

Abstract
Ab-dependent enhancement (ADE) of dengue virus (DENV) infection is mediated through the interaction of viral immune complexes with FcγRs, with notable efficiency of FcγRII. Most human dengue target cells coexpress activating (FcγRIIa) and inhibitory (FcγRIIb) isoforms, but their relative roles in ADE are not well understood. We studied the effects of FcγRIIa and FcγRIIb by transfecting cells to express each individual receptor isoform or through coexpression of both isoforms. We showed that although both isoforms similarly bind dengue-immune complexes, FcγRIIa efficiently internalized virus leading to productive cellular infection, unlike FcγRIIb. We next focused on the main discriminating feature of these isoforms: their distinct intracytoplasmic tails (FcγRIIa with an immunoreceptor tyrosine-based activation motif [ITAM] and FcγRIIb with an immunoreceptor tyrosine-based inhibitory motif [ITIM]). We engineered cells to express “swapped” versions of their FcγRII by switching the cytoplasmic tails containing the ITAM/ITIM motifs, leaving the remainder of the receptor intact. Our data show that both FcγRIIa and FcγRIIb comparably bind dengue immune complexes. However, wild type FcγRIIa facilitates DENV entry by virtue of the ITAM motif, whereas the swapped version FcγRIIa-ITIM significantly inhibited ADE. Similarly, replacing the inhibitory motif in FcγRIIb with an ITAM (FcγRIIb-ITAM) reconstituted ADE capacity to levels of the wild type activating counterpart, FcγRIIa. Our data suggest that FcγRIIa and FcγRIIb isoforms, as the most abundantly distributed class II Fcγ receptors, differentially influence Ab-mediated DENV infection under ADE conditions both at the level of cellular infection and viral production.
Introduction
Dengue virus (DENV) is a mosquito-borne, positive polarity, single-stranded RNA virus in the family Flaviviridae. The pathogenesis of complicated DENV infection is not fully understood, but viral, host, and immune factors likely influence disease severity (1, 2). Clinical DENV infection varies from asymptomatic or mild self-limited illness, dengue fever, to potentially life-threatening diseases such as dengue hemorrhagic fever and dengue shock syndrome (3). Ab-dependent enhancement (ADE) of DENV infection is often implicated in severe forms of DV infection (4–6). Dengue Abs likely bring the virus/Ab complex into close proximity with the cell surface Fc receptors, which in turn facilitate viral entry into the cells (7). Three classes of Fcγ receptors exist in humans: FcγRI (CD64), FcγRII (CD32), and FcγRIII (CD16). Each Fcγ receptor has a variety of isoforms with differing IgG affinities, tissue distribution, and expression levels (8). All human DENV target cells, including monocytes, macrophages, and dendritic cells (DCs), express Fcγ receptors. FcγRI and FcγRIIa have been shown to facilitate ADE in a human monocytic cell line (7, 9). In addition, FcγRIIa was found to be more efficient in DENV immune complex infectivity compared with FcγRIa in vitro in cell line–based transfection experiments (10, 11). The low-affinity activating Fcγ receptor FcγRIIa is unique to humans. It is the most widely distributed FcγR subclass expressed on many cell types, including monocytes, neutrophils, platelets, and DCs (12, 13). FcγRIIa preferentially binds IgG complexes and is the only Fc receptor that contains an immunoreceptor tyrosine-based activation motif (ITAM) motif in its cytoplasmic domain; therefore, it is the only Fc receptor that does not need an accessory associated subunit (i.e., γ-chain) to signal upon engagement of the Fc portion of immune complexes in its extracellular domain (8, 14). There is no identified murine equivalent of FcγRIIa (14); however, FcγRIIb is conserved in mice and humans and is the only known inhibitory FcγR. FcγRIIb transmits the inhibitory signal through an immunoreceptor tyrosine-based inhibitory motif (ITIM) within its cytoplasmic region as opposed to the activating receptor, FcγRIIa (15). The FcγRIIa isoform has been studied in ADE of dengue infection, but little is known about the role of FcγRIIb in ADE or its relative roles under typical coexpression conditions.
We previously demonstrated that the ADE effect observed in primary human mature DCs was mediated by FcγRIIa and blocking of this molecule abrogated ADE (16). DCs express both FcγRIIa and FcγRIIb and downregulate FcγRIIb upon maturation. The maturation status of DCs affects their susceptibility to both direct DV infection and ADE (16). These observations led us to investigate the function and influence of these two FcγRII isoforms on ADE of DV infection. In the current study, we tested the effects of FcγRII isoforms (FcγRIIa or FcγRIIb) individually or in combination on ADE by transiently expressing each receptor in different types of cell lines (i.e., mammalian, human) and further by genetic-swapping experiments of the relevant gene segments encoding the cytoplasmic tail domain of each respective FcγRII isoform. In this study, we demonstrate that FcγRIIa facilitates ADE of DENV infection, whereas FcγRIIb constrains it. By switching the ITAM- and ITIM-containing motifs between these two isoforms, we found that the intracellular portion of FcγRII is a major determinant of ADE infection.
Materials and Methods
Virus and cell lines
The DENV-2 isolate s16803 (origin: Thailand) was used for all experiments. Virus stock preparation and titration were described previously (16, 17). The non–Fc-bearing murine fibroblast cell line NIH 3T3 (American Type Culture Collection, Manassas, VA) were maintained in DMEM (Quality Biological, Gaithersburg, MD) with 10% heat inactivated FCS (Gemini Bio-Products, Sacramento, CA) supplemented with of 2 mM l-glutamine, 100 U/ml penicillin, and 10 ug/ml streptomycin (Quality Biological).
mAb and DV-immune serum
Intracellular DENV infection was determined using an mAb, 2H2 (provided by Dr. Robert Putnak, Walter Reed Army Institute of Research, Silver Spring, MD), and a monoclonal mouse anti-prM Ab (IgG2a) that detects all four serotypes, indicating de novo protein production (17). We used well-characterized polyvalent dengue-immune sera (Pediatric Dengue Vaccine Initiative) that was found to neutralize DENV serotypes 1–4 in a plaque reduction neutralization-50 assay.
ADE assay
DENV-immune serum was serially diluted 4-fold from 1:10 to 1:163,840 (8 dilutions). Virus at a multiplicity of infection (MOI) of 1, unless otherwise noted, was incubated with diluted Ab for 1h at 37°C with 5% CO2 to allow immune complex formation and then added to cells and incubated for 2 h. The exposed cells were washed with complete cell growth media to remove unbound immune complexes, resuspended, and incubated for an additional 48 h (17).
Flow cytometry
A FACSCalibur instrument (BD Biosciences, San Jose, CA) was used to determine cell-surface staining with Alexa Fluor 467 conjugated anti-human FcγRIIa mAb (Clone IV.3; ATCC) and FITC-conjugated anti-human FcγRIIb mAb 2B6 (MacroGenics, Rockville, MD). For detection of intracellular de novo DV protein production, cells were permeabilized with Cytofix/Cytoperm Solution (BD Biosciences) and stained intracellularly at 48 h after viral infection with the anti-DENV 2H2 mAb conjugated with Alexa Fluor 488 (Invitrogen, Carlsbad,CA). All flow cytometry experiments use blocking Abs in staining buffer to avoid nonspecific staining.
Knockdown of human FcγRIIa using siRNA
The K562 cells (5 × 106 cells per condition) were transfected with 10 g or 20 μg anti-CD32 (FcγRII) siRNA or scrambled negative control siRNA (Ambion; Life Technologies, Grand Island NY) via electroporation with the GenePulser XCell (Bio-Rad, Hercules CA). Following transfection, K562 cells were plated in complete media and incubated for 48 h at 37°C and 5% CO2.
Transfection
NIH 3T3 cells were transfected using the “empty” pCMV6-XL4 vector pCMV6-pCMV6-XL4 hFcγRIIa (NM_021642) or pCMV6-XL4-hFcγRIIb (NM_001002273) using Mega Tran 1.0 Transfection Reagent (OriGene Technologies, Rockville, MD), according to the manufacturer’s instructions. The FcγRIIa-ITIM and FcγRIIb-ITAM plasmids were generated by synthesizing the FcγRIIa-ITIM and FcγRIIb-ITAM genes. The genes were then inserted into pCMV6-XL4 using SgfI abd MluI sites. Efficiency of transfection as well as biological surface expression of these human FcγRII isoforms was confirmed by flow cytometry, and the transfected FcγRII genes were verified by DNA sequencing.
Viral RNA quantitation
Quantitative real-time PCR (QRT-PCR) was performed using primers, probes, RNA standards, and conditions described previously (16). Viral RNA was extracted from culture supernatants using the QIAamp viral RNA kit (Qiagen, Velencia, CA). Amplification was performed using an ABI prism 7500 detection instrument (Applied Biosystems, Foster City, CA). The reverse-transcription PCR thermal cycles were performed as follows: 50°C for 30 min and 95°C for 15 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. RNA copy numbers were calculated from a standard curve generated by an in vitro–transcribed RNA standard.
Binding and internalization assay
To measure DENV-2 immune complex cell surface binding/adsorption, the human FcγRIIa or FcγRIIb transfected 3T3 cells were exposed to DENV-2 at an MOI of 10 with or without sera, along with serum only and media only controls for 1 h at 4°C. Cells were washed three times at 4°C with 10 ml PBS containing 10% FCS. The number of viruses that bound to the cells was determined by QRT-PCR. To measure DENV-2 immune complex internalization, the cells were exposed to DV-2 at an MOI of 10 (with or without sera). The cells were washed three times with 10 ml PBS and resuspended with PBS containing 10% FCS. The cells were treated with 5 mg/ml of Pronase (Roche Applied Science, Indianapolis, IN) to remove excess virus on the cell surface. The number of internalized viruses was determined by QRT-PCR.
Plaque assay
The Vero cell plaque assay was performed as described previously (16). Briefly, 10-fold serial dilutions (10−1 to 10−6) of culture supernatants were inoculated onto six-well tissue culture plates containing confluent Vero cell monolayers in quadruplicate. After virus adsorption for 1 h at 37°C, the Vero cell monolayers were overlaid with complete minimal essential media (Cellgro, Manassas, VA) containing low-melting-point agarose (Invitrogen) to restrict dissemination of progeny virions. The infected overlaid cells were incubated for 5 d at 37°C and then overlaid with Neutral Red (Sigma-Aldrich, St.Louis, MO). Plaques were counted 24 h after Neutral Red overlay (6 d postinfection) to determine the number of PFUs per milliliter of culture supernatant.
Results
FcγRIIb transfection constrains ADE in human K562 cells
Most myeloid cells express more than one type of Fcγ receptor (15). To facilitate mechanistic studies and avoid confounding FcRγ types, we carefully selected cell lines to study the effects of FcγRIIa and IIb isoforms. First, we focused on K562 cells: a human erythroleukemia cell line that normally expresses a single type of FcγR (FcγRIIa) with an activating cytoplasmic ITAM motif (18, 19). K562 cells are widely used to study ADE of flaviviruses in vitro (20, 21). We used specific mAbs to block surface FcγRIIa and specific siRNA to knockdown FcγRIIa expression at the mRNA level in K562 cells. Each approach markedly inhibited ADE (Fig. 1A, 1B). Next, we transfected FcγRIIb cDNA (Origene, Rockville, MD) into K562 and observed nearly 40% FcγRIIb surface expression at 24 h posttransfection using flow cytometry (Fig.1C, lower right panel). We compared the FcγRIIb transfectants and wild type K562 transfected with an empty vector control (pCMV-XL4; Origene, Rockville MD) in the ADE assay (Fig. 1D). As shown, the presence of FcγRIIb resulted in a statistically significant 50% decrease in infection at enhancement titer, demonstrating a dominant inhibitory effect of coexpression of FcγRIIb on the K562 cells (Fig. 1D) and suggesting the two human FcγRII isoforms differentially contribute to ADE.
FIGURE 1.

Influence of FcγRIIa and FcγRIIb isoforms on ADE of dengue virus infection. (A) Representative phenotype of FcγRIIa expression on the cell surface of K562 cells following treatment with FcγRIIa (CD32a; red) or nonspecific siRNA control (green) compared with nontransfected K562 (blue) from at least three independent experiments. (B) ADE of dengue virus infection on wild type, FcγRIIa siRNA, or control siRNA–treated K562 cells (upper panel) and FcγRIIa specific Ab treated K562 cell (lower panel). (C) Expression of FcγRIIa and FcγRIIb on K562 cells transfected with hFcγRIIa cDNA or pCMV6-XL4 control plasmid. (D) Percent DV infection in FcγRIIb transfected and control transfected K562 cells in the absence or presence of dengue immune serum at enhancement titers. Data are shown as mean ± SE from n = 3 independent experiments.
FcγRIIa, but not FcγRIIb, supports ADE of dengue virus infection in NIH-3T3 murine cells
To investigate the effects of human FcγRIIa and FcγΡIIb independently, we transfected a non–Fc-bearing mouse fibroblast cell line, NIH-3T3, to express either FcγRIIa or FcγRIIb. It is important to note that NIH-3T3 cells are not permissive to dengue infection unless transfected with either DC-SIGN, a C-type lectin allowing direct infection, or FcγR for Ab mediated DENV infection. To compare the efficiency, we matched expression levels of FcγRIIa and FcγRIIb so that ∼40% of the 3T3 cells expressed either isoform with similar mean fluorescence intensity (Fig. 2A) and used them in the ADE assay. As expected, there was no infection in the wild type 3T3 cells transfected with empty vector control (pCMV6-XL4) under any condition (Fig. 2B, white bars). However, the 3T3-FcγRIIa cells showed significantly more infection than the 3T3-FcγRIIb cells at enhancement titers (Fig. 2B, right side; 10% versus 2.5%; p = 0.005 at 1:160 dilution and 3.5% versus 1.2%; p = 0.01 at 1:640 dilution). These data suggested that a postreceptor attachment process might contribute to ADE given the nearly identical extracellular domains and expression levels of these two isoforms (12, 22).
FIGURE 2.

FcγRIIa, but not FcγRIIb, support ADE of DENV infection. (A) Mean fluorescence intensity (MFI) of FcγRIIa and FcγRIIb on 3T3 cells following transfection with pCMV6-XL4 (control), FcγRIIa, and FcγRIIb. (B) Percent DENV infection in the absence or presence of dengue immune serum in transfected 3T3 cells from (A). Data are shown as mean ± SE from n = 3 independent experiments. *p < 0.1, **p < 0.001, Student t test.
FcγRII isoforms demonstrate similar DENV-immune complex binding but differences in internalization
To test our hypothesis that the isoforms had similar binding but different postreceptor processes, we established a real-time PCR-based binding assay to enumerate viral particles that bound and then entered into the different cell types via different FcγRII isoforms (Fig. 3A). Our data indeed showed comparable binding to FcγRIIa and FcγRIIb (Fig. 3B, white bars) as predicted, given the 92% homology of their extracellular domains. However, internalization via FcγRIIa was more efficient with a 2-log increase in viral copy number per million cells (Fig.3B, black bars; log106.8 versus 104.5 viral RNA copies; p = 0.01). It was shown previously that Ag endocytosed by the inhibitory receptor FcγRIIb accesses a nondegradative intracellular compartment that recycles to the cell surface (23, 24). Therefore, the efficiency of entry and postentry routing differences could contribute to overall retention of virus derived from immune complexes engaged with respective FcγRII isoforms.
FIGURE 3.

Binding and internalization of dengue immune complex via FcγRIIa and FcγRIIb. (A) Schematic diagram of RT-PCR–based binding/internalization assay. (B) Enumeration of viral RNA on the cell surface (binding: tube2-tube5-tube 1) and inside the cells (internalization: tube 4-tube 3-tube 5-tube 1) of FcγRIIa or FcγRIIb transfected 3T3 cells. Data are shown as mean ± SE from n = 3 independent experiments. *p < 0.01, Student t test. ns, not significant.
ITAM and ITIM motifs significantly modulate FcγR mediated ADE
With nearly identical extracellular domains (13) and comparable binding to the isoforms, we reasoned that postreceptor processes differentially directed the DV immune complex after attachment. We suspected that the ITIM/ITAM intracytoplasmic motifs were a deciding factor in dengue virus ADE. To address this question, we conducted a motif swapping experiment in which we transfected the 3T3 cells with either the FcγRIIa containing the ITIM motif or the FcγRIIb containing the ITAM motif and compared these swapped cells with the respective wild types in the ADE assay (Fig. 4). Despite similar expression levels of the Fcγ receptors on the 3T3 cells (Fig. 4C), the ability of the swapped cells to support ADE was markedly different from the wild types. The presence of the ITIM cytoplasmic tail, either in the wild type FcγRIIb cell or in the FcγRIIa-ITIM swapped cell, markedly limited ADE (Fig. 4D, FcγRIIa-ITIM [sw]). Interestingly, ADE was restored in 3T3 cells transfected with FcγRIIb containing the ITAM motif (Fig. 4D, FcγRIIb-ITAM [sw]). Finally, to assess productivity of infection, we tested the infectivity of the released DV virions in the supernatants from the different cell systems in the traditional Vero cell plaque assay. The cells containing an ITAM motif, regardless of the extracellular domain, showed the highest ADE mediated infectivity and viral output. In contrast, the presence of the ITIM motif, regardless of the extracellular receptor, markedly reduced or abrogated ADE entirely in these cell systems (Fig. 4E).
FIGURE 4.
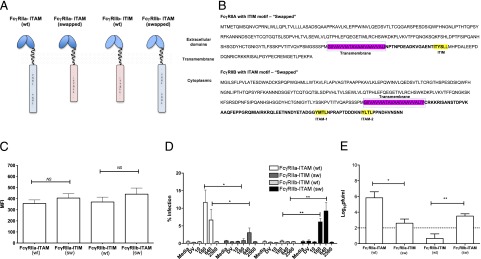
FcγRIIa and FcγRIIb cytoplasmic domains are critical for ADE of dengue virus infection. (A) Schematic diagram of cytoplasmic domain switching. (B) Amino acid sequences of FcγRIIa and FcγRIIb with swapped intracellular ITAM/ITIM motifs. (C) Cell surface expression of FcγRIIa and FcγRIIb on transfected 3T3 cells. (D) ADE of DV infection in FcγRIIa, FcγRIIb, FcγRIIa-ITIM, and FcγRIIb-ITAM transfected 3T3 cells. (E) The number of dengue infectious particles released from transfected 3T3 cells infected with DENV in the presence of dengue immune serum at peak enhancement titers (1:160). Data are shown as mean ± SE from n = 3 independent experiments. *p < 0.1, **p < 0.001, Student t test.
Discussion
The activatory FcγRII isoform, FcγRIIa, is uniquely expressed in humans and other primates (not rodents), and is a well-characterized receptor for Ab-enhanced dengue infection (7, 20, 25, 26). Among activatory Fcγ receptors, FcγRIIa (CD32) was recognized as the most efficient human FcγR compared with FcγRIa (CD64) in regard to Ab-mediated DENV infection in vitro (27, 28). In this study we focused on FcγRII isoforms. The human FcγRII (CD32) isoforms have different structural motifs in their cytoplasmic tail domains: ITAM motif in FcγRIIa and ITIM motif in FcγRIIb. Although it is widely held that FcγRII enables ADE, the relative roles of the activating and inhibitory isoforms, usually coexpressed on myeloid target cells, are not well understood in regard to viral infection. In this study, we transfected human FcγRIIa and FcγRIIb isoforms in their native (wild type) or cytoplasmic domain-swapped (mutant) forms into human and murine cell lines to address the relative contributions of each isoform specifically to DENV ADE in light of their notable extracellular domain homology, because the most abundantly expressed FcγR subclass and their codistribution on dengue target cells in vivo.
Most human or rodent myeloid cells contain multiple types of FcγRs, making it difficult to study the biology of individual FcγR types. We used human K562 cells in our initial characterizations of human FcγRIIa and FcγRIIb functions in regard to ADE of DENV infection. K562 cells, and many other cell types, are used for ADE studies of DENV in vitro. However, K562s have a distinct advantage because they express a single FcγR isoform: FcγRIIa. This approach allowed us to study FcγRIIa biology in isolation. Furthermore, the K562 FcγRIIa showed a bimorphic allotype at position 131 (H/R), thus minimizing any potential binding differences because IgG subclasses bind differently to human FcγRII allotypes/subtypes. The use of polyclonal anti-dengue serum for complexing with DENV2 was thought to minimize any potential binding differences because of IgG1-4 ligating FcγRII allotypes (11, 29). We used small inhibitory RNAs or a specific mAb directed to human FcγRIIa to inhibit DENV ADE and confirm its important role in DV infection of K562 cells (Fig. 1A, 1B). FcγRIIb is a unique class of FcγR with inhibitory capacity that is only expressed in higher order mammals. In contrast to FcγRIIa, ligation of the DENV immune complexes to FcγRIIb, which was previously shown to inhibit FcγR-mediated phagocytosis of large immune complexes, prevented ADE of dengue virus infection in FcγIIb knockdown and knock-in THP-1 cells (30). We confirmed the inhibitory effect of FcγRIIb on DENV ADE by coexpressing FcγRIIb on transfected K562 cells. As expected, we observed reductions in DENV infection at peak enhancement titers in FcγRIIb knock-in cells (Fig. 1D). These data strongly indicated that despite the extracellular homology of FcγRIIa and FcγRIIb (22), the intracytoplasmic signaling mediated by the respective cytoplasmic domain motifs or inhibition thereof contributed opposing effects on ADE of DENV2 infection. An interesting area to explore, not fully addressed here, is the precise inhibitory effects of FcγRIIb on ADE. For example, does ITIM directly inhibit ITAM activation, or is it better explained by postreceptor processing with differential routing as shown in other systems (22), or does FcγRIIb act as a “sink” competing for the available dengue immune complexes and effectively reduce the overall uptake by FcγRIIa, or some combination therein? The elucidation of the mechanistic details of potential interactions between FcγRIIa and FcγRIIb and signal integration in ADE warrants further investigation.
Building on our results, we separately investigated the influence of the cytoplasmic domains using human FcγRIIa and FcγRIIb transfected non–Fc-bearing and non–DENV-permissive 3T3 cells. Our findings also support the regulatory influence of FcγRII on ADE of DENV infection in cell types that coexpress FcγRII isoforms such as DCs (28).
We next discovered that despite comparable expression levels on transfected cells, human FcγRIIa supports ADE of DENV significantly greater than FcγRIIb (Fig. 2). Despite similar extracellular domains (12, 22) and immune complex binding capacity, we found markedly different influence of the two different human FcγRII isoforms on ADE of DENV2 infection (Fig. 3B). We hypothesized that DENV immune complex internalization after binding or ligation was a point of divergence for these isoforms (Fig. 3). It is possible that upon engagement of dengue immune complexes, FcγRIIa is delivered along with its ligand to lysosomal compartments for degradation, whereas FcγRIIb is dissociated from the ligand and routed separately into the recycling pathway, as suggested in a study using transfected cells and nonpathogenic particles (24). These postreceptor signaling and sorting differences in activatory versus inhibitory FcγRII isoforms could favor differential cellular activation, differential viral replication (when immune complexes engage one receptor type versus the other), and differential production in FcγRIIa-expressing cells versus FcγRIIb-expressing cells. As a counterbalance, the FcγRIIb pathway inhibits phagocytosis and signaling and could provide a means to control inflammatory responses after exposure to immune complexes (24). This activation and inhibition by FcγRIIa and FcγRIIb, respectively, in cells could be operating in regulating net ADE during DENV disease.
In this study, we specifically demonstrate that respective cytoplasmic domains of FcγRIIa and FcγRIIb largely determine ADE. Interchanging the intracellular portions of these receptors dramatically affected ADE. Strikingly, the FcγRIIa engineered to carry ITIM essentially no longer supported ADE (Fig. 4D), consistent with a previous report showing that targeted mutations of ITAM motifs in the cytoplasmic domain of FcγRIIa eliminated ADE of DENV infection (25, 27). However, the FcγRIIb containing ITAM restored ADE (Fig. 4D, 4E). Finally, entry via the wild type FcγRIIb-ITIM clearly restricted infection (Fig. 4D), which was confirmed by low numbers of infectious virus in culture supernatants (Fig. 4E).
Our findings highlight fundamental differences in the functions of human FcγRII isoforms and specifically identify the cytoplasmic tail as a major determinant of Ab-dependent enhancement of dengue virus infection. Swapping each of the isoform’s cytoplasmic tails revealed that the ITIM negatively regulated ADE while the ITAM facilitated ADE, regardless of which extracellular FcγRII domain was used for ligand binding. This finding raises the notion that the coexpression of FcγRIIa and FcγRIIb, which both bound DENV immune complexes comparably, serves a primary regulatory function in governing viral entry, net retention, and net virus production and replication (productive infection). Therefore, the relative expression levels of each FcγRII isoform on any given cell type or tissue in vivo could be an important determinant of infectious or inflammatory processes and, in turn, is likely another critical immunologic checkpoint. This work also draws attention to the variety of entry routes and cell types that factor into production of DENV from target cells. The inhibitory receptor isoform, FcγRIIb, might have evolved with its activatory counterpart as a mechanism of regulating the amount of immune complex intake by FcγRIIa during infections, as a means of controlling infection and modulating the immune response.
Given the wide cellular distribution of FcγRIIa, its restricted expression to humans and other primates, and its signaling capacity, it is tempting to speculate that relative overexpression of this activatory isoform could influence the severity of human dengue disease and pathogenesis. The next steps for dengue research could explore the role of FcγRIIa and FcγRIIb in human infections to further the understanding of signaling mechanisms and routing pathways mediated by engagement of DENV immune complexes and to study the effects of different viral serotypes complexed with different human or mouse IgG subclasses. Finally, these results support the development of a mouse model wherein all murine FcγRs are replaced with human FcγRs (31). The addition of FcγRIIa-expressing cells into mice could counterbalance the unopposed inhibitory murine FcγRIIb isoform, enable ADE, and generate a much needed small animal model to advance our understanding of complicated dengue disease.
Acknowledgments
We thank Dr. W.W. Shanaka I. Rodrigo for critical comments and helpful suggestions and MacroGenics for the mAbs.
This work was supported by the Pediatric Dengue Vaccine Initiative and in part by the cooperative agreement (W81XWH-07-2-0067) between the Henry M. Jackson Foundation for the Advancement of Military Medicine, and the U.S. Department of Defense.
The views expressed are those of the authors and should not be construed to represent the positions of the U.S. Army or the U.S. Department of Defense.
- ADE
- Ab-dependent enhancement
- DC
- dendritic cell
- DENV
- dengue virus
- ITAM
- immunoreceptor tyrosine-based activation motif
- ITIM
- immunoreceptor tyrosine-based inhibitory motif
- MOI
- multiplicity of infection
- QRT-PCR
- quantitative real-time PCR.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Gubler D. J. 1998. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 11: 480–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monath T. P. 1994. Dengue: the risk to developed and developing countries. Proc. Natl. Acad. Sci. USA 91: 2395–2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morens D. M., Fauci A. S. 2008. Dengue and hemorrhagic fever: a potential threat to public health in the United States. JAMA 299: 214–216 [DOI] [PubMed] [Google Scholar]
- 4.Hawkes R. A., Lafferty K. J. 1967. The enchancement of virus infectivity by antibody. Virology 33: 250–261 [DOI] [PubMed] [Google Scholar]
- 5.Halstead S. B., O’Rourke E. J. 1977. Antibody-enhanced dengue virus infection in primate leukocytes. Nature 265: 739–741 [DOI] [PubMed] [Google Scholar]
- 6.Halstead S. B. 2003. Neutralization and antibody-dependent enhancement of dengue viruses. Adv. Virus Res. 60: 421–467 [DOI] [PubMed] [Google Scholar]
- 7.Littaua R., Kurane I., Ennis F. A. 1990. Human IgG Fc receptor II mediates antibody-dependent enhancement of dengue virus infection. J. Immunol. 144: 3183–3186 [PubMed] [Google Scholar]
- 8.Dai X., Jayapal M., Tay H. K., Reghunathan R., Lin G., Too C. T., Lim Y. T., Chan S. H., Kemeny D. M., Floto R. A., et al. 2009. Differential signal transduction, membrane trafficking, and immune effector functions mediated by FcgammaRI versus FcgammaRIIa. Blood 114: 318–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kontny U., Kurane I., Ennis F. A. 1988. Gamma interferon augments Fc gamma receptor-mediated dengue virus infection of human monocytic cells. J. Virol. 62: 3928–3933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moi M. L., Lim C. K., Kotaki A., Takasaki T., Kurane I. 2011. Detection of higher levels of dengue viremia using FcγR-expressing BHK-21 cells than FcγR-negative cells in secondary infection but not in primary infection. J. Infect. Dis. 203: 1405–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodrigo W. W., Block O. K., Lane C., Sukupolvi-Petty S., Goncalvez A. P., Johnson S., Diamond M. S., Lai C. J., Rose R. C., Jin X., Schlesinger J. J. 2009. Dengue virus neutralization is modulated by IgG antibody subclass and Fcgamma receptor subtype. Virology 394: 175–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ravetch J. V., Kinet J. P. 1991. Fc receptors. Annu. Rev. Immunol. 9: 457–492 [DOI] [PubMed] [Google Scholar]
- 13.Hulett M. D., Hogarth P. M. 1994. Molecular basis of Fc receptor function. Adv. Immunol. 57: 1–127 [DOI] [PubMed] [Google Scholar]
- 14.Hogarth P. M. 2002. Fc receptors are major mediators of antibody based inflammation in autoimmunity. Curr. Opin. Immunol. 14: 798–802 [DOI] [PubMed] [Google Scholar]
- 15.Nimmerjahn F., Ravetch J. V. 2008. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 8: 34–47 [DOI] [PubMed] [Google Scholar]
- 16.Boonnak K., Slike B. M., Burgess T. H., Mason R. M., Wu S. J., Sun P., Porter K., Rudiman I. F., Yuwono D., Puthavathana P., Marovich M. A. 2008. Role of dendritic cells in antibody-dependent enhancement of dengue virus infection. J. Virol. 82: 3939–3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tassaneetrithep B., Burgess T. H., Granelli-Piperno A., Trumpfheller C., Finke J., Sun W., Eller M. A., Pattanapanyasat K., Sarasombath S., Birx D. L., et al. 2003. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 197: 823–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jefferis R., Lund J. 2002. Interaction sites on human IgG-Fc for FcgammaR: current models. Immunol. Lett. 82: 57–65 [DOI] [PubMed] [Google Scholar]
- 19.Allhorn M., Olin A. I., Nimmerjahn F., Collin M. 2008. Human IgG/Fc gamma R interactions are modulated by streptococcal IgG glycan hydrolysis. PLoS ONE 3: e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mady B. J., Kurane I., Erbe D. V., Fanger M. W., Ennis F. A. 1993. Neuraminidase augments Fc gamma receptor II-mediated antibody-dependent enhancement of dengue virus infection. J. Gen. Virol. 74: 839–844 [DOI] [PubMed] [Google Scholar]
- 21.Guy B., Chanthavanich P., Gimenez S., Sirivichayakul C., Sabchareon A., Begue S., Yoksan S., Luxemburger C., Lang J. 2004. Evaluation by flow cytometry of antibody-dependent enhancement (ADE) of dengue infection by sera from Thai children immunized with a live-attenuated tetravalent dengue vaccine. Vaccine 22: 3563–3574 [DOI] [PubMed] [Google Scholar]
- 22.Nimmerjahn F., Ravetch J. V. 2007. Fc-receptors as regulators of immunity. Adv. Immunol. 96: 179–204 [DOI] [PubMed] [Google Scholar]
- 23.Bergtold A., Desai D. D., Gavhane A., Clynes R. 2005. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity 23: 503–514 [DOI] [PubMed] [Google Scholar]
- 24.Zhang C. Y., Booth J. W. 2010. Divergent intracellular sorting of FcgammaRIIA and FcgammaRIIB2. J. Biol. Chem. 285: 34250–34258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moi M. L., Lim C. K., Takasaki T., Kurane I. 2010. Involvement of the Fc gamma receptor IIA cytoplasmic domain in antibody-dependent enhancement of dengue virus infection. J. Gen. Virol. 91: 103–111 [DOI] [PubMed] [Google Scholar]
- 26.Moi M. L., Lim C. K., Kotaki A., Takasaki T., Kurane I. 2010. Development of an antibody-dependent enhancement assay for dengue virus using stable BHK-21 cell lines expressing Fc gammaRIIA. J. Virol. Methods 163: 205–209 [DOI] [PubMed] [Google Scholar]
- 27.Rodrigo W. W., Jin X., Blackley S. D., Rose R. C., Schlesinger J. J. 2006. Differential enhancement of dengue virus immune complex infectivity mediated by signaling-competent and signaling-incompetent human Fcgamma RIA (CD64) or FcgammaRIIA (CD32). J. Virol. 80: 10128–10138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boonnak K., Dambach K. M., Donofrio G. C., Tassaneetrithep B., Marovich M. A. 2011. Cell type specificity and host genetic polymorphisms influence antibody-dependent enhancement of dengue virus infection. J. Virol. 85: 1671–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.García G., Sierra B., Pérez A. B., Aguirre E., Rosado I., Gonzalez N., Izquierdo A., Pupo M., Danay Díaz D. R., Sánchez L., et al. 2010. Asymptomatic dengue infection in a Cuban population confirms the protective role of the RR variant of the FcgammaRIIa polymorphism. Am. J. Trop. Med. Hyg. 82: 1153–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan K. R., Zhang S. L., Tan H. C., Chan Y. K., Chow A., Lim A. P., Vasudevan S. G., Hanson B. J., Ooi E. E. 2011. Ligation of Fc gamma receptor IIB inhibits antibody-dependent enhancement of dengue virus infection. Proc. Natl. Acad. Sci. USA 108: 12479–12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith P., DiLillo D. J., Bournazos S., Li F., Ravetch J. V. 2012. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc. Natl. Acad. Sci. USA 109: 6181–6186 [DOI] [PMC free article] [PubMed] [Google Scholar]