

Abstract
Objective
Parkinsonism has been occasionally reported as a consequence of infectious diseases. The present study examines the clinical and pathological correlates of parkinsonism across birth cohorts in relation to critical exposure to the encephalitis lethargica epidemic in the early 1900’s.
Methods
The study population consisted of 678 participants in the Nun Study, of whom 432 died and came to autopsy. Qualitative indices of SN depigmentation were verified in a subset of 40 randomly selected subjects using quantitative stereological techniques. Substantia nigra (SN) depigmentation, detected neuropathologically, was correlated with clinical parameters of PD, age, and birth cohort.
Results
SN depigmentation was detected in 57 (13.2%) of the cohort. While qualitative SN depigmentation correlated modestly with age (p=0.02), it correlated best with birth cohort (p=0.009) for women born in the years 1895–1899. Quantitative measures of SN depigmentation were increased in this birth cohort compared to age matched subjects from flanking birth cohorts 1890–1894 and 1900–1904 (p≤0.001). SN depigmentation correlated with speed of 6 and 50 foot walk (p<0.0001), “up and go” (p<0.0001), and hand coordination (p<0.0001).
Interpretation
Subjects in the birth cohort 1895–1899 would have been in their late teens and 20s at the onset and during the peak of the encephalitis lethargica epidemic. These were precisely the age ranges of persons that were most often affected by the illness. These data suggest the possibility that the coexistence of parkinsonism and SN depigmentation in this birth cohort may have resulted from the yet undetermined infectious agent responsible for encephalitis lethargica.
Keywords: Parkinson’s disease, substantia nigra, encephalitis lethargica
INTRODUCTION
While a number of genes have been identified as responsible for autosomal dominant and recessive inheritance of Parkinson’s disease (PD),1 the prevailing belief is that these are infrequently responsible for idiopathic PD and that the illness is consequence of an interaction of non-genetic factors with susceptibility genes.2 Among the contributing factors proposed for PD have been aging, genetic factors, endogenous and exogenous toxins, autoimmune disorders, and infectious diseases.3 A number of infectious diseases have been postulated as etiologies of PD. The proposed mechanism for the development of infection-related PD includes a loss of dopaminergic neurons from active infection or associated inflammation, a “hit and run” damage to the substantia nigra, and the development of autoimmunity with cross reactivity to the basal ganglia.
Encephalitis lethargica or Von Economo’s encephalitis was first recognized by von Economo in the wards of the Vienna Psychiatric Clinic in late 19164 although its initial appearance may have been in April and May 1915 in Rumania.5 By 1919, it had spread worldwide.6 The disease manifested in a number of fashions, but typically included the triad of fever, somnolence, and oculomotor paresis. For unexplained reasons, the disease had a predilection for young adults. Nearly 50% of infected patients were between the ages of 10 and 30 years old.7 The etiology of the disorder has never been identified. In most circumstances, the disease predated the influenza pandemic 8 and modern studies have failed to amplify influenza DNA from the brain tissues of affected individuals.9, 10
Parkinsonism was a frequent sequelae of this disorder. Parkinsonism could develop acutely or after more than a decade. A hiatus of 45 years from the time of encephalitis lethargica to parkinsonism has been reported.8 In contrast to idiopathic PD, these patients often demonstrated oculogyric crises, catatonia, cataplexy, paradoxical movements, and unique speech and respiratory problems.6 Examination might reveal pupillary changes, torsion spasms, and corticospinal tract abnormalities.6 In 1961, Poskanzer and Schwab proposed that nearly all cases of idiopathic PD were the consequence of clinical or subclinical encephalitis lethargica.11 Subsequent population-based studies, failed to confirm that hypothesis.12 To further investigate the possibility of a primary or contributory role of encephalitis lethargica in the causation of idiopathic PD, we examined the clinical and neuropathological features of 432 women participating in the Nun’s Study in birth cohorts spanning the influenza outbreak in the early 20th century that were at risk for critical disease exposure.
Methods
Subjects
Nun study participants were Catholic sisters from the Notre Dame congregation located in the United States. All participants were born before 1917 and a total of 678 sisters were enrolled. A total of 432 women were autopsied at the time of data analysis for the present study. No subjects were excluded for any reason. Informed consent was obtained on all participants and the Nun Study was approved by the University of Kentucky’s institutional review board.
Clinical analysis
Subjects were evaluated annually using the Mini-Mental State Examination, “Timed up and go” test (TUGO)13 in which participants were asked to stand from a seated position and walk for 6 feet around a stop sign and return to a sitting position in the chair, timed in seconds, “6 foot walking test” and “50 foot walking test” also measured in seconds. In these tests, the subjects were instructed to walk as fast as they could without running and time to complete the indicated test was measured in seconds. Hand coordination was timed at a maximum allowed time of 45 seconds. Participants were asked to correctly turn a doorknob and open a cabinet door that was placed in front of them. All participants were genotyped for APOE gene as previously described.14
Neuropathological evaluation
We examined a total of 432 post mortem brains. The methodology has been described in detailed elsewhere.15 One neuropathologist performed the gross and microscopic exam of the midbrain, after fixation in 10% buffered formaldehyde. Tissues blocks were cut at 10 um after being processed and embedded in paraffin. Tissues were further stained with hematoxylin-eosin (H-E) and the Hirano-silver method.16 Silver stains were used to determine CERAD plaque scores17 and Braak stage of neurofibrillary degeneration18. Qualitative assessment of substantia nigra depigmentation (pigmentary incontinence, reflecting extracellular deposits of normally intracellular pigmentation, that serve as as the tombstone” for neuronal loss in pigmented nuclei such as the substantia nigra) was recorded for each midbrain section. Midbrain sections were also stained with α-synuclein antibody (Novacaster, Newcastle, United Kingdom) to identify Lewy body pathology.
In order to validate the qualitative assessment of pigmentary incontinence (extracelluar pigment reflecting nigral neuronal loss; in this manuscript we use the terms “pigmentary incontinence”, “depigmentation”, and “nigral neuronal loss” synonomously), we randomly selected 40 samples from the total 432 subjects included in the present study, including the birth cohort of interest (1895–1899, n=6) and flanking birth cohorts (1890–1894, n=17 and 1900–1904, n=17), for quantitative stereological analysis. Tissue sections from the midbrain (including the substantia nigra) were evaluated using an Olympus BX51 microscope, digital DP70 camera, automated Proscan II microscope stage, and the CAST stereology software system (Olympus, Danmark A/S). Each section was visualized at 40x magnification and regions of interest were generated by digitally mapping areas of inclusion and exclusion in the tissue field that included all pigmented neurons and areas of incontinence (extracellular pigment) in the tissue section. Automated uniform random sampling of each tissue section was performed to allow unbiased estimates of incontinent and intact pigmented neurons. The ratio of incontinent areas to intact pigmented neurons was compared across birth cohorts to validate the semi-quantitative assessment used to examine the entire study sample.
Statistical analysis
Subjects were grouped by birth cohort in 5 year intervals for all analyses. Standard statistical analyses including ANOVA, Chi-square, and Kruskal-Wallis tests were performed for comparison of demographic, genetic and clinical variables across birth cohorts where appropriate. Kaplan-Meier analysis was used to compare mortality between subjects with pigmentary incontinence and those with an intact substantia nigra. Age adjusted linear mixed modeling was used for analysis of qualitative pigmentary incontinence and for motor performance measures across birth cohorts.
RESULTS
Demographic, genetic, clinical and pathological data are presented in Table 1. Birth cohorts differed in regards to age, MMSE and Braak staging, but were not different in terms of ApoE allele status, CERAD amyloid plaques scores or Lewy body pathology.
Table 1.
Description of the study sample by birth cohort including demographic, genetic, clinical and pathological variables.
| Birth Cohort | 1890–1894 (n=7) | 1895–1899 (n=36) | 1900–1904 (n=86) | 1905–1909 (n=137) | 1910–1914 (n=128) | 1915–1919 (n=36) | p-value |
|---|---|---|---|---|---|---|---|
| Age (mean years ± sd) | 102.3 ± 3.3 | 98.3 ± 2.8 | 93.7 ± 2.9 | 90.0 ± 3.0 | 85.8 ± 3.2 | 82.2 ± 2.9 | <0.0001† |
| ApoE4 (%) | 28.6 | 16.7 | 28.0 | 23.0 | 22.3 | 14.3 | n.s.‡ |
| MMSE (mean ± sd) | 16.3 ± 11.4 | 13.6 ± 10.4 | 14.5 ± 10.0 | 17.5 ± 10.9 | 19.3 ± 10.5 | 21.5 ± 9.8 | <0.002† |
| Date from last evaluation to death (mean years ± sd) | 0.80 ± 0.33 | 0.84 ± 0.47 | 0.76 ± 0.45 | 0.80 ± 0.55 | 0.89 ± 0.56 | 0.80 ± 0.40 | n.s.† |
| CERAD (mean ± sd) | 1.7 ± 1.5 | 1.6 ± 1.1 | 2.0 ± 1.0 | 1.8 ± 1.2 | 1.6 ± 1.2 | 1.2 ± 1.2 | n.s.¥ |
| Braak (mean years ± sd) | 3.6 ± 1.3 | 2.8 ± 1.8 | 3.8 ± 1.8 | 3.4 ± 1.8 | 2.9 ± 1.8 | 2.5 ± 1.9 | <0.001¥ |
| LBD pathology (%) | 0.0 | 0.0 | 18.0 | 28.4 | 19.8 | 15.4 | n.s.‡ |
ANOVA;
Chi-square;
Kruskal-Wallis
Fifty seven (13.2%) subjects had evidence for qualitative SN depigmentation. Age-adjusted odds ratio for SN depigmentation was highest (OR 21.2) for the birth cohort 1895–1899 (Figure1). Kaplan-Meier survival plot showed no difference in survival times between our substantia nigra depigmented group and normals (Figure 2).
Figure 1.

Age-adjusted odds-ratio for pigmentary incontinence by birth cohort demonstrating a a maximal risk for the birth cohort 1895–1899 that would have been at a critical age during the influenza epidemic for possible development of Von Economo’s encephalitis. The birth cohort 1915–1919 was used as reference.
Figure 2.
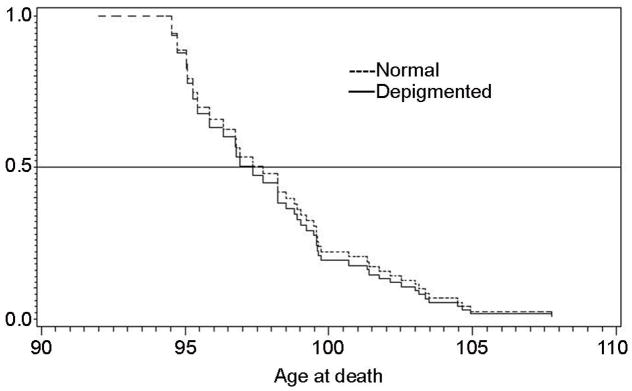
Kaplan-Meier survival curve for subjects with and without pigmentary incontinence demonstrates no difference in survival rates based on this variable.
Qualitative analysis of pigmentary incontinence was validated through quantitative stereological analysis of a random subset of cases from birth cohorts 1895–1899, 1890–1894, and 1900 to 1904 (Figure 3). This quantitative approach increased statistical validity of the qualitative estimate of SN depigmentation in the birth cohort of interest (p= 0.009 to p=0.001).
Figure 3.

Quantitative ratios of incontinent to intact pigmented neurons in the substantia nigra demonstrates a significant increase in nigral degeneration for the birth cohort 1895–1899 compared to flanking birth cohorts 1890–1894 and 1900–1904.
On motor measures, the 1895–1899 birth cohort had increased median times on the TUGO (p<0.0001), 50 and 6 foot walking tasks (p<0.0001), and hand coordination tasks (p<0.0001) (Figure 4).
Figure 4.

Motor tasks of pigmentary incontinent group compared to normals.
DISCUSSION
The etiology of idiopathic PD remains poorly understood. It is quite possible that the disorder arises as a consequence of the interplay of a number of factors, including inflammation and infection.19 The data presented demonstrate an increased risk of nigral degeneration and clinical parkinsonian signs in the birth cohort potentially exposed to the influenza virus at a critical age for development of Von Economo’s encephalitis, although none had documented clinical infection at this time. Recently, Rohn and colleagues have discovered the influenza A virus on macrophages in addition to the presence of T cells within the substantia nigra pars compacta (SNpc) from postmortem PD brain sections20, further supporting the possible link between idiopathic PD and viral inflammatory causes.
In animal studies, the potential importance of inflammation is illustrated in a mouse model. The intranigral injection of lipopolysaccharides (LPS) results in an inflammatory reaction followed by dopaminergic neurodegeneration.21, 22 This model is potentially useful for the study of PD. LPS appears to induce these changes through caspase-11.21 This same mechanism accompanies certain environmental toxins that produce Parkinsonism, such as, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) which results in selective dopaminergic neurodegeneration.23 Furthermore, the exposure of prenatal rats to LPS results in a subsequent progressive loss of dopaminergic neurons during aging.24
Parkinsonism was a well recognized consequence of encephalitis lethargica, typically developing 1 to 5 years after the acute illness.6 This illness differed from idiopathic PD in several respects as patients often displayed paradoxical movements, oculogyric crises, catatonia, and unusual speech and respiratory problems.6 Dale and colleagues have recently reported 20 cases of encephalitis lethargica and maintain that the illness persists in the modern population.25 Occasional cases of selective involvement of the substantia nigra and parkinsonism following an acute encephalitic illness from identified viruses are recognized as well. Among these viruses are coxsackie B426, Epstein-Barr virus27–29, and Japanese B encephalitis.30 HIV may also cause a clinical picture that masquerades as PD31–33 and the basal ganglia appears to be a selective target for HIV with greater rates of viral replication, associated with loss of dopaminergic neurons, decrease CSF dopamine, central atrophy exceeding cortical atrophy.34 Parkinsonism may also follow other infectious illnesses including gram negative sepsis,35 and cryptococcal meningoencephalitis.36
While the confound of idiopathic Parkinson”s disease should be considered as an alternative explanation for the nigral neuronal loss and clinical signs of parkinsonism seen in the cohort, α-synuclein pathology was conspicuously absent in the older birth cohorts demonstrating the association between critical exposure to Von Economo’s encephalitis, clinical parkinsonism in late life, and substantia nigra depigmentation at autopsy. This data likely reflects a survival bias in those predisposed to the development of α-synuclein pathology as has been demonstrated previously.37 Additionally, this data suggests that the environmental agent (infectious or toxic) responsible for Von Economo’s encephalitis and the development of late-life parkinsonism and nigral neuronal loss in this cohort, exerts its effects through α-synuclein-independent mechanisms. Further work defining such causative agents and mechanisms for selective nigral degeneration are warranted.
The findings from this study are consistent with the hypothesis that suggests those women who were in their late teens and 20s, the precise ages that were most at risk for development of clinical illness during the height of the pandemic of encephalitis lethargica, had a greater risk of developing substantia nigra degeneration and exhibiting features of PD. The clinical correlates of nigral degeneration are evident in our analysis of timed motor measures across birth cohorts. While the study does not prove causality, the association seen between birth cohort and the development of clinical and pathological features of parkinsonism provides a tantalizing clue to the pathogenesis of α-synuclein-independent, non-Lewy body, idiopathic PD.
Acknowledgments
This study was funded by grants from the U.S. National Institute on Aging: R01AG09862, P30 AG028383, and grants from the Abercrombie Foundation, the Kleberg Foundation, and the Sanders-Brown Center on Aging. This research would not have been possible without the generous support of the School Sisters of Notre Dame congregation.
References
- 1.Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson’s disease and parkinsonism. Ann Neurol. 2006 Oct;60(4):389–98. doi: 10.1002/ana.21022. [DOI] [PubMed] [Google Scholar]
- 2.de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006 Jun;5(6):525–35. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 3.Stoessl AJ. Etiology of Parkinson’s disease. Can J Neurol Sci. 1999 Aug;26( Suppl 2):S5–12. doi: 10.1017/s0317167100000032. [DOI] [PubMed] [Google Scholar]
- 4.von Economo C. Encephalitis lethargica: Its sequelae and treatment. London: Oxford University Press; 1931. [Google Scholar]
- 5.Urechia CL. Dix cas d’encephalite epidemique avec autopsie. Arch Inter Neurol. 1921;2:65–78. [Google Scholar]
- 6.Berger JR, Glitza IC. Von Economo’s Encephalitis. In: Nath A, Berger JR, editors. Clinical Neurovirology. New York: Marcel Dekker, Inc; 2003. pp. 523–42. [Google Scholar]
- 7.Wilson SAK. Epidemic encephalitis. Neurology. 1940;1:99–144. [Google Scholar]
- 8.Berger JR, Glitza I. Von Economo’s encephalitis. In: Nath A, Berger JR, editors. Clinical Neurovirology. New York, NY: Marcel Dekker; 2003. pp. 523–42. [Google Scholar]
- 9.McCall S, Henry JM, Reid AH, Taubenberger JK. Influenza RNA not detected in archival brain tissues from acute encephalitis lethargica cases or in postencephalitic Parkinson cases. J Neuropathol Exp Neurol. 2001 Jul;60(7):696–704. doi: 10.1093/jnen/60.7.696. [DOI] [PubMed] [Google Scholar]
- 10.Jellinger KA. Influenza RNA not detected in archival brain tissues from acute encephalitis lethargica cases or in postencephalitic Parkinson cases. J Neuropathol Exp Neurol. 2001 Nov;60(11):1121–2. doi: 10.1093/jnen/60.11.1121. [DOI] [PubMed] [Google Scholar]
- 11.Poskanzer DC, Schwab RS. Studies in the epidemiology of Parkinson’s disease predicting its disappearance as a major clinical entity by 1980. Trans Am Neurol Assoc. 1961;86:234–5. [PubMed] [Google Scholar]
- 12.Lipczynska-Lojkowska W. Etiology of parkinsonism. A verification of Poskanzer’s and Schwab’s hypothesis. Neurol Neurochir Pol. 1979 Nov-Dec;13(6):593–9. [PubMed] [Google Scholar]
- 13.Morris S, Morris ME, Iansek R. Reliability of measurements obtained with the Timed “Up & Go” test in people with Parkinson disease. Phys Ther. 2001 Feb;81(2):810–8. doi: 10.1093/ptj/81.2.810. [DOI] [PubMed] [Google Scholar]
- 14.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990 Mar;31(3):545–8. [PubMed] [Google Scholar]
- 15.Iacono D, Markesbery WR, Gross M, et al. The Nun study: clinically silent AD, neuronal hypertrophy, and linguistic skills in early life. Neurology. 2009 Sep 1;73(9):665–73. doi: 10.1212/WNL.0b013e3181b01077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamoto T, Hirano A. A comparative study of modified Bielschowsky, Bodian and thioflavin S stains on Alzheimer’s neurofibrillary tangles. Neuropathol Appl Neurobiol. 1986 Jan-Feb;12(1):3–9. doi: 10.1111/j.1365-2990.1986.tb00677.x. [DOI] [PubMed] [Google Scholar]
- 17.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991 Apr;41(4):479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 18.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82(4):239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 19.Jang H, Boltz DA, Webster RG, Smeyne RJ. Viral parkinsonism. Biochim Biophys Acta. 2009 Jul;1792(7):714–21. doi: 10.1016/j.bbadis.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohn TT, Catlin LW. Immunolocalization of influenza A virus and markers of inflammation in the human Parkinson’s disease brain. PLoS One. 2011;6(5):e20495. doi: 10.1371/journal.pone.0020495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arai H, Furuya T, Mizuno Y, Mochizuki H. Inflammation and infection in Parkinson’s disease. Histol Histopathol. 2006 Jun;21(6):673–8. doi: 10.14670/HH-21.673. [DOI] [PubMed] [Google Scholar]
- 22.Liu B, Gao HM, Hong JS. Parkinson’s disease and exposure to infectious agents and pesticides and the occurrence of brain injuries: role of neuroinflammation. Environ Health Perspect. 2003 Jun;111(8):1065–73. doi: 10.1289/ehp.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furuya T, Hayakawa H, Yamada M, et al. Caspase-11 mediates inflammatory dopaminergic cell death in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. J Neurosci. 2004 Feb 25;24(8):1865–72. doi: 10.1523/JNEUROSCI.3309-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carvey PM, Chang Q, Lipton JW, Ling Z. Prenatal exposure to the bacteriotoxin lipopolysaccharide leads to long-term losses of dopamine neurons in offspring: a potential, new model of Parkinson’s disease. Front Biosci. 2003 Sep 1;8:s826–37. doi: 10.2741/1158. [DOI] [PubMed] [Google Scholar]
- 25.Dale RC, Church AJ, Surtees RA, et al. Encephalitis lethargica syndrome: 20 new cases and evidence of basal ganglia autoimmunity. Brain. 2004 Jan;127(Pt 1):21–33. doi: 10.1093/brain/awh008. [DOI] [PubMed] [Google Scholar]
- 26.Cree BC, Bernardini GL, Hays AP, Lowe G. A fatal case of coxsackievirus B4 meningoencephalitis. Arch Neurol. 2003 Jan;60(1):107–12. doi: 10.1001/archneur.60.1.107. [DOI] [PubMed] [Google Scholar]
- 27.Roselli F, Russo I, Fraddosio A, et al. Reversible Parkinsonian syndrome associated with anti-neuronal antibodies in acute EBV encephalitis: a case report. Parkinsonism Relat Disord. 2006 May;12(4):257–60. doi: 10.1016/j.parkreldis.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Dimova PS, Bojinova V, Georgiev D, Milanov I. Acute reversible parkinsonism in Epstein-Bbarr virus-related encephalitis lethargica-like illness. Mov Disord. 2006 Apr;21(4):564–6. doi: 10.1002/mds.20742. [DOI] [PubMed] [Google Scholar]
- 29.Hsieh JC, Lue KH, Lee YL. Parkinson-like syndrome as the major presenting symptom of Epstein-Barr virus encephalitis. Arch Dis Child. 2002 Oct;87(4):358. doi: 10.1136/adc.87.4.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murgod UA, Muthane UB, Ravi V, Radhesh S, Desai A. Persistent movement disorders following Japanese encephalitis. Neurology. 2001 Dec 26;57(12):2313–5. doi: 10.1212/wnl.57.12.2313. [DOI] [PubMed] [Google Scholar]
- 31.Hersh BP, Rajendran PR, Battinelli D. Parkinsonism as the presenting manifestation of HIV infection: improvement on HAART. Neurology. 2001 Jan 23;56(2):278–9. doi: 10.1212/wnl.56.2.278. [DOI] [PubMed] [Google Scholar]
- 32.Mattos JP, Rosso AL, Correa RB, Novis SA. Movement disorders in 28 HIV-infected patients. Arq Neuropsiquiatr. 2002 Sep;60(3-A):525–30. [PubMed] [Google Scholar]
- 33.Nath A, Jankovic J, Pettigrew LC. Movement disorders and AIDS. Neurology. 1987 Jan;37(1):37–41. doi: 10.1212/wnl.37.1.37. [DOI] [PubMed] [Google Scholar]
- 34.Berger JR, Arendt G. HIV dementia: the role of the basal ganglia and dopaminergic systems. J Psychopharmacol. 2000;14(3):214–21. doi: 10.1177/026988110001400304. [DOI] [PubMed] [Google Scholar]
- 35.Alasia DD, Asekomeh GA, Unachuku CN. Parkinsonism induced by sepsis: a case report. Niger J Med. 2006 Jul-Sep;15(3):333–6. doi: 10.4314/njm.v15i3.37243. [DOI] [PubMed] [Google Scholar]
- 36.Wszolek Z, Monsour H, Smith P, Pfeiffer R. Cryptococcal meningoencephalitis with parkinsonian features. Mov Disord. 1988;3(3):271–3. doi: 10.1002/mds.870030312. [DOI] [PubMed] [Google Scholar]
- 37.Jicha GA, Parisi JE, Dickson DW, et al. Age and apoE associations with complex pathologic features in Alzheimer’s disease. J Neurol Sci. 2008 Oct 15;273(1–2):34–9. doi: 10.1016/j.jns.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]