

Abstract
Because of the very short half-life of factor VII, prophylaxis in factor VII deficiency is considered a difficult endeavor. The clinical efficacy and safety of prophylactic regimens, and indications for their use, were evaluated in factor VII-deficient patients in the Seven Treatment Evaluation Registry. Prophylaxis data (38 courses) were analyzed from 34 patients with severe factor VII deficiency (<1-45 years of age, 21 female). Severest phenotypes (central nervous system, gastrointestinal, joint bleeding episodes) were highly prevalent. Twenty-one patients received recombinant activated factor VII (24 courses), four received plasma-derived factor VII, and ten received freshfrozen plasma. Prophylactic schedules clustered into “frequent” courses (three times weekly, n=23) and “infrequent” courses (≤2 times weekly, n=15). Excluding courses for menorrhagia, “frequent” and “infrequent” courses produced 18/23 (78%) and 5/12 (41%) “excellent” outcomes, respectively; relative risk, 1.88; 95% confidence interval, 0.93-3.79; P=0.079. Long term prophylaxis lasted from 1 to >10 years. No thrombosis or new inhibitors occurred. In conclusion, a subset of patients with factor VII deficiency needed prophylaxis because of severe bleeding. Recombinant activated factor VII schedules based on “frequent” administrations (three times weekly) and a 90 μg/kg total weekly dose were effective. These data provide a rationale for long-term, safe prophylaxis in factor VII deficiency (clinicaltrials.gov: NCT01269138).
Introduction
Inherited factor VII (FVII) deficiency is the most common of the rare autosomal recessive bleeding disorders, with an estimated prevalence of one case per 300,000 population in European countries.1,2 The prevalence of the severe forms of FVII deficiency is, however, likely to be higher in those countries in which consanguineous marriages are frequent.3
Affected patients display a wide range of clinical phenotypes, from an asymptomatic condition to serious and potentially fatal hemorrhagic episodes such as central nervous system or gastrointestinal bleeding episodes.4 Symptomatic patients can be divided into two major categories: those with a mild to moderate bleeding tendency, and those with a very clear bleeding tendency that may be more severe than that in hemophilia A or B. The former group of patients mainly experience mucosal bleeding, a clinical picture that mimics that of a platelet disorder and does not call for prophylaxis. In contrast, for the most severely affected patients, a group whose condition is characterized by life- or limb-threatening hemorrhages, long-term prophylactic FVII replacement therapy should be considered.
Patients with clinically severe FVII deficiency commonly become symptomatic at a young age (e.g. soon after birth or when toddlers), have low to very low levels of residual FVII, and usually have severe causative F7 gene lesions.4 Attention is increasingly focusing on this cohort of patients with severe symptoms, who account for 10-15% of patients with FVII deficiency.1,4,5 Prophylaxis appears to be the most appropriate therapeutic option for these patients, but little is known about its use in FVII deficiency in particular, or in rare congenital bleeding disorders in general.6
The introduction of prophylaxis as a therapeutic modality in congenital FVII deficiency has been hampered by the fact that FVII and activated FVII (FVIIa) have very short halflives (<3 hours), a feature that is particularly apparent in children.7 It would, therefore, appear difficult to use the same schedules in FVII deficiency as those adopted in hemophilia, a bleeding disorder in which prophylaxis has become common practice.8-11 Clinical experience with prophylaxis in FVII deficiency has been limited to individual case reports, mostly not published in full.12-29 The Seven Treatment Evaluation Registry (STER, www.targetseven.org) has collected information on prophylaxis from a worldwide series of well-characterized FVII-deficient patients. Here we report an analysis of prophylaxis in these patients, focusing on the indications, clinical efficacy and safety of these prophylactic regimens.
Design and Methods
STER prospectively collected data on bleeding episodes, surgery or prophylaxis in patients with FVII deficiency following strictly controlled data collection procedures established by the International FVII Deficiency Study Group.1,4 We used the structured and detailed data captured in STER to examine the management of patients with FVII deficiency. In particular, our objectives were to identify patients for whom prophylaxis is advisable, to define clinical settings in which prophylaxis is necessary, and to identify effective and safe dosing schedules. The STER protocol is published on clinicaltrials.gov (NCT01269138).
Information in STER was collected using web-based forms. At enrollment, a blood sample was drawn for centralized plasma inhibitor determination, and a standardized inhibitor determination30 was performed before and 30 days after administering one or more replacement therapies. In the treatment section, information was collected on the prophylactic regimen used (type of product, dosage and schedule), treatment outcomes (bleeding episodes recorded during prophylaxis), complications and adverse events. Two categories of prophylaxis were employed: primary (early) prophylaxis (i.e. when regular infusions of factor concentrates were started after the first bleeding episode and/or before 2 years of age) and secondary prophylaxis (i.e. regular administrations of replacement therapy started after 2 years of age or after two or more bleeding episodes). Short-term prophylaxis courses were arbitrarily defined as those lasting <12 months. Investigators were asked to report bleeding episodes that occurred during the period of prophylaxis.
The registry database was examined in September 2011. The prophylactic regimens and types of replacement therapy used [fresh-frozen plasma (FFP), plasma-derived FVII concentrates (pd-FVII), recombinant FVIIa (rFVIIa)] reflected the clinical judgment of the physician enrolling a patient. Dosing was expressed as follows: (i) rFVIIa in μg/kg; (ii) pd-FVII in IU/kg; and (iii) FFP in IU/kg, where 1 IU corresponds to 1 mL of FFP. Patients in whom prophylaxis was started before the online registry was active were also enrolled into STER, provided that prophylaxis was still ongoing and was planned to be continued with the same dose and schedule. The research proposed by the STER Study Group was approved by the Ethics Committee of L'Aquila University (coordinator's institution) and, in parallel, by the committees of the other institutions involved. The STER protocol is publicly available (http://www.targetseven.org). Informed consent was obtained from all patients at enrollment.
Efficacy was evaluated according to the following criteria: “excellent” (i.e. no bleeding episodes reported while on prophylaxis, especially in target sites), “partly effective” (i.e. reduction in bleeding events by >50% and no bleeding in a target site) or “ineffective” (<50% reduction in the number of bleeding events). Data on gene mutations were only reported in terms of zygosity for privacy reasons.
Statistical analysis
Data were obtained from the STER database and analyzed following a data quality and consistency check according to the STER data-management plan. The statistical analysis was based on descriptive measures of the distribution, such as mean and median for the position parameter, and range for the variability. Two different clinical approaches, expressed in terms of treatment frequency (frequent versus infrequent), were compared using relative risks (RR), which examined the ratio of cases having a positive outcome in the two groups, and is reported together with their associated z-statistic, P values and confidence intervals (CI). Analyses were performed using MedCalc® software version 7.4.1.2 (http://www.medcalc.org).
Results
Information was recorded in the STER database from 34 patients with FVII deficiency receiving prophylaxis in 13 hemophilia centers (11 countries). Of these patients, 21 were female (Table 1) and the majority (n=19) were Caucasian. The remainder were Asian (n=12) or Latino (n=3). Of the 34 patients receiving prophylaxis, 33 had no inhibitor, while one had an inhibitor to FVII, diagnosed before enrollment into the study (patient 25; high responder). The mean age at which a course of prophylaxis was started was 11.1 years (median 7 years, range, <1-45 years). The clinical spectrum and the number of different bleeding episodes observed prior to prophylaxis are shown in Table 1, as are FVII coagulant activity (FVIIc) levels. Fourteen of the 20 patients for whom FVII gene zygosity data were available were compound heterozygotes and six were homozygotes.
Table 1.
Clinical characteristics of the patients prior to prophylaxis.
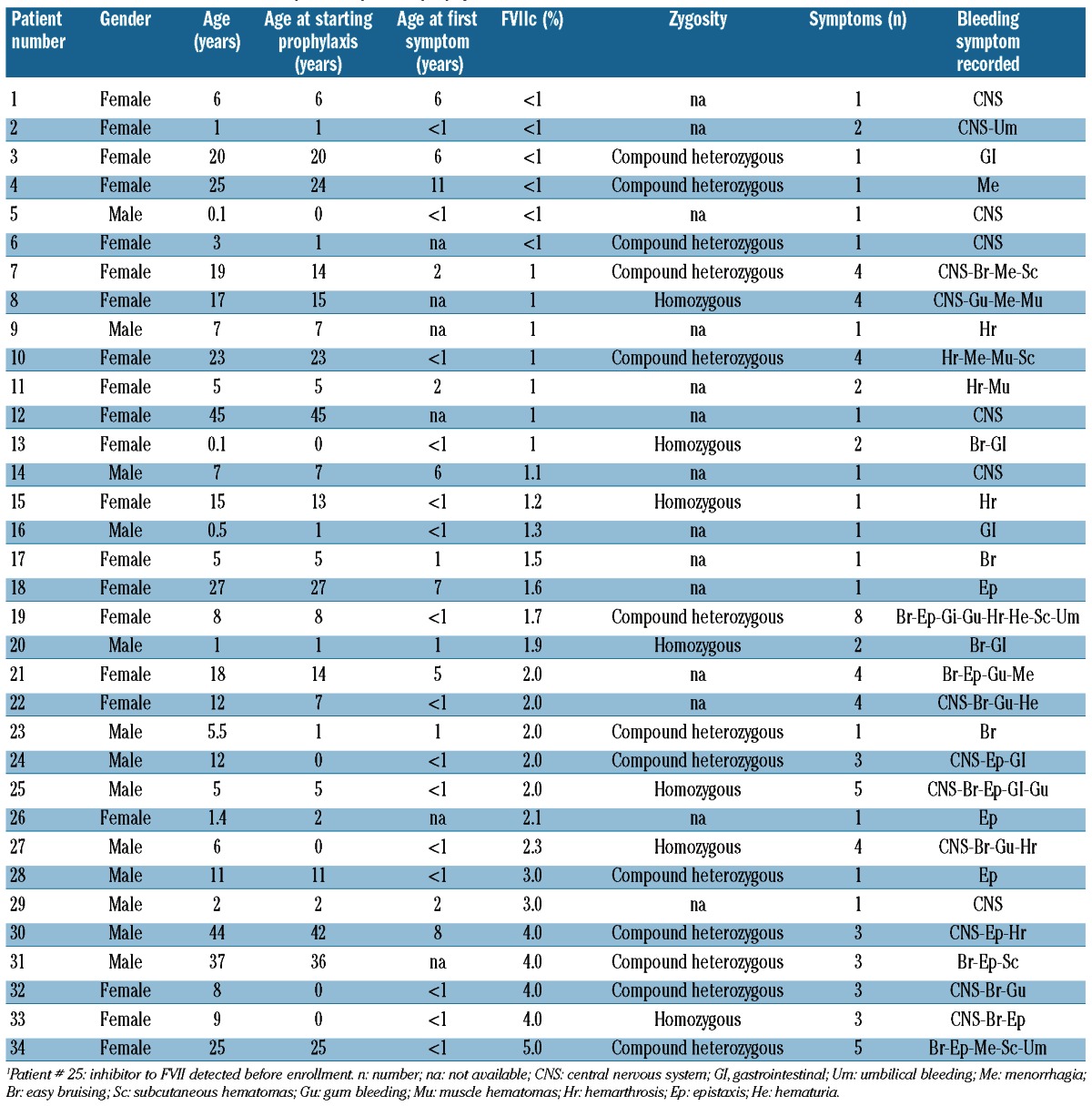
The reasons for initiating prophylaxis and the treatment regimens used varied among the patients analyzed (see Table 2 for details of course duration, schedules, dosing, efficacy evaluation and side effects). Thirty-eight courses were recorded altogether (patients 9, 10, 19 and 28 being recorded twice). Of these, 28 courses lasted at least 12 months, while ten were of short duration (<12 months; range, 2-10 months). Overall, ten patients received courses of “primary” prophylaxis (Table 2).
Table 2.
Prophylaxis treatment schedules.
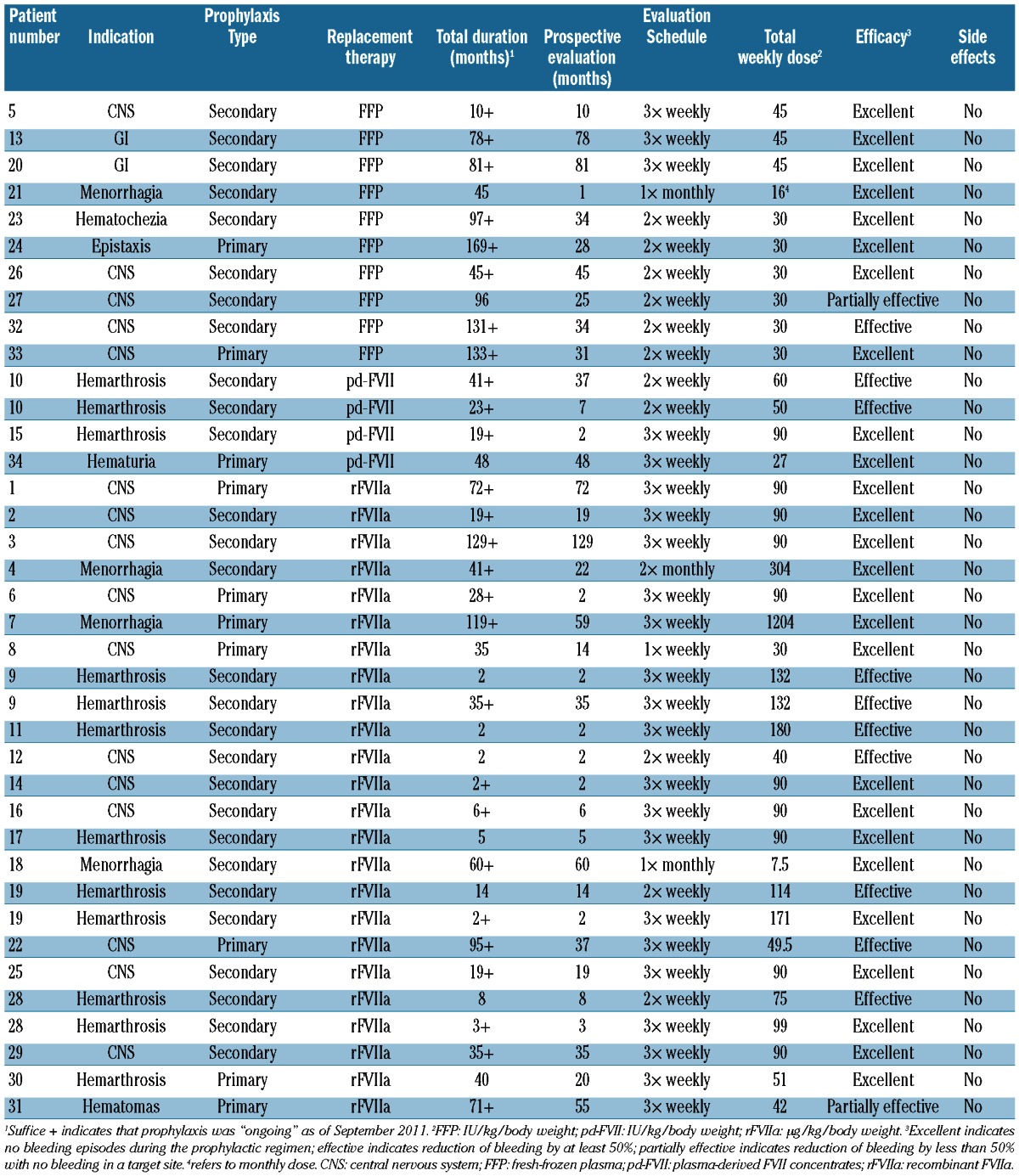
Twenty-one patients (Table 2) received rFVIIa (NovoSeven®, Novo Nordisk A/S, Denmark) for prophylaxis (24 courses), three patients (four courses) were given pd-FVII concentrates (Facteur VII®, LFB, Courtaboeuf, France or Provertin-UM TIM 3, Baxter-Immnuno, Vienna, Austria), and ten children received FFP, mostly soon after birth because of severe “spontaneous” bleeding episodes. One patient had a high titer inhibitor (maximum titer: 59 Bethesda Units), which appeared at 1 year of age after treatment with FFP, prior to the study. Prophylaxis with rFVIIa was initiated in this patient because of severe symptoms (recurrent central nervous system bleeding).
The total duration of prophylaxis ranged from 2 months to 169 months (median 37.5), while the prospective duration ranged from 1 to 129 months (median 21). The “prospective” portion of the courses (1,085 months; mean 31.9 months/patient) amounted to 58.3% of the “total” duration (1,860 months; mean 54.7 months/patient).
Recombinant activated factor VII
Three types of schedules were reported in the 21 patients receiving rFVIIa prophylaxis (24 courses). Excluding two patients with severe, refractory menorrhagia (>80 mL discharge/menses with profound iron-deficient anemia since the menarche, who were given a “personalized” prophylactic regimen on the first and second days of menses), the frequency of rFVIIa administration was categorized as “frequent” (three times per week, n=18) or “infrequent” [n=4; twice (n=3) or once (n=1) weekly].
With reference to the evaluation of prophylaxis, 14/22 courses were reported as having an “excellent” outcome (13 “frequent” and one “infrequent” administration), five were “effective” (four “frequent” and one “infrequent”) and two “partly effective” (“frequent”). The total weekly dose of rFVIIa administered was similar for the “excellent” (mean, 85.7 μg/kg; median, 90 μg/kg; range, 30-120 μg/kg) and “effective” (mean, 106.8 μg/kg; median, 132 μg/kg; range, 40-189 μg/kg) outcomes. The patient whose efficacy evaluation was reported as “partly effective” received 42 μg/kgrFVIIa.
Plasma-derived factor VII concentrates
Four courses of prophylaxis with pd-FVII concentrates were given to three patients (age range, 5-25 years). The outcome was “excellent” in two patients given pd-FVII three times per week, whereas, prophylaxis given twice weekly did not eliminate, but only reduced, the occurrence of bleeding episodes in the other young adult, with either course of prophylaxis administered.
Fresh-frozen plasma
FFP was given at a median dose of 15 IU/kg twice or three times per week in nine children (mostly neonates or toddlers) and one young female (aged 18 years) resulting in “excellent” (n=8), “effective” (n=1) and “partly effective” (n=1) outcomes.
Differences between the outcomes of patients receiving “frequent” and “infrequent” prophylactic schedules did not reach formal statistical significance, although a “trend” was evident (RR, 1.88; 95% CI, 0.93-3.79; P=0.079); comparison between the effects of different replacement therapy dosages did not reveal statistically significant differences (RR, 0.93; 95% CI, 0.60-1.44; P=0.75).
Three females received “personalized” prophylactic courses for menorrhagia (one with FFP and two with rFVIIa), comprising two doses on the first 2 days or one dose on the first day of their cycles. All three females had normal menses (<50 mL) after prophylaxis was started.
Safety
No thrombotic events of any kind were reported with any product, nor were new inhibitors recorded.
Discussion
In this paper, we report an analysis of data on the long and short-term use of prophylaxis in FVII deficiency collected in the STER. Considering the rarity of the disease and, in particular, of the severe forms (10-15%), this series of patients is especially important because it examined a number of controversial issues concerning the efficacy and safety of repeated administrations of replacement therapy. In essence, the objectives of our study were to identify patients for whom prophylaxis is advisable, to define clinical settings in which prophylaxis is necessary and, finally, to identify effective and safe dosing schedules.
This cohort of patients had severe disease characterized by life-threatening and crippling bleeding symptoms (e.g. central nervous system bleeding episodes, hemarthrosis and gastrointestinal bleeding episodes) and accounted for 26% of those patients in the STER with FVIIc levels less than 5%, and 89% of patients with FVIIc levels less than 1%. Importantly, central nervous system bleeding was the most frequent indication for prophylaxis (16/34 individuals, 47%) followed by hemarthrosis [12/34 (35%)], a finding consistent with the literature.13-16,18-23,26 The severity of the clinical picture is also highlighted by the age at disease presentation [within the first year of life in 18/34 (53%) cases] and the high number of different bleeding symptoms reported in many of the patients. Clotting phenotypes were consistent with the clinical phenotypes, as indicated by the low to very low residual FVIIc levels.
A recent, seminal study performed in young children with hemophilia confirmed the superiority of long-term prophylaxis in the prevention of chronic arthropathy, in comparison with on-demand treatment.8 These results were confirmed by a randomized Italian study.10
In the current publication, the first prospective study on prophylaxis in FVII deficiency, a variety of schedules, doses and treatment modalities (rFVIIa, pd-FVII, FFP) were used, which may partly reflect the choices of the treating physician, but may also be a consequence of the lack of evidence-based publications and guidelines. Overall, prophylaxis yielded “excellent” results in 68% of the courses. As expected, our analysis suggested that schedules based on more frequent administrations (three times weekly) were associated with the best outcomes, although the number of courses evaluated was too small to yield a statistical significance. The most commonly used schedule of rFVIIa yielding “excellent” outcomes was a weekly dose of 90 μg/kg.
The widely known low concentration/high infusion volume problem related to FFP administration may lead to less optimal treatment than with factor concentrates, especially in FVII deficiency, in which a “rare” protein with a short half-life is lacking. Replacement therapy with FFP, the only treatment available in particular settings (e.g. Southeast Asia), was employed as primary prophylaxis in children less than 2 years old, who otherwise would have had little chance of survival. Outcomes with FFP, although difficult to compare directly with those of pd-FVII or rFVIIa concentrates, did not appear to be different, probably because many of the patients were less than 1 year of age. Although FFP may not secure full hemostasis during prophylaxis, it should be recognized that it can reduce the risk of severe, spontaneous or traumatic bleeding episodes, thereby substantially lowering the risk of death in these babies;13,26 however, as patients grow, their increased body weight becomes the most apparent limitation to the continuation of prophylaxis with FFP.
It is difficult to explain how prophylaxis schedules similar to those used in hemophilia A8,11 can be effective in FVII deficiency, a disease characterized by the lack of a protein with a rapid plasma clearance, especially in children.7 One explanation for this apparent paradox is offered by the possible presence of infused FVII bound extravascularly to pericytes, which may allow FVIIa to be available for a longer time than expected.31 Another possibility is that suggested by a recent study demonstrating binding of excess rFVIIa to activated protein C receptor on undamaged endothelial cells,32,33 which saturates the receptor and may keep excess rFVIIa in circulation for longer, or may allow a late subsequent release of the factor.
In our cohort of patients, as reported in the literature,12,19,20,25-27 the indications for prophylaxis were clear, as most patients (27/37) had types and clusters of bleeding that are considered the hallmarks of a severe bleeding disorder.4 We have also shown that the age of our patients at disease presentation fitted within the age ranges that we have previously reported:4 for central nervous system and gastrointestinal bleeding a median of 0.1 years, and for hemarthrosis 1.8 years.4 Our data are also consistent with those of an important study on FVII deficiency, in which a high prevalence of central nervous system bleeding (16%) and consanguinity were reported.34 Hemarthroses affected a smaller proportion of FVII-deficient patients (16% in the STER database), but joint damage in those affected by recurrent hemarthoses did not differ clinically from those in patients with FVIII deficiency.
Taking these observations into account, we suggest that “primary” prophylaxis is mandatory in babies less than 2 years old and in toddlers in whom a high bleeding risk has been ascertained. Prophylaxis should, therefore, be proposed (possibly within the frame of a prenatal diagnosis program) to those families who already have an affected child bearing a “null” F7 gene lesion associated with severe bleeding.35,36 In addition, early prophylaxis should be started in severely affected children when central nervous system and gastrointestinal bleeding episodes occur. We have recently published a paper on the treatment of “spontaneous” bleeding episodes in FVII deficiency and found that “on-demand” treatment of these severe bleeds is a suboptimal therapeutic choice.37 Recombinant FVIIa is the only product available for FVII-deficient patients that does not carry a risk of virus or prion transmission, the avoidance of such risk being especially important for very young children.
The first inhibitor to FVII was detected in an infant who had accidentally received very high doses (20-40 times higher than the recommended dose) of rFVIIa, and the occurrence of a few other inhibitors have been reported in previously published studies from STER.25,30,38,39 The one patient in this series with an inhibitor developed it before the initiation of prophylaxis.25 Nevertheless, prophylaxis with rFVIIa was started because of persisting, severe bleeding episodes and proved to be very effective in preventing recurrent central nervous system hemorrhages.
Thrombosis is another potential side effect of replacement therapy which warrants consideration. Although thrombosis is a rare complication, replacement therapy repeated for a long time could reveal a latent risk for thrombosis, even in patients with a clotting disorder such as FVII deficiency.40 In the literature, only catheter-related thrombotic complications in two FVII-deficient patients receiving high doses of rFVIIa (58 and 118 μg/kg daily) have been reported.6 In our cohort, despite the large number of infusions, no thrombotic events of any kind were reported during the follow-up period, either in adults or elderly patients, a finding consistent with the experience reported in a recent surgical study of FVII-deficient patients.41
Limitations to our study include the efficacy evaluation performed by the investigators, the retrospective part of the follow-up and also the small number of patients, which may have hampered a more precise definition of the efficacy of the prophylactic schedule. Nevertheless, for the patients with “excellent” results, no breakthrough bleeding episodes were reported in the database, in contrast to the less favorable outcomes in which bleeding episodes were reported. The retrospective part of the follow-up does not appear to be a critical issue considering the rarity of the disease, the fact that the outcomes from the retrospective follow-up were confirmed by the prospective part of the prophylaxis and, finally, that most of the followup (approximately 60%) was prospective.
In conclusion, our analysis suggests that prophylaxis programs should be established to prevent central nervous system and gastrointestinal bleeding episodes and/or their recurrences in infants with FVIIc levels less than 1%, and should also be adopted in young children with recurrent hemarthroses. Our data also show that a weekly dose of 90 to 100 mg/kg/body weight per week, split into three administrations, can be efficacious in preventing bleeding and was not associated with any side effects.
Supplementary Material
Acknowledgments
The authors would like to thank Prof. F. Bernardi and Prof. M. Pinotti for their assistance with the mutation analysis (some of the mutations were characterized in their laboratory at the University of Ferrara, Italy). The STER operations were run by Quintiles, who provided the following services: global project management (Late Phase Global Project Management – Dr. J. L. Merot, Quintiles-Benefit SNC, 3-5 rue Maurice Ravel, 92594 Levallois-Perret, Cedex, France), site start-up, remote site monitoring and data cleaning by the Project Coordination Centre. The authors are grateful to Mehran Karimi (Iran), Arlette Ruiz de Saez (Venezuela), Uriel Martinowitz and Gili Kenet (Israel), Nuria Bermejo (Spain), and Dragan Micic (Serbia).
MN and MG-B contributed equally to this manuscript.
Funding
This work was supported by institutional research organizations and unrestricted funding from Novo Nordisk and charities, administered by the Internal Medicine Department of the University of L'Aquila. GM, as coordinator of the International FVII Study Group and the STER, collected the financial support. Editorial assistance to the authors during the preparation of this manuscript was provided by Sharon Eastwood (medical writer, PAREXEL) and financially supported by Novo Nordisk A/S, in compliance with international guidelines for good publication practice.
Footnotes
Authorship and Disclosures: Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Mariani G, Bernardi F. Factor VII deficiency. Semin Thromb Hemost. 2009;35 (4):400-6 [DOI] [PubMed] [Google Scholar]
- 2.Mariani G, Dolce A. Congenital factor VII defiency. In:Lee CA, Berntorp EE, Hoots WK. Textbook of Haemophilia. 2nd ed Oxford: Wiley-Blackwell;2010:341-7 [Google Scholar]
- 3.Borhany M, Pahore Z, Ul Qadr Z, Rehan M, Naz A, Khan A, et al. Bleeding disorders in the tribe: result of consanguineous in breeding. Orphanet J Rare Dis. 2010;5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mariani G, Herrmann FH, Dolce A, Batorova A, Etro D, Peyvandi F, et al. Clinical phenotypes and factor VII genotype in congenital factor VII deficiency. Thromb Haemost. 2005;93 (3):481-7 [DOI] [PubMed] [Google Scholar]
- 5.Bernardi F, Dolce A, Pinotti M, Shapiro AD, Santagostino E, Peyvandi F, et al. Major differences in bleeding symptoms between factor VII deficiency and hemophilia B. J Thromb Haemost. 2009;7 (5):774-9 [DOI] [PubMed] [Google Scholar]
- 6.Todd T, Perry DJ. A review of long-term prophylaxis in the rare inherited coagulation factor deficiencies. Haemophilia.2010;16 (4):569-83 [DOI] [PubMed] [Google Scholar]
- 7.Berrettini M, Mariani G, Schiavoni M, Rocino A, Di Paolantonio T, Longo G, Morfini M. Pharmacokinetic evaluation of recombinant, activated factor VII in patients with inherited factor VII deficiency. Haematologica. 2001;86 (6):640-5 [PubMed] [Google Scholar]
- 8.Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med.2007;357 (6):535-44 [DOI] [PubMed] [Google Scholar]
- 9.Manco-Johnson M. Comparing prophylaxis with episodic treatment in haemophilia A: implications for clinical practice. Haemophilia.2007;13 (Suppl 2):4-9 [DOI] [PubMed] [Google Scholar]
- 10.Gringeri A, Lundin B, von MS, Mantovani L, Mannucci PM. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost.2011;9(4):700-10 [DOI] [PubMed] [Google Scholar]
- 11.Fischer K, van den Berg HM. Prophylaxis. In:Lee CA, Berntorp EE, Hoots WK.Textbook of Hemophilia. 2nd ed Oxford: Wiley-Blackwell;2010:38-43 [Google Scholar]
- 12.Mariani G, Konkle BA, Ingerslev J. Congenital factor VII deficiency: therapy with recombinant activated factor VII – a critical appraisal. Haemophilia.2006;12(1): 19-27 [DOI] [PubMed] [Google Scholar]
- 13.Chuansumrit A, Visanuyothin N, Puapunwattana S, Chaivisuth A, Rasmidat P, Charoenkwan P, Chiemchanya S. Outcome of intracranial hemorrhage in infants with congenital factor VII deficiency. J Med Assoc Thai.2002;85 (Suppl 4): S1059-64 [PubMed] [Google Scholar]
- 14.Cohen LJ, McWilliams NB, Neuberg R, Zinkham W, Bauer K, Gribble TJ, et al. Prophylaxis and therapy with factor VII concentrate (human) immuno, vapor heated in patients with congenital factor VII deficiency: a summary of case reports. Am J Hematol.1995;50(4):269-76 [DOI] [PubMed] [Google Scholar]
- 15.Farah RA, Hamod D, Melick N, Giansily-Blaizot M, Sallah S. Successful prophylaxis against intracranial hemorrhage using weekly administration of activated recombinant factor VII in a newborn with severe factor VII deficiency. J Thromb Haemost. 2007;5(2):433-4 [DOI] [PubMed] [Google Scholar]
- 16.Halsey C, Singer JD, Manning R, Laffan M, Layton M. The use of prophylactic recombinant factor VII to prevent intracranial haemorrhage in an infant with severe factor VII deficiency. Br J Haematol. 2002;117 (Suppl 1):11-2 [Google Scholar]
- 17.Holve S, Stuart R, Recht M. Prophylaxis of recurrent epistaxis and menorrhagia using recombinant factor VIIa in severe congenital factor VII deficiency. Pediatr Res. 2001;49(4):203A11158514 [Google Scholar]
- 18.Huth-Kühne A, Loges P, Zimmermann R. Regular prophylaxis with recombinant factor VIIa in a patient with severe congenital FVII deficiency. Haemostaseologie.2008;28(Suppl 1):S55 [Google Scholar]
- 19.Jahromi BJ, Karimi M. Long-term follow-up of prophylaxis with recombinant activated factor VII in patients with congenital factor VII deficiency. Haemophilia. 2011;17(4): 713-5 [DOI] [PubMed] [Google Scholar]
- 20.Karimi M, Shafieian R. Prophylactic effect of recombinant factor VIIa with congenital factor VII deficiency. Haemophilia.2008;14 (4):851-2 [DOI] [PubMed] [Google Scholar]
- 21.Karayalcin G, Hart D. Prophylactic effect of recombinant factor VIIa with congenital factor VII deficiency. Pediatr Res.2002;51(4):259A-60A [Google Scholar]
- 22.Kuperman AA, Livnat T, Braester A, Amar N, Kenet G. Rationale for tailored prophylaxis with recombinant FVIIa in severe congenital factor VII deficiency. J Thromb Haemost 2011;9(Suppl 2):695 Abstract 55513. [Google Scholar]
- 23.Recht M, Holve S, Stuart R, Hu D, Moul S. Prophylaxis of bleeding using recombinant factor VIIa in an infant with severe congenital factor VII deficiency. Pediatr Res. 2002;51(4):260A [Google Scholar]
- 24.Salcioglu Z, Akcay A, Sen HS, Aydogan G, Akici F, Tugcu D, et al. Factor VII deficiency: a single-center experience. Clin Appl Thromb Haemost. 2012; 18(6):588-93 [DOI] [PubMed] [Google Scholar]
- 25.Tokgoz H, Caliskan U, Lavigne-Lissalde G, Giansily-Blaizot M. Successful prophylactic use of recombinant activated factor VII (rFVIIa) in a patient with congenital FVII deficiency and inhibitors to FVII. Haemophilia. 2012;18(1):e25-e27 [DOI] [PubMed] [Google Scholar]
- 26.Kankirawatana S, Mahasandana C, Veerakul G, Seeloem J, Suwantol L, Tanphaichitr V, Suvatte V. Successful prophylaxis of intracranial hemorrhage in infants with severe congenital factor VII deficiency. Southeast Asian J Trop Med Public Health. 2000;31(4):795-800 [PubMed] [Google Scholar]
- 27.Mathijssen NC, Masereeuw R, Verbeek K, Lavergne JM, Costa JM, van Heerde WL, Nováková IR. Prophylactic effect of recombinant factor VIIa in factor VII deficient patients. Br J Haematol. 2004;125(4):494-9 [DOI] [PubMed] [Google Scholar]
- 28.Michaels LA, Philipp CS, Eisele J, Pappas H, Saidi P. Prophylactic treatment of a small child with severe factor VII deficiency using repeat dosing from a single vial of recombinant activated factor VII. Pediatr Blood Cancer. 2007;49(5):736-9 [DOI] [PubMed] [Google Scholar]
- 29.Tcheng WY, Donkin J, Konzal S, Wong WY. Recombinant factor VIIa prophylaxis in a patient with severe congenital factor VII deficiency. Haemophilia.2004;10(3):295-8 [DOI] [PubMed] [Google Scholar]
- 30.Ingerslev J, Christiansen K, Sorensen B. Inhibitor to factor VII in severe factor VII deficiency: detection and course of the inhibitory response. J Thromb Haemost. 2005;3(4):799-800 [DOI] [PubMed] [Google Scholar]
- 31.Hoffman M, Colina CM, McDonald AG, Arepally GM, Pedersen L, Monroe DM. Tissue factor around dermal vessels has bound factor VII in the absence of injury. J Thromb Haemost. 2007;5(7):1403-8 [DOI] [PubMed] [Google Scholar]
- 32.Nayak RC, Sen P, Ghosh S, Gopalakrishnan R, Esmon CT, Pendurthi UR, Rao LV. Endothelial cell protein C receptor cellular localization and trafficking: potential functional implications. Blood.2009;114(9): 1974-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sen P, Gopalakrishnan R, Kothari H, Keshava S, Clark CA, Esmon CT, et al. Factor VIIa bound to endothelial cell protein C receptor activates protease activated receptor I and mediates cell signaling and barrier protection. Blood. 2011;117(11):3199-208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ragni MV, Lewis JH, Spero JA, Hasiba U. Factor VII deficiency. Am J Hematol. 1981;10 (1):79-88 [DOI] [PubMed] [Google Scholar]
- 35.Hennewig U, Eisert S, Wulff K, Herrmann FH, Schneider DT, Gobel U. Long-term FVII substitution in a preterm infant with severe gastrointestinal bleeding and FVII deficiency due to a homozygous donor splice mutation IVS4+1G-->A. Pediatr Hematol Oncol. 2006;23(2):129-33 [DOI] [PubMed] [Google Scholar]
- 36.Giansily-Blaizot M, Aguilar-Martinez P, Briquel ME, d'Oiron R, De Maistre E, Epelbaum S, Schved J F.Two novel cases of cerebral haemorrhages at the neonatal period associated with inherited factor VII deficiency, one of them revealing a new nonsense mutation (Ser52Stop). Blood Coagul Fibrinolysis. 2003;14(2):217-20 [DOI] [PubMed] [Google Scholar]
- 37.Mariani G, Napolitano M, Dolce A, Pérez Garrido R, Batorova A, Karimi M, et al. Replacement therapy for bleeding episodes in factor VII deficiency: a prospective evaluation. Thromb Haemost. 2013;109(2):238-47 [DOI] [PubMed] [Google Scholar]
- 38.Pruthi RK, Rodriguez V, Allen C, Slaby JA, Schmidt KA, Plumhoff EA. Molecular analysis in a patient with severe factor VII deficiency and an inhibitor: report of a novel mutation (S103G). Eur J Haematol. 2007;79(4):354-9 [DOI] [PubMed] [Google Scholar]
- 39.Nicolaisen EM. Long-term follow-up with regard to potential immunogenicity: clinical experience with NovoSeven (recombinant factor VIIa). Haemostasis. 1996;26 (Suppl 1):98-101 [DOI] [PubMed] [Google Scholar]
- 40.Mariani G, Herrmann FH, Schulman S, Batorova A, Wulff K, Etro D, et al. Thrombosis in inherited factor VII deficiency. J Thromb Haemost. 2003;1 (10):2153-8 [DOI] [PubMed] [Google Scholar]
- 41.Mariani G, Dolce A, Batorova A, Auerswald G, Schved JF, Siragusa S, et al. Recombinant, activated factor VII for surgery in factor VII deficiency: a prospective evaluation - the surgical STER. Br J Haematol. 2011;152(3):340-6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.