endocrine disorders in TM syndromes:
How to measure, interpret, and analyze growth data
How to assess, interpret, and analyze pubertal development and fertility problems
Symptoms and signs of glycemic abnormalities
Symptoms and signs of calcium abnormalities
Imaging abnormalities
Defining growth and pubertal abnormalities in TM including their prevalence per age period (childhood, puberty, adolescence, and adulthood)
Defining glycemic abnormalities and prevalence in TM
Defining calcium abnormalities and prevalence in TM
Defining thyroid and parathyroid abnormalities and prevalence in TM
Identifying proper use and interpreting different laboratory findings in TM including:
RBC and hematologic findings, bilirubin, and hepatic functions
Serum iron and transferrin levels, transferrin saturation and ferritin concentrations and their relation, especially high ferritin, to endocrine disorders
Hepatitis screening and liver function tests and their relation to endocrine abnormalities (growth and glycemic abnormalities)
- When and how to investigate and interpret results related to:
- Growth hormone–Insulin like growth factor axis (GH-IGF-I) axis,
- Pituitary gonadal axis,
- Glucose homeostasis,
- Pituitary–thyroid functions,
- Calcium homeostasis,
- Skeletal changes and bone mineral density, and
- Reviewing and interpreting imaging findings in TM or TM syndromes with different endocrine disorders
GUIDELINES
Monitoring of growth, pubertal development, reproductive ability, and endocrine functions in general are essential to achieve a good quality of life in TM.[17]
The position statement, outlined in this document, applies to transfusion-dependent patients with TM. The following recommendations agreed upon and reviewed by the I-CET members are based on the current evidence-based literature and existing guidelines, in order to bring uniform, consistently good care to patients affected by TM.
OUTLINE FOR ENDOCRINE SCREENING
Growth assessment
Growth retardation is commonly reported in children and adolescents with TM. Although some patients show normal growth and development, many have growth abnormalities during late childhood in addition to failure or attenuation of their pubertal growth spurt.[13,18,19] The pathogenesis of growth failure is multifactorial.[13,18,19] Key contributing factors to stunted growth in patients with TM include chronic anemia,[12] transfusion-related iron overload,[18–21] and chelation toxicity.[22,23] Other contributing factors include hypothyroidism, hypogonadism, GH deficiency/insufficiency, zinc deficiency, chronic liver disease, under nutrition, and psychosocial stress.[13] Malnutrition is an significant cause of growth retardation in thalassemic children living in poor countries. In these children, inadequate nutrient intake (zinc, folic acid, vitamin D, carotenoids, and retinol binding proteins) contribute significantly to their growth impairment.[24]
Patients are assessed for growth every 6 months to one year by
Recording of parental heights at the first visit (calculate mid-parental height).
Accurate measurement of standing and sitting heights, weight, and head at each visit; measurement of the head circumference especially during the first 2 years of life
Plotting growth data on ethnically adjusted charts or international (WHO) adjusted charts
Calculating annual growth velocity (GV), body mass index (BMI) and upper/lower segment ratio.
-
Interpreting growth data for early detection of growth delay.
Note: The average height velocity varies at different developmental phases:
- Infancy: 23-28 cm/year
- Childhood: 5-6.5 cm/year
- Puberty: 8.3 cm/year (girls), 9.5 cm/year (boys)
Comparing growth data of the patient (height SDS or centiles) with the population data as well as with the mid-parental height (SDS or centiles).
Evaluating pubertal status according to Tanner stages (breast development (girls) and testicular volume (boys))
Child is considered short if
Height is less than the 3rd percentile or 2 SD below the mean height for age and sex;
Height is within normal but GV is consistently <25th percentile over 6-12 months; or
The patient is excessively short for his/her mid-parental height, though his absolute height may be within the normal percentiles.
Annual endocrine screening
This should be started from the age of 9 years, or earlier if clinically indicated (e.g., short stature or decreased GV/year).
The following tests and assessments are recommended annually:
Serum TSH and free T4.
Serum calcium, ionized calcium, inorganic phosphate, magnesium, and alkaline phosphatase.
Fasting glucose/insulin semi-annually, for the assessment of Homeostasis Model Assessment (HOMA index), which is based on the product of the fasting plasma insulin and glucose concentrations (insulin × glucose/22.5), and oral glucose tolerance testing (OGTT) in case of impaired fasting glucose (IFG).
Serum IGF-I and IGF BP-3 in growth screening are useful indicators of growth hormone secretion and nutrition, putting in mind that chronic liver diseases and malnutrition may interfere with their secretion.
Serum or hair zinc (in selected cases).
Bone age (X-ray of wrist and hand).
Radiographs of tibia and spine should be evaluated in patients with TM who have body disproportion to exclude the presence of platylospondylosis or metaphyseal cartilaginous dysplasia changes.
LH, FSH, and sex steroids, in the pubertal age group.
Notes
An abnormal upper to lower segment ratio may be seen not only in pre-pubertal but also in pubertal patients with TM leading to the conclusion that delayed puberty is not the only cause of relative truncal shortening. Furthermore, truncal shortening has been observed in children with good chelation therapy compliance and low ferritin levels due to platyspondylosis induced by DFO toxicity. In the severe form of DFO toxicity, there is significant slowing of linear growth with a short trunk, moderate sternal protrusion, swelling of wrist and knees, and genu valgum of variable severity. In some patients, back pain, bone pain, and limitations in movement are also present.[13,19]
The normal range of plasma zinc concentration using atomic absorption spectrophotometry is 11–19 μmol/l. In mild deficiency, it is between 7 and 10 μmol/l and in severe deficiency, it is less than 7 μmol/l.
Pancreatic MRI may identify high-risk patients before irreversible pancreatic damage
SPECIFIC ENDOCRINOPATHIES: TESTING AND EVALUATION
GROWTH HORMONE DEFICIENCY
Referral to endocrinologist for determination and treatment of growth hormone deficiency (GHD) is required if there is fall-off from the growth curve (slow GV), height <3rd %ile or >2 SD below the mean for age and sex in the reference population, or 1 SD below the mid-parental height SDS.
Early diagnosis of GHD and judicious management before and during puberty is recommended to attain best results.
Investigations
The evaluation for patients with TM and short stature should include:
Dietary assessment preferably by a registered dietician.
-
Laboratory tests:
- Biochemical: Urine analysis, serum albumin, serum glucose, complete and differential blood count, liver function tests, calcium and phosphate, alkaline phosphatase, creatinine, electrolytes, and ferritin concentrations.
- Endocrine: Serum free T4, TSH, cortisol, and IGF-I concentrations
Bone age assessment (X-ray of wrist and hand)
Assessment of GH secretion: Significant GH insufficiency may be diagnosed by a reduced response of GH to two provocative tests (GH peak <10 ng/ml) in children and adolescents and 3 ng/ml in adults)[19–21] or reduced response of GH in one test plus low IGF-I and IGFBP3 concentrations.
In those with poor GH response, basal and IGF-1 generation test may be advised to exclude a partial GH insensitivity (GHI).[25]
Short children with normal GH secretion to stimulation tests and low IGF-I levels potentially may benefit from IGF1 or GH-IGF-I treatment. However, the treatment is expensive and the results in TM patients remain undetermined.
MRI of the hypothalamic–pituitary region is useful to evaluate pituitary iron overload as well as the size of pituitary gland (atrophy).
Notes
Refer to endocrinology for GH determination and treatment of GHD.
Priming with sex steroids is necessary if the bone age is 10 years or greater. An intramuscular injection of testosterone ester (Sustanon) 100 mg is given 7 days before GH stimulation tests in boys. Ethinylo estradiol 50 μg is given orally for 3 days before GH stimulation tests in girls with bone age of 9 years or greater. Girls whose bone age is less than 9 years and boys whose bone age is less than 10 years do not need priming.
Management
Treat underlying cause (hypothyroidism, uncontrolled diabetes mellitus, chronic illnesses).
For children suspected to be GHD, refer to pediatric endocrinologist for initiation of GH.
Psychological support for non-treatable causes (genetic/familial short stature; constitutional delay of growth and puberty)
GH TREATMENT
Children and adolescents
rhGH dose: 0.025-0.05 mg/kg/day (0.5-1.0 units/kg/wk) SC daily at night.
rhGH treatment should start with low doses and be titrated according to clinical response (growth rate) and IGF-I levels. Side effects are uncommon, but should be monitored, especially glucose intolerance.
Notes
rhGH treatment is not always as effective as expected in non thalassemic children with GHD and may result in decreased insulin sensitivity and abnormal glucose tolerance.[13]
During GH treatment, patients should be monitored at 3-monthly intervals with a clinical assessment (growth parameters, compliance) and an evaluation for parameters of GH response and adverse effects.
Continue treatment until the child reaches near final height, defined as a height velocity <2 cm/year over at least 9 months (or bone age >13 years in girls and >14 years in boys)
Treat other pituitary hormone deficiencies such as hypothyroidism, hypogonadism, hypocortisolism.
Adults
At present, there are no guidelines in the literature for the use of GH treatment in adult patients with TM and GHD. The American Association of Clinical Endocrinologists (AACE) recommends that one GH stimulation test is sufficient if clinical suspicion is high, such as in patients with at least one other pituitary hormone deficiency and low or low-normal IGF-1 level. However, in patients with TM and chronic liver disease, IGF-1 levels are low and may have a low diagnostic sensitivity for GHD.[23]
HYPOTHYROIDISM
Annual investigation of thyroid function is recommended beginning at the age of 9 years (unless symptomatic hypothyroidism is observed). Measuring serum T4 and TSH levels easily makes the diagnosis. The stages of hypothyroidism are presented in Table 2.
Table 2.
Primary hypothyroidism and its treatment
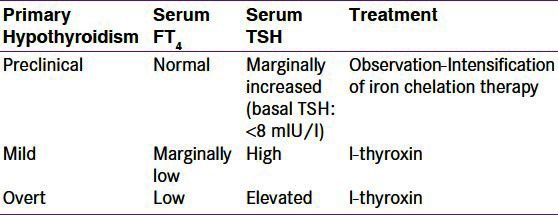
Symptoms and signs of primary hypothyroidism in children and adolescents, such as growth retardation and weight increase, may present.[13,23] The majority of patients have primary thyroid dysfunction. Secondary hypothyroidism (low-normal TSH, low FT4) is very rare.
Notes: Refer to endocrinology and start replacement therapy as indicated.
Management
Good compliance with chelation therapy may prevent or improve hypothyroidism, Sub-clinical hypothyroidism—basal TSH 5 to 7 mUI/ml—requires regular follow-up and optimizing chelation therapy.[13,23] In patients with mild or overt hypothyroidism, L-thyroxine is given.[13,26]
Notes: In patients with TM, subclinical hypothyroidism, and cardiomyopathy, treatment with amiodarone may result in a rapid progression of severe thyroid disease and deterioration of cardiac function.[23]
HYPOPARATHYROIDISM
This uncommon complication presents after the age of 16 years equally in both sexes. The majority of the patients present with mild hypocalcemia and rarely, with tetany and cardiac failure.[10,20] Hypoparathyroidism is thought to be the consequence of iron deposition in the parathyroid glands or due to suppression of parathyroid secretion induced by bone reabsorption resulting from increased hematopoiesis secondary to the chronic anemia.[10,20]
A majority of patients have mild disease and present with only paraesthesia. However, severe cases may present with tetany, seizures, or cardiac failure.[21] The diagnosis is based on low serum calcium, high phosphate and low PTH levels.[26] If hypoparathyroidism is suspected, there is always a risk of hypocalcemia which must be treated without delay.[23]
Notes: Refer to endocrinology and start therapy with vitamin D
Management
Calcitriol (0.25-2.0 μg/day) and calcium (1 g of calcium/day) are the treatment of choice, with frequent monitoring of serum and urine calcium levels. Diet rich in calcium and low in phosphorus is advised.[26]
Notes: Recently, abnormal cerebral CT findings (calcifications) have been reported in relation to hypoparathyroidism in patients with TM.[27]
CALCIUM HOMEOSTASIS AND BONE DISEASE (OSTEOPENIA AND OSTEOPOROSIS)
Osteopenia and osteoporosis are the most common bone complications of TM, despite regular transfusions and iron chelation therapy.[28] The etiology of TM-induced bone mineral loss is multi-factorial.[28] Medullary expansion due to anemia, patient age, duration of the disease, chronic liver disease, vitamin D deficiency, hypogonadism, concurrent hypothyroidism, and other endocrine-associated complications are significant contributory factors.
Investigations
Serum calcium, phosphorus, alkaline phosphatase
25-hydroxy vitamin D
Parathyroid hormone
Osteocalcin
C-terminal telopeptide
24-h urinary calcium
Spinal radiograph (AP and lateral views)
MRI scan of the spine may be considered for patients with severe back pain to exclude a intervertebral disc degeneration
Currently, dual-energy X-ray absorptiometry (DEXA) scan is the method of choice to assess bone mineral density (BMD)
Notes: Annual evaluation of calcium metabolism and parathyroid function is recommended. If the patient has pubertal delay or failure, FSH, LH, testosterone, or estrogen should be measured.
Definitions
The absolute value of bone density varies but the accepted definitions for adult patients are:
Bone density T-score >1.5 SD below the mean is diagnostic of osteopenia.
Bone density T-score >2.5 SD below the mean is diagnostic of osteoporosis.
The diagnosis of osteoporosis in children should not be made on the basis of densitometric criteria alone. The diagnosis of osteoporosis requires the presence of both a clinically significant fracture history and low bone mineral density. Terminology such as “low bone density for chronological age” may be used if the Z-score is below –2.0.
Notes: DEXA or other accepted methods of measurement should be performed at 10-12 years for girls and boys annually and every 2 years thereafter. BMD should be related to the age (and the height in short subjects) and sex.
The same method of bone density measurement should be used for subsequent evaluations.
Treatment
Supplement with calcium 500-1000 mg/day starting at age 12 years.
Follow nutritional status and vitamin D levels. Supplement with vitamin D 400-800 units/day for patients with low levels or those at high risk to develop vitamin deficiency.
For patients with established osteoporosis, treatment with bisphosphonates should be considered.
Endocrine referral is recommended for hormone replacement (estrogen for females, testosterone for males), calcium supplementation, exercise program, and consideration of bisphosphonates.[28]
DIABETES MELLITUS
Diabetes mellitus is seen after the age of 10 years.[18,20,21,26] The etiology of DM is multifactorial (genetic factors, insulin deficiency, insulin resistance, and liver dysfunction secondary to viral hepatitis).
Insulin-dependent diabetes in TM presents with some unusual characteristics[18,26] compared with type 1 diabetes:
Ketoacidosis is a rare presenting symptom
Renal glucose threshold is high
Islet cell antibodies are negative
There is no association with HLA haplotypes B8-DR3, BW15, and DR4.
Patients usually present with impaired glucose tolerance, mostly due to Insulin resistance and subsequently develop insulin deficiency.[18,20,21,26]
A 2-h oral glucose tolerance testing (OGTT), preferably combined with insulin secretion determination, should be performed at 10, 12, 14, and 16 years of age and annually thereafter. If fasting serum glucose is >110 mg/dl, OGTT is indicated.
The diagnostic criteria for glucose tolerance are as follows
Fasting blood glucose >126 mg/dl is diagnostic of diabetes mellitus
OGTT serum glucose at 2 h >140 <200 mg/dl indicates glucose intolerance
OGTT serum glucose at 2 h > 200 mg/dl is diagnostic of diabetes mellitus
Therapeutic approaches
The patient should be referred to an endocrinologist for management of diabetes mellitus or glucose intolerance.[18,20,21,26]
In cases where impaired glucose tolerance is present the patients are advised to follow a proper diet and lose weight if they are obese.
Intensive iron chelation therapy with deferoxamine and deferiprone seems to be associated with an improvement in glucose intolerance in terms of glucose and insulin secretion, particularly in patients in early stages of glucose intolerance.[29]
The use of oral anti-diabetic agents in DM (metformin and glibenclamide) remains undetermined.
Insulin therapy is considered in the stage of insulin deficiency, where all other measures fail. Insulin dose is adjusted based on frequent glucose monitoring.
Patients with DM require periodical monitoring of metabolic control and possible complications.
Notes
Metabolic control may be difficult to achieve. Insulin requirements (0.15-1.72 U/kg) differ due to a wide variation in pancreatic beta-cell function as determined by C-peptide level
-
Because the HbA1C test is based on normal hemoglobin, hemoglobinopathies can affect the reliability of the test in three ways:[30]
- Altering the normal process of glycation of HbA to HbA1C
- Causing an abnormal peak on chromatography, making estimation of A1C unreliable,
- Making the red blood cell more prone to hemolysis, thereby decreasing the time for glycosylation to occur and producing a falsely low A1C result.
Fructosamine testing may be used as a target for metabolic control
Investigation of kidney function and imaging of the fundi should be carried out to evaluate the presence and degree of diabetic complications. However, the incidence of retinopathy and nephropathy in thalassemic patients with diabetes is lower than in patients affected by juvenile diabetes. It is suggested that this may be due to normal or below normal serum levels of cholesterol and triglycerides and to the frequent presence of hypogonadism in TM.[29]
ADRENAL INSUFFICIENCY
Secondary hemochromatosis due to excess iron has the potential to disrupt adrenal function by affecting the hypothalamic-pituitary adrenal axis at the hypothalamic or pituitary and/or adrenal level.[30]
Several studies reported a significant prevalence of “biochemical” adrenal insufficiency, ranging from 0% to 45%, in patients with TM, due to different patient characteristics and different diagnostic tests used. Clinical adrenal insufficiency, i.e., adrenal crisis, on the other hand, is extremely rare.[30,31]
In primary adrenal insufficiency, cortisol, aldosterone, and androgen secretion are affected. Secondary or tertiary adrenal insufficiency (pituitary or hypothalamic affected) causes no mineralocorticoid defect.[32]
Patients are usually asymptomatic. Adrenal androgen levels might be decreased explaining the poor development of pubic and axillary hair observed in thalassemic adolescents.[30,31] Manifestations of mild adrenal hypofunction might be masked by symptoms commonly complained of by thalassemic patients, such as asthenia, muscle weakness, arthralgias, and weight loss.
Investigations
Measurement of both basal serum cortisol level and cortisol response to stimulation by ACTH or insulin stimulation (ITT) can be used for assessment of adrenal function.
Baseline cortisol [morning baseline cortisol level (at 8-9 AM)] can be used to detect subtle adrenal insufficiency. Patients with abnormal screening are referred to the endocrinologist for further evaluation and replacement therapy.
ACTH stimulation test should be done in patients with an abnormal baseline cortisol level. A peak cortisol level of <500 nmol/L (18 μg/dL) 30-60 min after ACTH stimulation is abnormal.
Even though the insulin tolerance test (ITT) has been considered the gold standard to disclose impaired adrenal function, the ACTH stimulation test has been used for many years as an acceptable alternative with the advantage of lacking relevant adverse effects.[30,31,33] Either the low-dose (1 μg) or the standard high dose (250 μg/1.75 m2 surface area) cosyntropin are used.[30,31]
Treatment
It is advised to test adrenal function every 1-2 years, especially in GHD patients during rhGH therapy.
Clinical adrenal insufficiency and adrenal crisis are very rare. On the other hand, subclinical impairment of adrenocortical function in patients with TM is not uncommon, but of little or no clinical impact under basal conditions; however, it may have relevance during stressful events. Accordingly, glucocorticoid treatment coverage might be advised only for stressful conditions.[30,31]
HYPOGONADISM
Delayed puberty and hypogonadism are the most obvious clinical consequences of iron overload.[13,18] Iron deposition in the pituitary gonadotrophic cells leads to disruption of gonadotropin (LH and FSH) production. In the majority of well-chelated patients, the function of gonads is normal; however, gonadal iron deposition occasionally occurs. TM patients with a favorable genotype manifest less severe gonadal dysfunction, due to less iron loading.[34]
Delayed puberty is defined as the complete lack of pubertal development in girls by the age of 13 years and in boys by the age of 14 years. Hypogonadism is defined by the absence of testicular enlargement (less than 4 ml) in boys, and by the absence of breast development in girls by the age of 16 years.[35]
Adolescent patients with TM may present with delayed puberty or slowly progressive puberty. Arrested puberty is a relatively common complication in moderately or grossly iron overloaded patients with TM. This is characterized by the lack of pubertal progression over a year or longer. In such cases, the yearly GV is either markedly reduced or completely stops.[13,18]
Most women with TM present with primary amenorrhea (PA), whereas secondary amenorrhea (SA) will invariably develop with time, especially in patients poorly compliant to chelation therapy. Ovarian function of these women is normal as they produce the expected number of ova after stimulation therapy. Damage of the ovaries by iron deposition is rare and is more likely to appear in women of 25-30 years of age because of high vascular activity in the ovaries at this age.[36]
Investigations
Tanner staging should be determined every 6 months starting from the age of 12 years.
- Girls without evidence of puberty by 13 years and boys by 14 years require screening with measured serum levels of LH, FSH, and estradiol/testosterone:
- Low FSH and LH for age infer hypogonadotropic hypogonadism (HH) (hypothalamic-pituitary lesion) and MRI of the pituitary (T2*) is recommended.
- Elevated FSH and LH suggests primary hypogonadism (very rare)
If LH and/or FSH are low, performing GnRH stimulation with poor or flat LH and FSH responses confirms the diagnosis of HH.
Bone age evaluation is useful for prediction of the final adult height of these patients.
Pelvic ultrasound is useful in assessing ovarian and uterine maturation.
Induction of puberty in boys: At 14 years or BA >12 years
The treatment of delayed or arrested puberty and HH depends on factors such as age, severity of iron overload, degree of damage to the hypothalamic-pituitary-gonadal axis, presence of chronic liver disease, and the psychological problems resulting from hypogonadism in these patients. The treatment of pubertal disorders in TM is a complex issue. Collaboration between endocrinologists and other physicians, nurses, psychologists, dieticians, and social workers is critical due to the many associated hematological, cardiac, hepatic, skeletal, and psychological disorders in these patients. Therefore, each patient has to be assessed individually.[18,20,21,26]
In boys, treatment is usually initiated with IM depot testosterone in doses of 50-100 mg per month. This is followed by hormonal re-assessment. In patients with HH, low dose testosterone therapy (50 mg) can be continued until the growth rate wanes. The fully virilizing dose is 75-100 mg of depot-testosterone esters every 10 days administered intramuscularly. The same effects can be achieved with topical testosterone gel.
For pubertal arrest, the treatment is similar to that of delayed puberty if growth potential is present or to that of HH when growth is completed.
Induction of puberty in girls: At 13 years or BA >11 years
For girls, therapy may begin with the oral administration of ethinyl estradiol (EE) 2.5-5 μg daily for 6 months, followed by hormonal reassessment. If spontaneous puberty does not occur within 6 months after stopping the treatment, oral estrogen is re-introduced with gradually increasing dosages of EE from 5 to 10 μg daily for another 12 months and to 20 μg for additional 12 months. Norethisterone or medroxyprogesterone acetate 5 mg/day 10-21 day and E2 for 21 days, once EE reaches 20 μg/day or if breakthrough bleeding occurs.
Transdermal administration of estrogens can also be used.[18,20,24,26]
The transdermal use of estradiol and norethisterone is the ideal substitution treatment in the hypogonadal female, because of its proven beneficial effect on bones and its fewer side effects.
Management of fertility and pregnancy in ί-thalassemia
The desire of the thalassemic woman to become a mother is always viewed with special caution and sensitivity. Ambitions of this sort pose numerous medicolegal and ethical issues that need to be addressed prudently if the patients' quality of life is to be optimized.[37–47] Therefore, the desire of the female thalassemic to procreate should to be viewed with special caution and sensitivity by all physicians who are involved in her medical care.
Gynecological consultation is recommended in women for fertility evaluation.
Each patient should be assessed regarding suitability to embark on pregnancy with optimum outcome both for the mother and the fetus.[37–47]
Cardiac and liver functions are important issues, as well as the risk of vertical transmission of viruses. Ascertaining the hemoglobinopathy status of the partner is important.[37–39]
Women with TM, who are regularly transfused and are well chelated, can become pregnant either spontaneously or by inducing ovulation. The presence of gonadal dysfunction can be overcome with proper combination treatment. It is necessary that all pregnant women with TM be followed up very closely.[37–39]
Apart from the routine pregnancy follow-up, the thalassemic pregnant woman needs additional medical care. Hemoglobin levels should be maintained at 10 g/dL and careful monitoring of vital signs during transfusion is required. Ferritin levels should also be measured and observed to avoid iron overload. Careful monitoring of the transfusion regime and regular evaluation of cardiac function should be done in all pregnant thalassemic women to prevent fluid overload. Cardiac function should be evaluated periodically by a cardiologist.[37–39]
Iron chelation therapy, due to its possible teratogenic effects, is withheld as soon as the pregnancy is planned or identified. It has been assumed that pregnancy is an efficient chelator of iron due to its hemodilution effect and the fetal consumption of free iron. Although therapy with DFO and deferasirox has not been implicated in any deleterious effect on the fetus, the current recommendation is its discontinuation, both once pregnancy is identified[39] and during the induction period.
Chronic maternal anemia in the thalassemic pregnant woman may result in fetal hypoxia, which predisposes to premature labor, intrauterine growth retardation (IUGR), and death.[39]
Induction of spermatogenesis in males by HCG/HMG should only be undertaken by a specialist. Cryopreservation of sperm should be recommended for all thalassemic patients after induction of spermatogenesis.[39,40]
CONCLUSION
The practical objectives of I-CET are to encourage and guide endocrinological follow up of multi-transfused patients in developing countries, to promote and help collaborative research in this field, and to educate and train more endocrinologists and other pediatricians/physicians to prevent and improve management of the growth and endocrine complications in these patients.[41]
The following recommendations are an effort, based on current evidence and existing guidelines,[23,42–47] to bring uniform, consistently good care to patients affected by TM. Such comprehensive care is best provided in by coordinated, multidisciplinary teams working in expert centers. Physical, emotional, educational, and vocational aspects must be addressed.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Weatherall DJ, Clegg JB. Thalassemia-a global public health problem. Nat Med. 1996;2:847–9. doi: 10.1038/nm0896-847. [DOI] [PubMed] [Google Scholar]
- 2.Lukens JN. The thalassemia and related disorders. In: Pine JW Jr, editor. Wintrobe's clinical hematology. 10th. USA: Williams and Wilkins; 1999. pp. 1405–49. [Google Scholar]
- 3.Porter JB. Practical management of iron overload. Br J Haematol. 2001;115:239–52. doi: 10.1046/j.1365-2141.2001.03195.x. [DOI] [PubMed] [Google Scholar]
- 4.Modell B. Total management of thalassaemia major. Arch Dis Child. 1977;52:489–500. doi: 10.1136/adc.52.6.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gabutti V, Piga A. Results of long term chelation therapy. Acta Haematol. 1996;95:26–36. doi: 10.1159/000203853. [DOI] [PubMed] [Google Scholar]
- 6.Clarke GM, Higgins TN. Laboratory investigation of hemoglobinopathies and thalassemias: Review and update. Clin Chem. 2000;46:1284–90. [PubMed] [Google Scholar]
- 7.Galanello R, Origa R. Pathophysiology of beta thalassaemia. Pediatr Endocrinol Rev. 2011;8:263–70. [PubMed] [Google Scholar]
- 8.Forget BG, Pearson HA. Hemoglobin synthesis and the thalassemias. In: Hoffman R, Benz EJ Jr, Shattil SJ, editors. Basic Principles and Practice. 3rd ed. NY: Churchill Livingstone; 2000. p. 1525. [Google Scholar]
- 9.Pippard MJ, Callender ST, Warner GT, Weatherall DJ. Iron absorption and loading in beta-thalassaemia intermedia. Lancet. 1979;2:819–21. doi: 10.1016/s0140-6736(79)92175-5. [DOI] [PubMed] [Google Scholar]
- 10.Hershko C, Link G, Cabantchik I. Pathophysiology of iron overload. Ann N Y Acad Sci. 1998;30:191–201. doi: 10.1111/j.1749-6632.1998.tb10475.x. [DOI] [PubMed] [Google Scholar]
- 11.Andrews NC, Schmidt PJ. Iron homeostasis. Annual Review of Physiology, 2007;69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- 12.Magro S, Puzzonia P, Consarino C, Galati MC, Morgione S, Porcelli D, et al. Hypothyroidism in patients with thalassaemia syndromes. Acta Haematol. 1990;84:72–6. doi: 10.1159/000205032. [DOI] [PubMed] [Google Scholar]
- 13.De Sanctis V. Growth and puberty and its management in thalassaemia. Horm Res. 2002;58:72–9. doi: 10.1159/000064766. [DOI] [PubMed] [Google Scholar]
- 14.Olivieri NF, Brittenham GM. Iron-chelation therapy and the treatment of thalassemia. Blood. 1997;89:739–61. [PubMed] [Google Scholar]
- 15.Meerpohl JJ, Antes G, Rücker G, Fleeman N, Motschall E, Niemeyer CM, et al. Deferasirox for managing iron overload in people with thalassaemia. Cochrane Database Syst Rev. 2012;2:CD007476. doi: 10.1002/14651858.CD007476.pub2. [DOI] [PubMed] [Google Scholar]
- 16.Farmaki K, Tzoumari I, Pappa C. Oral chelators in transfusion-dependent thalassemia major patients may prevent or reverse iron overload complications. Blood Cells Mol Dis. 2011;47:33–40. doi: 10.1016/j.bcmd.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 17.De Sanctis V, Elawwa A, Angastiniotis M, Eleftheriou A, Kattamis C, Karimi M, et al. Highlights from the first thalassaemia forum on growth and endocrine complications in thalassemia Doha, (October 2-3, 2011) Pediatr Endocrinol Rev. 2012;9:672–9. [PubMed] [Google Scholar]
- 18.Wonke B, De Sanctis V. Clinical aspects of transfusional iron overload. Clin Exp Hematol. 2001;12:322–34. [Google Scholar]
- 19.Soliman AT, Khalafallah H, Ashour R. Growth and factors affecting it in thalassemia major. Hemoglobin. 2009;33:S116–26. doi: 10.3109/03630260903347781. [DOI] [PubMed] [Google Scholar]
- 20.De Sanctis V, Vullo C, Bagni B, Chiccoli L. Hypoparathyroidism in beta-thalassemia major. Clinical and laboratory observations in 24 patients. Acta Haematol. 1992;88:105–8. doi: 10.1159/000204662. [DOI] [PubMed] [Google Scholar]
- 21.De Sanctis V, Borsari G, Brachi S, Gubellini E, Gamberini MR, Carandina G. A rare cause of heart failure in iron-overload thalassaemic patients-primary hypoparathyroidism. Georgian Med News. 2008;156:111–3. [PubMed] [Google Scholar]
- 22.De Sanctis V, Savarino L, Stea S, Cervellati M, Ciapetti G, Tassinari L, et al. Microstructural analysis of severe bone lesions in seven thalassaemic patients treated with desferrioxamine. Calcif Tissue Int. 2000;67:128–33. doi: 10.1007/s00223001122. [DOI] [PubMed] [Google Scholar]
- 23.Skordis N. Endocrine investigation and follow up in thalassaemia. Time for specific guidelines. Thalassemia Reports. 2011;1(s2):e22. [Google Scholar]
- 24.Fuchs GJ, Tienboon P, Linpisarn S, Nimsakul S, Leelapat P, Tovanabutra S, et al. Nutritional factors and thalassaemia major. Arch Dis Child. 1996;74:224–7. doi: 10.1136/adc.74.3.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chrysis DC, Alexandrides TK, Koromantzou E, Georgopoulos N, Vassilakos P, Kiess W, et al. Novel application of IGF-I and IGFBP-3 generation tests in the diagnosis of growth hormone axis disturbances in children with beta-thalassaemia. Clin Endocrinol (Oxf) 2001;54:253–9. doi: 10.1046/j.1365-2265.2001.01198.x. [DOI] [PubMed] [Google Scholar]
- 26.De Sanctis V, Wonke B. Rome: Mediprint; 1998. Growth and endocrine complications in thalassaemia. [Google Scholar]
- 27.Karimi M, Rasekhi AR, Rasekh M, Nabavizadeh SA, Assadsangabi R, Amirhakimi GH. Hypoparathyroidism and intracerebral calcification in patients with beta-thalassemia major. Eur J Radiol. 2009;70:481–4. doi: 10.1016/j.ejrad.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Skordis N, Loannou YS, Kyriakou A, Savva SC, Efstathiou E, Savvides I, et al. Effect of bisphosphonate treatment on bone mineral density in patients with thalassaemia major. Pediatr Endocrinol Rev. 2008;6:144–8. [PubMed] [Google Scholar]
- 29.Smaldone A. Glycemic control and hemoglobinopathy: When A1C may not be reliable. Diabetes Spectr. 2008;21:46–9. [Google Scholar]
- 30.Poomthavorn P, Isaradisaikul B, Chuansumrit A, Khlairit P, Sriphrapradang A, Mahachoklertwattana P. High prevalence of “biochemical” adrenal insufficiency in thalassemics: Is it a matter of different testings or decreased cortisol binding globulin? J Clin Endocrinol Metab. 2010;95:4609–15. doi: 10.1210/jc.2010-0205. [DOI] [PubMed] [Google Scholar]
- 31.Elsedfy HH, El Kholy M, Tarif R, Hamed A, Elalfy M. Adrenal function in thalassemia major adolescents. Pediatr Endocrinol Rev. 2011;8:295–9. [PubMed] [Google Scholar]
- 32.Pasqualetti P, Colantonio D, Collacciani A, Casale R, Natali G. Circadian pattern of circulating plasma ACTH, cortisol, and aldosterone in patients with beta-thalassemia. Acta Endocrinol (Copenh) 1990;123:174–8. doi: 10.1530/acta.0.1230174. [DOI] [PubMed] [Google Scholar]
- 33.Incorvaia C, Parmeggiani F, Mingrone G, Sebastiani A, De Sanctis V. Prevalence of retinopathy in diabetic thalassaemic patients. J Pediatr Endocrinol Metab. 1998;11:879–83. [PubMed] [Google Scholar]
- 34.Raiola G, Galati MC, De Sanctis V, Caruso Nicoletti M, Pintor C, De Simone M, et al. Growth and puberty in thalassemia major. J Pediatr Endocrinol Metab. 2003;16:259–66. [PubMed] [Google Scholar]
- 35.Skordis N, Gourni M, Kanaris C, Toumba M, Kleanthous M, Karatzia N, et al. The impact of iron overload and genotype on gonadal function in women with thalassaemia major. Pediatr Endocrinol Rev. 2004;2:292–5. [PubMed] [Google Scholar]
- 36.Toumba M, Kanaris C, Simamonian K, Skordis N. Outcome and management of pregnancy in women with thalassaemia in Cyprus. East Mediterr Health J. 2008;14:628–35. [PubMed] [Google Scholar]
- 37.Skordis N, Christou S, Koliou M, Pavlides N, Angastiniotis M. Fertility in female patients with thalassaemia. J Pediatr Endocrinol Metab. 1998;11:935–43. [PubMed] [Google Scholar]
- 38.Skordis N, Petrikkos L, Toumba M, Hadjigavriel M, Sitarou M, Kolnakou A, et al. Update on fertility in thalassaemia major. Pediatr Endocrinol Rev. 2004;2:296–302. [PubMed] [Google Scholar]
- 39.De Sanctis V, Vullo C, Katz M, Wonke B, Nannetti C, Bagni B. Induction of spermatogenesis in thalassaemia. Fertil Steril. 1998;50:969–75. doi: 10.1016/s0015-0282(16)60382-5. [DOI] [PubMed] [Google Scholar]
- 40.Soliman AT, Nasr I, Thabet A, Rizk MM, El Matary W. Human chorionic gonadotropin therapy in adolescent boys with constitutional delayed puberty vs. those with beta-thalassemia major. Metabolism. 2005;54:15–23. doi: 10.1016/j.metabol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 41.De Sanctis V, Soliman AT, Angastiniotis M, Eleftheriou A, Kattamis Ch, Karimi M, et al. International network on endocrine complications in thalassaemia (I-CET): An opportunity to grow. Georgian Med News. 2012;205:52–7. [PubMed] [Google Scholar]
- 42.2nd ed. United Kingdom: Thalassaemia Society; 2008. Standards for the clinical care of children and adult with thalassaemia in UK; pp. 1–120. Available from: http://www.ukts.org. 2008. [Google Scholar]
- 43.2nd ed. Thalassaemia International Federation; 2008. Guidelines for the clinical management of thalassaemia; pp. 1–202. Available from: http://www.thalassaemia.org.cy. 2007. [PubMed] [Google Scholar]
- 44.Children's Hospital and Research Center Oakland; 2009. Standards of care guidelines for thalassaemia; pp. 1–38. Available from: http://www.thalassaemia.org. 2009. [Google Scholar]
- 45.Guidelines for the clinical care of patients with thalassemia in Canada. 2009. pp. 1–84. Available from http://www.thalassemia.ca. 2011.
- 46.Caruso Nicoletti M, De Sanctis V, Anastasi S. Linee guida per il follow-up auxologico del paziente talassemico. Riv Ital Pediatr. 1997;23:305–7. [Google Scholar]
- 47.De Sanctis V, Caruso Nicoletti M, Raiola G, Pintor C. Proposta di linee guida per la diagnosi ed il trattamento delle complicanze endocrine nel paziente talassemico. Riv Ital Pediatr. 1999;25:1132–7. [Google Scholar]