

Abstract
Membrane proteins (MPs) mediate a variety of cellular responses to extracellular signals. While MPs are intensely studied for their values as disease biomarkers and therapeutic targets, in situ investigation of binding kinetics of MPs with their ligands has been a challenge. Traditional approaches isolate MPs and then study them ex situ, which does not accurately reflect their native structures and functions. We present here a label-free plasmonic microscopy method to map the local binding kinetics of MPs in their native environment. This new analytical method can perform simultaneous plasmonic and fluorescence imaging, thus making it possible to combine the strengths of both label-based and label-free techniques in one system. Using this method, we have determined the distribution of MPs on the surface of single cells, and the local binding kinetic constants of different MPs. Furthermore, we have studied the polarization of the MPs on the cell surface during chemotaxis.
Membrane proteins (MPs) are a large group of proteins attached to or embedded in the lipid bi-layer of the cell surface and are responsible for many basic cellular processes, such as cell signaling and communications, as well as transportation of substances in and out of cells.1,2 MPs are also the most popular drug targets, accounting for more than half of all the present drug targets, including both large antibody-based and small molecule drugs.3 In addition, increasing number of MPs has been identified as biomarkers for various diseases, such as the breast cancer, lymphoma and colorectal cancer.4 The basic cellular and therapeutic processes usually start with the binding of ligands or drugs to MPs. Studying the binding activities of MPs is thus critical to the understanding of their biological functions, and also to the discovery of new drugs.4,5
Despite its importance, measuring the binding kinetics of MPs has been a great challenge. Fluorescence-based technology has been used to study MPs, however, like other label-based technologies, it is mainly an end point assay by measuring changes in fluorescence signals before and after binding, which is not suitable for obtaining the binding kinetics. Label-free techniques, such as surface plasmon resonance (SPR)6, can measure protein binding kinetics by immobilizing purified proteins onto a SPR chip. This approach involves multiple experimental steps, including extraction and purification of proteins and immobilization of the proteins onto the SPR chip, which is not only laborious but also changes the native environment of the proteins. The latter is particularly problematic for integral MPs that require amphiphilic lipid environment to keep their native conformations intact.7
Various methods have been developed to minimize disruptions in the native conformations of MPs. Examples include coating a sensor chip with membranes to mimic the native environment8 and depositing MPs embedded in liposome or native membrane patches onto the chip.9,10 These methods have contributed to the understanding of MPs, however, a capability that can directly study the binding kinetics of MPs in living cells is highly desired because it will not only validate the ex situ methods but also reveal novel features.
One of the important features of MPs is that they often distribute heterogeneously across the cell membrane and change dynamically in order to regulate different cellular behaviors.11,12 Understanding MPs thus requires an analytical tool with spatial and temporal resolutions and monitor the MPs activities in situ. Atomic force microscopy (AFM) can provide high spatial resolution for studying MPs,13 but it is not suitable for studying binding kinetics.14 Fluorescence-based microscopy (FLM) can image MPs interactions in individual cells,15–19 but it has also several drawbacks. First, it requires careful development of proper labels and strategies for different target proteins. Second, it is, in general, an end point assay, not for following fast kinetics.17 Finally, labeling may affect the activities of the host protein.17
Here we present a label-free imaging method that can measure the binding kinetics of MPs in single living cells without extracting the proteins from the cell membranes, thus ensuring intact native conformations of the MPs. On the basis of three applications, we further show that the distribution and local binding activities of MPs in the membrane of each cell can be mapped and quantified, and demonstrate that such spatial resolution can resolve polarized redistribution of MPs during chemotaxis. The method is based on SPR microscopy (SPRM),20,21 but it also allows for simultaneously optical and fluorescence imaging of the same sample, making it possible to combine the strengths of both label-based and label-free detection technologies in a single setup.
Adherent cells are cultured on a gold film, which is placed in an inverted microscope for simultaneous bright field, fluorescence and SPRM imaging (Fig. 1a).20 Fig. 1b shows recorded bright field, fluorescence and SPRM images of a cell. When a ligand solution flows over the cell surface (see Supplementary Information Section 1 for details of the flow system), the binding of the ligand to its respective MP receptors on the cell surface occurs, which is monitored by SPRM with millisecond temporal resolution, and sub-micron to micron spatial resolution (limited by optical diffraction and SPR propagation length). To demonstrate the capability of the SPRM method for studying local binding kinetics of MPs in single cells, we have studied the specific interactions between glycoproteins (membrane protein) and lectin (ligand), and the binding activity and spatial distribution of nicotinic acetylcholine receptors on the membrane of a single cell. We have also applied the method to study glycoprotein polarization during chemotaxis. We present the findings below.
Fig. 1. Surface plasmon resonance microscopy (SPRM).
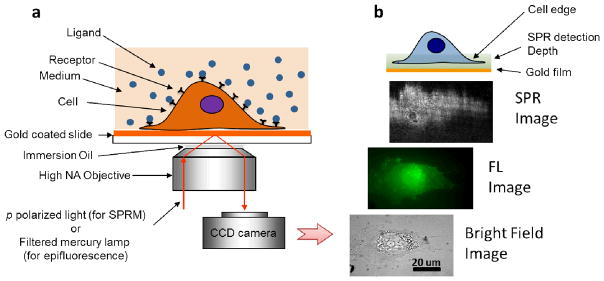
(a) Schematic illustration of the experimental setup. A p-polarized laser beam is directed onto a gold-coated glass coverslip via an oil immersion objective to create surface plasmon resonance (SPR) on the gold surface, which is imaged with a CCD camera. A mercury lamp and appropriate optical filters are used to perform epifluorescence measurements. Conventional bright field images of the same sample can also be taken via illumination from the top of the sample. Cells to be studied are cultured on the coverslip. (b) From bottom to top, example of bright field, fluorescence and SPR images of a cell stained by Alexa Fluor 488 labeled wheat germ agglutinin (WGA). The top cartoon illustrates that the entire cell bottom membrane and partial cell top membrane in the cell edge regions are located within the typical detection depth of the present SPRM.
Results and discussions
Lectin-glycoprotein interaction
Glycoproteins are proteins that have sugar chains attached to them and most MPs are glycosylated.22 The extracellularly extended sugar groups of glycoproteins are abundant and flexible, which play a central role in the cell recognition and communication via the specific interactions with neighboring cells, pathogens, proteins, hormones and other signaling molecules.23 An important example of the specific interactions is between lectins (proteins that bind to and recognize specific sugar structures) and glycoproteins on the cell membranes. Investigation of the binding activities between lectins and glycoproteins provide information about the nature, distribution and biological role of sugar groups on the cell surface, especially those that may serve as disease biomarkers and therapeutic targets.24
We used wheat germ agglutinin (WGA), a lectin that can specifically recognize N-acetylglucosamine (GlcNAc) and to a small extent N-acetylneuraminic acid (sialic acid) sugar residues of glycan chains, to study the cell surface glycosylation of SH-EP1 human epithelial cells. Exposure of WGA to the cells results in a local increase in SPR intensity indicating the presence of these sugar residues on the glycan chains of the MPs where the lectin are bound as shown in Fig. 2d.
Fig. 2. Glycoprotein-lectin interaction.

The bright field (a) and SPRM (b) images of a SH-EP1 cell. (c) The epifluorescence image of the same cell stained with Alexa Fluor 555 labeled WGA with focusing on the bottom cell membrane portion. White arrows indicate the borderline between the thick cell body (in the center) and the thin cell membrane (in the edge). (d) SPR sensorograms of the entire cell region (black curve), cell edge region (red curve) and cell central region (blue curve) during the binding and dissociation of 80 μg/mL WGA. (e) SPR sensorograms of the cell edge region (black curves) and the global fitting (red curves) with WGA solutions of different concentrations (from bottom to top: 5, 10, 40, 80, 200 μg/mL). (f) The equilibrium-based dissociation constant (KD) was determined to be 0.32 μM by plotting the concentration dependent equilibrium responses.
Figs. 2a and 2b show the bright field and SPRM images of a SH-EP1 cell adherent on the gold film, respectively. The PBS buffer was running over the cells continuously for 50 seconds at a flow rate of 350 μL/min while the SPRM images were recorded at a speed of 20 frames per second until signals were stabilized. A detailed description of the flow system and its capability for rapid switching between buffer and sample solution injection is given in the Supplementary Information Section 1. At the 50th second, 80 μg/mL WGA prepared in PBS buffer solution was injected and flowed over the cells for 300 seconds, during which WGA interacted and associated with the glycoproteins on the individual cells, and resulted in an increase in the local SPR signal. At the 350th second, the sample solution was stopped and the buffer solution was injected again to allow the bound WGA to dissociate from the cell surface, which was measured as a decrease in the SPR signal. The dissociation process was monitored for 5 minutes to avoid possible system drift during hours’ recording since 5 minutes was long enough to enable the fitting to the dissociation curve (Supplementary Information Section 2.8). The association and dissociation processes were recorded continuously and presented in Supplementary Information as a movie in order to show the spatially resolved binding activities of a single cell (Movie S1 and Supplementary Information Section 2.1). Additional evidence for the binding was provided by the epifluorescence image of the same cell as shown in Fig. 2c, in which fluorescence labeled WGA was applied to stain the cell after regeneration. Plotting the average SPR intensity in a whole cell region vs. time leads to a SPR sensorogram, providing quantitative information about the association and dissociation kinetics of WGA binding to the glycoproteins of a single cell (black curve of Fig. 2d). Note that the cultured cells were fixed prior to the measurement in order to minimize possible cell micromotions, migration and detachment. While there was no significant difference in the association rate constant (ka), the dissociation rate constant (kd) in living cells was found to be about one time smaller than that of the fixed ones, indicating a slower dissociation when cells were alive (Supplementary Information Section 2.6). Such results might be related with the WGA uptake by living cells through a receptor mediated endocytosis, which was eliminated when the cells were fixed.25
Similar measurements on the cells were performed with the addition of different WGA concentrations incrementally. Before each new concentration of WGA was injected, 50 mM GlcNAc solution was introduced for 100 seconds to regenerate the cell surface.26 Free GlcNAc has a high affinity for WGA and binds competitively to WGA previously attached to the cell surface leading to a regeneration of the membrane glycoprotein receptors. Note that the large increase in the SPR signal in the regeneration step was due to the bulk refractive index of the concentrated GlcNAc solution (650th – 750th seconds in Fig. 2d). The data in Fig. 2e shows that the WGA binding signal increases with the increased solution concentration of WGA, which is expected for a first order binding kinetic process.
We carried out negative and positive control experiments to further validate the observation and interpretation. We injected WGA pre-mixed with 50 mM GlcNAc, and observed no binding signal (Supplementary Information Section 2.2), which supports that the SPR responses in Figs. 2d and 2e are due to the binding of WGA to GlcNAc of the MPs on the cell surface. We repeatedly measured the binding kinetics of the same cell by regenerating the GlcNAc residues on the glycan chains of MPs and re-exposing the cell to WGA (Supplementary Information Section 2.3). Following each regeneration step, the SPR signal completely returned to the level prior to the binding of WGA, showing that the regeneration is effective without disturbing the cells, which is critical for the concentration dependence measurement shown in Fig. 2e. Furthermore, the interaction between WGA and poly-L-lysine was investigated since the gold chips were modified by poly-L-lysine in prior to the cell culture. The results showed that poly-L-lysine modification did not affect the observed SPR sensorograms (Supplementary Information Section 2.4).
We also monitored WGA binding onto multiple cells simultaneously by increasing the field of view of the imaging system (Supplementary Information Section 2.5). The overall sensorograms are similar for all the cells, but the SPR signal varies from cell to cell with a standard derivation of 14%. Since SPR is proportional to the amount of WGA that binds to the cell membrane, such cell-to-cell variation in the SPR signal reflects different abundance of WGA binding sites, indicating a need for single cell studies, a capability provided by the present method.
At a given ligand (WGA) concentration (80 μg/mL), the SPR signal at equilibrium (in the plateau region of the association process of the sensorogram) reflects the distribution of the GlcNAc-containing MPs. Fig. 3a is such a map showing the distribution of GlcNAc-containing MPs on the surface of the cell. As supported by the local SPR sensorograms at the cell edge (Fig. 2d, red curve) and center (Fig. 2d, blue curve), only the edge regions of the cell show binding activities, which is because the evanescent field associated with SPR penetrates into the cell volume over a distance ~200 nm, such that the top surface of cell near the center is not measured by SPR. This feature is shared by total internal reflection fluorescence microscopy (TIRFM), which also relies on the evanescent field. The fluorescence image of the same cell in Fig. 2c shows the thick central region and thin edge region divided by a borderline pointed by white arrows, which supports the above interpretation.
Fig. 3. Mapping glycoprotein distribution and binding kinetics.
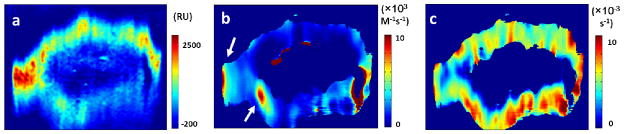
(a) The distribution of N-acetyl glucosamine-containing glycoproteins was quantified by the SPR signal increment after WGA binding. By fitting the local SPR sensorograms pixel by pixel with a first order kinetics model, the local association (b) and dissociation (c) rate constants were mapped. The white arrows point relatively fast binding regions.
Since the entire bottom surface of the cell was within the SPR detection range, the fact that no binding signal was observed in the central portion of the cell indicates that the binding of WGA primarily took place on the top surface of the cell. This conclusion is further supported by the following two facts. First, the SPR response started simultaneously at different regions of the cell. If the binding took place at the bottom surface, the diffusion of WGA from the edge towards center would have led to a time delay in the SPR signal at locations that are far away from the edge. Second, the binding kinetics followed the first order kinetics with no sign of mass limited transport associated with diffusion of WGA in the sensorograms. In general, whether a macromolecule can access the bottom surface of a cell depend on its size, charge, polarity, hydrophilicity and other properties.27 Although it is possible for certain marcomolecules to diffuse through the gap between the bottom of the cell and substrate to bind on the bottom surface of the cell, it has been reported that some molecules cannot reach the bottom membrane surface of the cell, and WGA likely belongs to the latter case.27
Despite the limited penetration length of the evanescent field, the present imaging system can resolve the local binding kinetics of WGA on single cells, as we will show below. The binding kinetics of WGA to MPs averaged over the entire edge regions in a single cell can be determined by fitting the sensorograms of different WGA concentrations via a global fitting based on a 1:1 Langmuir model (Fig. 2e). The corresponding association (ka) and dissociation rate constants (kd) were found to be 5.2×103 M−1s−1 (ka) and 1.2×10−3 s−1, respectively, from which the dissociation constant (KD) was determined to be 0.23 μM. In addition to the kinetic analysis, equilibrium analysis was carried out at different ligand concentrations, which led to a KD value of 0.32 μM, as shown in Fig. 2f (Supplementary Information Section 2.11). The good agreement between kinetic and equilibrium analysis shows that the mass transport and WGA rebinding effects are minimal in the present work. This conclusion was further supported by the fact that a decrease in the flow rate from 350 to 200 μL/min did not affect the dissociation rate constant (see Supplementary Information Section 2.7).
The KD value obtained via the present in situ approach is about 1 to 2 orders of magnitude greater than those determined by ex situ methods.26 It has been reported that the lectin-glycoprotein interaction was greatly influenced by the conformation, density and the chemical environment of the target sugar residues.7,28 For instance, the interaction of WGA with pure GlcNAc oligosaccharides showed up to 20 times difference in binding kinetics depending on the number of GlcNAc units.26 In another ex situ study, 2 orders of magnitude variation in binding affinity was also observed among five glycoproteins when interacted with the same lectin ligand.29 Moreover, significantly different binding affinity of membrane protein between in situ and ex situ measurements have also been reported by fluorescence and enzyme-linked immunosorbent assay, implying the great influence of biological environment on the binding behaviors of MPs.30,31
The subcellular imaging capability allows us to map the local binding constants of single cells by fitting local sensorograms pixel by pixel. Figs. 3b and 3c show the obtained ka and kd maps, respectively, which show significant spatial variations in the binding kinetics of the glycoproteins across the cell surface. Statistical analysis of kd values in Fig. 3c shows Gaussian distribution, with an average of 5×10−3 s−1 and a modest standard deviation of 2×10−3 s−1 (Supplementary Information Section 2.12). Although the distribution of GlcNAc (Fig. 3a) and maps of ka and kd (Figs. 3b and 3c) also show spatial variations, the variations are uncorrelated, indicating the independent kinetic and thermal equilibrium parameters.
It is accepted that the distribution of MPs and associated glycans are heterogeneous across the cell membrane surface but the molecular level-mechanism for the heterogeneous binding kinetics remains unclear. Previous ex situ studies have suggested that the binding kinetics of the same lectin to different glycoproteins vary up to 100 times even if this lectin recognizes the same sugar group, because the type of glycoproteins greatly affect the lectin binding kinetics.29 It is thus possible that the local variations in the binding kinetics shown in Figs. 3b and 3c are due to heterogeneous distribution of different types of glycoproteins in the membrane of the cell. Further studies are clearly needed for a better understanding of the phenomenon, and the unique capability of the present imaging system is anticipated to provide detailed data for one to achieve the goal.
Glycoprotein polarization in chemotaxis
Many cellular processes, such as cell migration32,33 and immune recognition,16,34 involve polarization or redistribution of glycoproteins in the cell membrane. Studying the polarization of glycoproteins is critical for a better understanding of these important cellular processes. Previously, glycoprotein polarization during chemotaxis has been studied with fluorescence microscopy34 and with transmission electron microscopy (TEM) by labeling the glycoprotein with ferritin to enhance TEM contrast.35 We demonstrate below that the current method allows us to map the MPs redistribution in a single living cell during chemotaxis. It is label-free and non-invasive, and more importantly, monitors the spatial response of glycoproteins in the native membrane environment of living cells.
The chemotaxis of live SH-EP1 cells was validated using fetal bovine serum (FBS) as a chemoattractant according to the protocol previously described in literature36 (Supplementary Information Movie S2). Cells were serum-starved by culturing them in serum-free media for 3 hours followed by exposure to serum introduced via a pipette placed near the cell (Fig. 4a). The slow diffusion of serum from the tip of the pipette creates a serum concentration gradient (~10%) and induces migration of the cells towards the pipette tip (Supplementary Information Section 3.2). Fig. 4b shows the SPRM image of a cell before introducing the chemoattractant and Fig. 4c indicates the binding pattern of WGA at the leading edge of the cell, which reflects the heterogeneous glycoprotein distribution in the cell.
Fig. 4. Glycoprotein polarization during chemotaxis.

(a) A micropipette tip filled with fetal bovine serum is located near the target cell and induce the cell migration towards the chemoattractant. Another perfusion tube is located at the other side of the cell and introduces the WGA solution in order to obtain the distribution map of glycoprotein. The SPRM (b, d, f) and distribution images (c, e, g) were obtained in the beginning (b, c), after 20 minutes waiting without any treatment (d, e) and at another 20 minutes after the chemoattractant was applied (f, g), respectively.
A negative control experiment in the absence of chemoattractant was carried out to evaluate the spontaneous glycoprotein re-distribution, in which the same cell was exposed to WGA solution again after 20 minutes without any treatment. The images (Figs. 4d and 4e) are nearly identical before and after the 20 min-waiting period, demonstrating that the cell remained stable, and the distribution of the WGA binding sites stayed. Note that the cell surface was regenerated by removing bound WGA after each WGA introduction. Subsequently, a pipette filled with FBS was placed in the left upper corner of the target cell and kept there for 20 minutes before another SPR image was captured (Fig. 4f). A filopodium pointing to the pipette tip is indicated by the white arrow in image Fig. 4f, showing the migration of the cell towards the chemoattractant. Such a behavior is common in the early stage of a chemotaxis process. WGA was then re-introduced to map the glycoproteins distribution after the chemotaxis process (Fig. 4g). Compared with Fig. 4c, a 28% increase in the average glycoproteins density in the leading edge of the cell was observed as indicated by the white arrows, demonstrating a redistribution of the glycoproteins took place even before the leading edge of the cell migrated (Supplementary Information Section 3.3). The negative result of the control experiment supports the conclusion that the glycoprotein polarization in the leading edge was the consequence of the chemotaxis response to the chemoattractant. This study demonstrates a method for non-invasively mapping the dynamic redistribution of glycoproteins during the chemotaxis.
Mapping local distribution and binding activities of nicotinic acetylcholine receptor (nAChR)
Nicotinic acetylcholine receptor (nAChR) is one of the most studied membrane receptors because of its critical role in neurotransmission and nicotine addiction. The expression level and the subcellular distribution of nAChR, especially in neurons, are critical not only for basic neuroscience and electrophysiology, but also for the clinical evaluation of nicotine addiction.37 The conventional immunofluorescence method to map the nAChR distribution in cells is based on fluorescence-labeled secondary antibody. As mentioned earlier, the label-based detection is an end-of-point method, which does not provide direct kinetic information. More importantly, the fluorescence-based method is prone to false positives partially because the secondary antibody can often bind to multiple different targets. In contrast, our label-free imaging technology directly measures the binding of the primary antibody to nAChR without the secondary antibody, which is not only simpler, but also overcomes the drawbacks associated with the use of labeled secondary antibody.
Engineered SH-EP1 cells that express α4β2 nAChR38 were used to examine the binding kinetics of nAChR to its anti-α4 primary antibody, while the wildtype SH-EP1 cells served as a control. Figs. 5a and 5b show the bright field and SPR images of a fixed engineered cell, respectively. Upon injection of the primary antibody, the SPR signal averaged over the entire cell increases rapidly and then reaches a plateau, indicating the binding of the primary antibody to the cell surface (red line in Fig. 5e). Flushing the cell surface with buffer solution results in a decrease in the SPR signal, which is due to the dissociation of the primary antibody from the cell surface. The red curve in Fig. 5e represents the average response of five individual cells while the pink background indicates the standard deviation. In order to investigate the possible influence of non-specific immunoglobulins in the antibody solution, the sensorograms of anti-nAChR alpha4 antibody solution and isotype rat IgG solution were recorded in the same engineered cells that were incubated with normal rat serum for 1 hour before the experiments. A clear sign of specific interaction was observed for anti-nAChR alpha4 antibody, while the isotype rat IgG injection led to a simple bulk effect with no sign of specific binding (Supplementary Information Section 4.1).
Fig. 5. Mapping nicotinic acetylcholine receptors (nAChR).

(a) and (b) Bright field and SPRM images of an engineered SH-EP1 cell that expresses nAChR receptors. (c) The immunofluorescence image of the same cell obtained by sequentially incubating with primary and Alexa Fluor 488 labeled secondary antibody. (d) Distribution of nAChR of the same cell resolved via the binding of primary antibody to the cell. (e) SPR sensorograms of engineered (red curve) and wildtype (black curve) SH-EP1 cells during primary antibody binding. Five cells were examined in each case and variation is shown by the pink and grey curves. (f) The sensorogram (black dots) after the correction of bulk refractive index effect (by taking the wildtype as reference), where the red curve is fitting the first-order kinetics model. The fitting residuals are shown in the lower portion of the figure.
In addition to engineered cells, we have also studied multiple wildtype cells using the same procedure. The black curve in Fig. 5e shows the average sensorogram of five wildtype cells, and the gray background shows the standard deviation. The α4β2 nAChR expression is known to be a major difference between the engineered and the wildtype cells, and such a difference is clearly shown in the SPR sensorograms (red and black curves). Note that the SPR sensorograms for both the engineered and wildtype cells contain bulk refractive index effect, which can be subtracted out by taking the difference of the two sensorograms. The corrected sensorogram is shown in Fig. 5f, which was fit with the first order kinetic equations (red line). The extracted binding constants, ka and kd, were 6.4×104 M−1s−1 and 2.8×10−3 s−1, respectively, from which the dissociation constant, KD, was found to be 45 nM.
The distribution of nAChR on the membrane surface was obtained by subtracting the initial SPR image before association from the one taken 50 seconds after the buffer was injected in dissociation process (Fig. 5d). The 50 seconds delay minimized the influence of the bulk effect and revealed the SPR increase mainly due to specific binding. The fluorescence image of the same cell was also obtained by sequentially incubating with primary antibody and fluorescence-labeled secondary antibody, which is shown in Fig. 5c for comparison. While the spatial distribution of nAChR from conventional immunofluorescence approach (Fig. 5c) shared some features with the one obtained from SPRM measurement (Fig. 5d), they were generally different since the SPRM images focused the MPs located in the depth of evanescent wave instead of the entire cell membrane. Control experiments showed that no fluorescence was observed for the wildtype cells or the engineered cells without primary antibody (Supplementary Information Section 4.2), and therefore, validated the quality of both primary antibody and the nAChR expression in the engineered cells.
The flow system of the present setup provides rapid switching between buffer and sample solution (1–2 seconds), which allows for the study of fast dissociation and association processes. The noise level of the current setup is ~10 RU on a single cell level, corresponding to ~80 antibody binding site per μm2 (Supplementary Information Sections 2.9 and 2.13). While the sensitivity could be further improved by tens of times since the commercial SPR instrument can reach a noise level of 0.1 RU, the present system is adequate for many biologically significant MPs. For instance, many MPs are overexpressed in the cancer cells and act as the biomarkers for disease diagnosis and clinical treatment. The expression level could be as high as 1~3 million receptors per cell,39 which is ~10 times higher than the detection limit in the present method. Moreover, the local enrichment is one of the most important features that affect the membrane receptor function in the cell signaling.40 The imaging feature of the present approach enables one to measure the local binding kinetics even if the average receptor density is not high enough to be detected. The potential application could be further broadened with the help of gene transfection, which is now a widely used technique to overexpress certain membrane proteins in the cells so that the SPR signal could be greatly enhanced.
Conclusions
We have demonstrated a plasmonic-based imaging system for measuring the binding kinetics of MPs in single live cells in real time without labeling. It allows us, for the first time, to determine kinetic constants of MPs in their native membrane environment, and to map the distribution of MPs in the cell membrane, and local association and dissociation rate constants of MPs to different ligands. The system can also perform simultaneous optical transmission and fluorescence microscopy of the same sample, thus making it possible to combine the strengths of both labeled and label-free detection and imaging technologies in one integrated setup.
By taking advantages of the new capabilities, we have studied the specific interactions between glycoproteins and lectin in single cells and binding activity and spatial distribution of nicotinic acetylcholine receptors on the membrane of single cells. We have also studied glycoprotein polarization during chemotaxis. We anticipate that the SPRM approach will have a broad impact on the study of the biological activities of MPs in their native states, and in the discovery of drugs that target MPs including both monoclonal antibody drugs and small molecule drugs via competitive assay (Supplementary Information Section 2.14). We also expect that the approach find applications in the study of heterogeneous surface binding and interactions.
Methods
Materials
Wheat germ agglutinin (WGA) and N-acetyl-glucosamine (GlcNAc) were purchased from Sigma-Aldrich (St. Louis, MO). WGA conjugated with Alexa Fluor 555 was obtained from Invitrogen (Carlsbad, CA). Primary monoclonal antibody against neuronal nicotinic acetylcholine receptor α4-subunit was obtained from Covance (Princeton, NJ). Alexa Fluor 488-AffiniPure Goat Anti-Rat IgG from Jackson ImmunoResearch (West Grove, PA) was used as the secondary antibody. Normal rat serum and isotype rat IgG were purchased from Invitrogen.
1X Phosphate buffered saline (PBS) was used as buffer in all the binding experiments for fixed cells. The extracellular fluid (ECF) buffer contained NaCl (120 mM), KCl (3 mM), CaCl2 (2 mM), MgCl2 (2 mM), D-Glucose (25 mM), HEPES (15 mM) was used as buffer for the chemotaxis experiments in which live cells were used. A proper amount of NaOH was added to bring the ECF buffer pH to 7.4. All the reagents were analytical grade, purchased from Sigma-Aldrich, except otherwise stated. Deionized (DI) water was used to prepare all the buffers.
Cell culture
The human epithelial SH-EP1 cells were cultured in a humidified incubator at 37 °C with 5% CO2 and 70% relative humidity. Cells were grown in Dubelco’s modified eagle’s medium (DMEM; Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA) with penicillin and streptomycin (BioWhittaker, Basel, Switzerland). Cells were passaged with 0.05% trypsin and 0.02% ethylenediamine-tetraacetic acid (EDTA) in Hank’s balanced salt solution (HBSS; Sigma-Aldrich, St. Louis, MO) when they were approximately 75% confluent. The fixation to cells was performed by incubating with 4% paraformaldehyde solution for 30 minutes at room temperature.
SPRM setup
The optical system comprises of a fiber coupled 680 nm super-LED light source (Qphotonics LLC, Ann Arbor, MI), an inverted microscope (Olympus X81) with total internal reflection fluorescence (TIRF) imaging attachment, and a charge-coupled device (CCD) camera (Pike F-032). The sensor chips were BK-7 glass coverslips coated with 32 nm chromium followed by 347 nm gold. Each chip was washed with water and ethanol followed by hydrogen flame annealing to remove surface contamination before each experiment. A Flexi-Perm silicon chamber (Greiner Bio-One) was placed on top of the gold chip to serve as a cell culture well. The gold surface was modified to promote cell adhesion by adding 80 μL of 100 μg/mL poly-L-lysine solution in the chamber and incubating overnight. Chips were rinsed by DI water twice prior to cells seeding. ~5,000 SH-EP1 cells in 300-μL growth medium were seeded to the chip with a Flexi-Perm chamber attached. After overnight incubation to allow the cells to attach and grow, the growth medium was replaced by ECF buffer or PBS buffer solution before each experiment.
Epifluorescence microscopy and immunofluorescence microscopy
The same SPRM platform as described above was used to perform immunofluorescence measurements as an epifluorescence microscope. A 150W mercury lamp was used as the light source. For WGA staining, the fixed cells were regenerated by GlcNAc solution and then incubated with 100 μg/mL WGA-Alex555 for 10 minutes at room temperature. The fluorescence image was captured after the well was rinsed twice by PBS buffer. A set of optical filters (Ex535/Em565) was applied for WGA staining. For nAChR immunofluorescence, the fixed cells were thoroughly washed by PBS buffer for 20 minutes before incubated with 1% fetal bovine serum (FBS) for 1 hour at room temperature to block the cell surface. Subsequently the primary antibody was added and incubated for 30 minutes at temperature, before the Alexa Fluor 488-labeled secondary antibody was added and incubated for another 30 minutes. The FL image was captured after the well was rinsed twice by PBS buffer. A set of optical filter (Ex420–480/Em515) was applied for nAChR immunofluorescence.
Flow system
A gravity-based multi-channel drug perfusion system (SF-77B, Warner Instruments, CT) was used to control the local solution surrounding the target cell. The typical transition time between different solutions was about 1–2 seconds (see Supplementary Information Section 1 for more details).
Data analysis
The global fitting shown in Fig. 2e and the kinetics fitting shown in Fig. 5f were carried out in BIAevaluation software by applying a 1:1 Langmuir model. The curve fitting provided ka, kd and KD(=kd/ka). The fitting in Fig. S10 was carried out with a Matlab program using first order kinetic equations. To create the ka and kd maps shown in Fig. 3, global fitting of the association curves at different WGA concentrations from 10 to 200 μg/mL were performed. From the fitting parameter, kobs=ka*CWGA+kd, ka and kd were extracted with a linear regression (slope and intercept, respectively).
Supplementary Material
Acknowledgments
We thank NIH (R21RR026235) for support.
References
- 1.Cho WH, Stahelin RV. Membrane-protein interactions in cell signaling and membrane trafficking. Annu Rev Biophys Biomol Struc. 2005;34:119. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- 2.Marinissen MJ, Gutkind JS. G-protein-coupled receptors and signaling networks: emerging paradigms. Trend Pharm Sci. 2001;22:368. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- 3.Hopkins AL, Groom CR. The druggable genome. Nature Rev Drug Disc. 2002;1:727. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 4.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nature Biotech. 2005;23:1147. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 5.Rees DC, Congreve M, Murray CW, Carr R. Fragment-based lead discovery. Nature Rev Drug Disc. 2004;3:660. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]
- 6.Salamon Z, Macleod HA, Tollin G. Surface plasmon resonance spectroscopy as a tool for investigating the biochemical and biophysical properties of membrane protein systems. 2 Applications to biological systems. Biochim Biophys Acta. 1997;1331:131. doi: 10.1016/s0304-4157(97)00003-8. [DOI] [PubMed] [Google Scholar]
- 7.Lee AG. How lipids affect the activities of integral membrane proteins. Biochim Biophys Acta. 2004;1666:62. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Fruh V, Ijzerman AP, Siegal G. How to catch a membrane protein in action: A review of functional membrane protein immobilization strategies and their applications. Chem Rev. 2011;111:640. doi: 10.1021/cr900088s. [DOI] [PubMed] [Google Scholar]
- 9.Bally M, et al. Liposome and lipid bilayer arrays towards biosensing applications. Small. 2010;6:2481. doi: 10.1002/smll.201000644. [DOI] [PubMed] [Google Scholar]
- 10.Holden MA, et al. Nature Chem Bio. 2006;2:314. doi: 10.1038/nchembio793. d. [DOI] [PubMed] [Google Scholar]
- 11.Dykstra M, et al. Location is everything: Lipid rafts and immune cell signaling. Annu Rev Immun. 2003;21:457. doi: 10.1146/annurev.immunol.21.120601.141021. [DOI] [PubMed] [Google Scholar]
- 12.Sato TK, Overduin M, Emr SD. Location, location, location: Membrane targeting directed by PX domains. SCience. 2001;294:1881. doi: 10.1126/science.1065763. [DOI] [PubMed] [Google Scholar]
- 13.Li GY, Xi N, Wang DH. Probing membrane proteins using atomic force microscopy. J Cell Biochem. 2006;97:1191. doi: 10.1002/jcb.20753. [DOI] [PubMed] [Google Scholar]
- 14.Muller DJ, Engel A. Atomic force microscopy and spectroscopy of native membrane proteins. Nature Protocols. 2007;2:2191. doi: 10.1038/nprot.2007.309. [DOI] [PubMed] [Google Scholar]
- 15.Groves JT, Parthasarathy R, Forstner MB. Fluorescence imaging of membrane dynamics. Annu Rev Biomed Eng. 2008;10:311. doi: 10.1146/annurev.bioeng.10.061807.160431. [DOI] [PubMed] [Google Scholar]
- 16.Schwarzenbacher M, et al. Micropatterning for quantitative analysis of protein-protein interactions in living cells. Nature Methods. 2008;5:1053. doi: 10.1038/nmeth.1268. [DOI] [PubMed] [Google Scholar]
- 17.Johnson AE. Fluorescence approaches for determining protein conformations, interactions and mechanisms at membranes. Traffic. 2005;6:1078. doi: 10.1111/j.1600-0854.2005.00340.x. [DOI] [PubMed] [Google Scholar]
- 18.Wallrabe H, Periasamy A. Imaging protein molecules using FRET and FLIM microscopy. Curr Opin Biotech. 2005;16:19. doi: 10.1016/j.copbio.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2:764. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- 20.Wang W, et al. Single cells and intracellular processes studied by a plasmonic-based electrochemical impedance microscopy. Nature Chem. 2011;3:249. doi: 10.1038/nchem.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang B, Yu F, Zare RN. Surface plasmon resonance imaging using a high numerical aperture microscope objective. Anal Chem. 2007;79:2979. doi: 10.1021/ac062284x. [DOI] [PubMed] [Google Scholar]
- 22.Kadurin I, et al. Differential effects of N-glycans on surface expression suggest structural differences between the acid-sensing ion channel (ASIC) 1a and ASIC1b. 2008;412:469. doi: 10.1042/BJ20071614. [DOI] [PubMed] [Google Scholar]
- 23.Dell A, Morris HR. Glycoprotein structure determination mass spectrometry. Science. 2001;291:2351. doi: 10.1126/science.1058890. [DOI] [PubMed] [Google Scholar]
- 24.Durand G, Seta N. Protein glycosylation and diseases: Blood and urinary oligosaccharides as markers for diagnosis and therapeutic monitoring. Clin Chem. 2000;46:795. [PubMed] [Google Scholar]
- 25.Liu SL, et al. Visualizing the endocytic and exocytic processes of wheat germ agglutinin by quantum dot-based single-particle tracking. Biomaterials. 2011;32:7616. doi: 10.1016/j.biomaterials.2011.06.046. [DOI] [PubMed] [Google Scholar]
- 26.Vila-Perello M, Gallego RG, Andreu D. A simple approach to well-defined sugar-coated surfaces for interaction studies. ChemBioChem. 2005;6:1831. doi: 10.1002/cbic.200500125. [DOI] [PubMed] [Google Scholar]
- 27.Gingell D, Todd I, Bailey J. Topography of cell-glass apposition revealed by total internal reflection fluorescence of volume markers. J Cell Biol. 1985;100:1334. doi: 10.1083/jcb.100.4.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato Y, et al. High Mannose-binding Lectin with Preference for the Cluster of 1–2-Mannose from the Green Alga Boodlea coacta Is a Potent Entry Inhibitor of HIV-1 and Influenza Viruses. J Biol Chem. 2011;286:19446. doi: 10.1074/jbc.M110.216655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katrlik J, Skrabana R, Mislovicova D, Gemeiner P. Binding of D-mannose-containing glycoproteins to D-mannose-specific lectins studied by surface plasmon resonance. Colloid Surf A. 2011;382:198. [Google Scholar]
- 30.Rathanaswami P, Babcook J, Gallo M. High-affinity binding measurements of antibodies to cell-surface-expressed antigens. Anal Biochem. 2008;373:52. doi: 10.1016/j.ab.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 31.Troise F, et al. Differential binding of human immunoagents and Herceptin to the ErbB2 receptor. FEBS J. 2008;275:4967. doi: 10.1111/j.1742-4658.2008.06625.x. [DOI] [PubMed] [Google Scholar]
- 32.Lehmann S, et al. An endogenous lectin and one of its neuronal glycoprotein ligands are involved in contact guidance of neuron migration. Proc Nat Acad Sci USA. 1990;87:6455. doi: 10.1073/pnas.87.16.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zieske JD, Higashijima SC, Gipson IK. Con A-binding and WGA-binding glycoproteins of stationary and migratory corneal epithelium. Invest Ophth Vis Sci. 1986;27:1205. [PubMed] [Google Scholar]
- 34.Huppa JB, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kataoka M, Tavassoli M. Polarization of membrane-glycoproteins during monocyte chemotaxis. Exp Cell Res. 1984;153:539. doi: 10.1016/0014-4827(84)90622-0. [DOI] [PubMed] [Google Scholar]
- 36.Russo VC, et al. Insulin-like growth factor binding protein-2 binding to extracellular matrix plays a critical role in neuroblastoma cell proliferation, migration, and invasion. Endocrinology. 2005;146:4445. doi: 10.1210/en.2005-0467. [DOI] [PubMed] [Google Scholar]
- 37.Albuquerque EX, Pereira EFR, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol Rev. 2009;89:73. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eaton JB, et al. Characterization of human alpha 4 beta 2-nicotinic acetylcholine receptors stably and heterologously expressed in native nicotinic receptor-null SH-EP1 human epithelial cells. Mol Pharmacol. 2003;64:1283. doi: 10.1124/mol.64.6.1283. [DOI] [PubMed] [Google Scholar]
- 39.DeFazio-Eli L, et al. Quantitative assays for the measurement of HER1–HER2 heterodimerization and phosphorylation in cell lines and breast tumors: applications for diagnostics and targeted drug mechanism of action. Breast Cancer Res. 2011;13:R44. doi: 10.1186/bcr2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manz BN, Groves JT. Spatial organization and signal transduction at intercellular junctions. Nature Rev Mol Cell Biol. 2010;11:342. doi: 10.1038/nrm2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.