

Abstract
Obesity affects the cardiovascular system at many different levels, including the heart muscle itself. Clinical and experimental studies have shown an accumulation of triglycerides and other lipid species in cardiomyocytes. Analogous to hepatic steatosis, investigators have introduced the term “cardiac steatosis”. The present review addresses the complex relationships between cardiac fuel homeostasis, insulin resistance, and proposed mechanisms of damage to cardiomyocytes in different models of obesity, insulin resistance, and lipotoxicity. Specifically, the review weighs the evidence whether there is a heart muscle disorder in human obesity. It discusses how adipokines can modulate cardiac metabolism, and it focuses on the metabolic remodeling accompanying increased fatty acid supply in the heart of rodent models of lipotoxicity, with special attention to the role played by mitochondrial uncoupling and futile cycling. We stress the notion that, in spite of the many proposed mechanisms, cardiac lipotoxicity is still a hypothesis rather than an established pathophysiologic principle. Although the concept of a “lipotoxic cardiomyopathy” seems attractive, we propose instead a series of steps on a path from adaptation to maladaptation of the heart in obesity. A case in point is insulin resistance of the heart which may be both adaptive (protecting the heart from excess fuel) or maladaptive (associated with reactive oxygen species formation and activation of signaling pathways of programmed cell death). The present literature reflects an extraordinary complexity of the heart’s metabolic, functional and structural changes in obesity.
Keywords: cardiac metabolism, cardiac function, futile cycling, lipotoxicity
Introduction
Obesity appears to be a major cause of hypertension and associated cardiovascular pathophysiology, including cardiac dysfunction. However, obesity may lead to abnormal cardiac function through mechanisms that are independent of, or that act in concert with, hypertension. One hypothesis of obesity-induced cardiac dysfunction is that an oversupply of substrates leads first to adaptive changes and eventually to contractile dysfunction of the heart. We reason that increased supply of non-esterified fatty acids (NEFA) together with metabolic dysregulation in obesity, including an inadequate activation of fat oxidation, results in the accumulation of toxic lipid byproducts and subsequent contractile dysfunction. Although the phenomenon may have already been known to Virchow when he described “fatty metamorphosis” of the heart,1 the concept of cardiac “lipotoxicity” reemerged only recently with its description in the heart of the obese Zucker diabetic fatty (ZDF) rat.2 The concept is, however, still a hypothesis rather than an established physiological principle. In spite of the many investigations performed in rodent models, the mechanism(s) responsible for impaired contractile function of the heart is still obscure, and it is uncertain whether lipid metabolites contribute to “obesity cardiomyopathy” in humans. Our brief review is an attempt to understand the chronic regulatory effects of changes in systemic metabolism on cardiac function. In other words, we discuss current concepts of cardiac adaptation and maladaptation to a deranged metabolic environment.
Heart Muscle Disease in Human Obesity
Changes in cardiovascular function in the setting of clinically-severe obesity were first reported in obese volunteers undergoing cardiac catheterization. The patients demonstrated reduced left ventricular (LV) compliance and a decrease in stroke work index in the presence of increased LV end diastolic pressure that correlated with the severity of obesity.3 There is a significant correlation between obesity and LV mass, even after controlling for age and blood pressure4, and there is also a significant correlation between weight and impaired diastolic filling of the left ventricle.5 Both systolic and diastolic function are decreased in otherwise healthy obese young women.6 In the same population there is a decrease of cardiac efficiency.7 In severely obese patients with a body mass index (BMI) over 50 the serum concentrations of NEFA show a negative correlation with load-independent diastolic function.8 In addition to hemodynamic changes, obesity is also associated with increased risk of atrial fibrillation 9 and ventricular ectopic activity.10
The mechanisms of cardiac remodeling with obesity are complex.11,12 A major obstacle in any attempt to characterize “obesity cardiomyopathy” is the prevalence of comorbid disorders and confounding variables such as the metabolic syndrome,13 insulin resistance, hypertension, type 2 diabetes, and physical inactivity. It is of note that both increased blood pressure and increased BMI are independently associated with increased LV mass in obese individuals; the effects of hypertension seem to amplify those of sleep apnea and more severe obesity.14 In type 2 diabetes hepatic steatosis coexists with myocardial insulin resistance and endothelial dysfunction.15 Liver and heart share the characteristic of “first pass” organs into which fatty acids drain from a visceral adipose depot (the intra-abdominal and the epicardial fat). Not surprisingly, a cross sectional study performed on a small cohort of healthy men has shown an association between circulating free fatty acids levels and myocardial fat.16
Another line of reasoning refers to structural and functional changes of the heart in type 2 diabetes. Metabolic remodeling, or sustained changes in flux through a metabolic pathway, may precede functional and structural remodeling of the heart,17 including insulin resistant states.18 We have observed intramyocardial lipid accumulation in the failing heart of obese or diabetic patients.19 Intracytoplasmic lipid in cardiomyocytes of type 2 diabetic patients is accompanied by considerable loss of myofibrils.20 In patients without heart failure, increased myocardial triglycerides were associated with either impaired glucose tolerance or type 2 diabetes.21 Thus impaired metabolism of energy providing substrates and myocardial lipid accumulation are early events found in obese and insulin resistant individuals.
Obesity and Heart Failure
After correction for hypertension and other risk factors for heart failure, obesity still increases the risk of heart failure approximately twofold.22 At the same time overweight patients with symptomatic heart failure have a better prognosis than non-obese individuals.23,24 In essence, obesity is a risk factor for developing heart failure, but after the onset of heart failure obesity is a positive predictor for survival.25,26 While visceral fat is considered a defining feature of the metabolic syndrome,27 a potential cardioprotective effect of perivascular and epicardial white adipose tissues has been proposed.28 The existence of this “obesity paradox” has led physicians to question whether obesity should be treated when associated to heart failure.
Obesity doubles the risk of premature death and increases the risk of death from cardiovascular disease fivefold.29 In contrast to drug therapies and weight loss programs,30 bariatric surgery seems to offer a more effective therapy for severely obese patients. It has been suggested that bariatric surgery improves heart function and survival,31 even in patients with cardiomyopathy.32 Interestingly, weight reduction is a better predictor of changes in LV structure (decreased wall thickness and mass) than the concomitant decrease in blood pressure for patients undergoing bariatric surgery.33 More studies are needed to determine whether the surgery is feasible, safe, and effective for heart failure patients.
Adipokines as Possible Causes for Cardiac Lipotoxicity
Dysregulation of adipokine signaling is likely to play a role in the maladaptation of the heart and skeletal muscle to a high fat environment.34 Under normal conditions, white adipose tissue protects non-adipose organs from lipid overload by storing the excess of NEFA in the form of triglycerides. Adipose tissue also protects non-adipose organs by leptin-regulated central and peripheral effects.35 A state of lipotoxicity may thus be promoted by the state of leptin resistance associated with obesity.36
Adiponectin has beneficial effects on the vasculature and cardiac hypertrophy,37 and regulates overall energy homeostasis. In liver, adiponectin improves insulin sensitivity, decreases NEFA uptake while increasing their oxidation, and reduces neoglucogenesis.38 In skeletal muscle, adiponectin stimulates both glucose and fatty acid utilization.38 Whether adiponectin directly regulates cardiac metabolism is not clear, although in vitro experiments suggest that adiponectin can accelerate fatty acid oxidation in the heart.39 However, decreased adiponectinemia in the state of obesity is likely to promote insulin resistance, which will consequently affect heart metabolism.
Another adipokine, serum retinol-binding protein 4 (RBP4), is correlated to the severity of insulin resistance in human subjects.40 Interestingly, RBP4, like leptin and adiponectin, is related to ectopic fat accumulation,41 but a putative action of RBP4 on cardiac metabolism is unknown. Other adipokines, also linked to insulin resistance, like resistin, 42 or visfatin,43 still need to be investigated for their possible involvement in cardiac adaptation or maladaptation to obesity. Lastly, pro-inflammatory cytokines released by adipose tissue in the state of obesity may have the ability to promote tissue synthesis of ceramide,44 a critical molecule in the lipotoxic process. The appreciation of adipokine signaling and direct effects of adipokines on cardiac metabolism, especially leptin and adiponectin, are well established,37, 45 but the pathways of adipocyte-heart crosstalk are still under investigation.
Lipotoxicity in Rodent Models of Obesity
While the role of adipokines in the development of lipotoxicity is still debated, there is already good evidence for cardiac dysfunction in rodent models of obesity.2, 46-48 Most models, such as the ob/ob mouse or the ZDF rat, are the result of single gene mutations which alter leptin signaling. Other models rely on the cardiac-specific overexpression of key regulatory enzymes of either fatty acid metabolism or fatty acid partitioning.49 Several studies in rodents have used an excess supply of dietary calories (usually in the form of fat) to define metabolic changes in the heart.50-54 But while the hearts of genetically-engineered animals are prone to maladaptation, it is more difficult to define why a normal heart fails to adapt to a chronic change in its metabolic environment. The situation is complicated by discrepancies among studies.51, 53, 54 The composition of the diet is of importance: a Western diet (high fat and high carbohydrate meal) seems detrimental for cardiac function, while high fat diet is not.54 A brief recapitulation of myocardial fatty acid metabolism will help to understand why an unmitigated oversupply of lipids may adversely affect cardiac function.
Fatty Acid Uptake and Activation
Free fatty acids enter the myocardial cell via diffusion, or via fatty acid specific transporters like fatty acid translocase (FAT/CD36).55 Fatty acid uptake seems critical because FAT/CD36 deficiency is sufficient to reverse the lipotoxic phenotype in MHC-PPARα mice.56 Inside the cell, free fatty acids bound to fatty acid binding proteins, are activated to fatty acyl-CoA and directed into different metabolic pathways: β-oxidation, binding to transcription factors, cellular signaling (via direct interaction with signaling proteins), conversion to lipid-based signaling molecules (e.g. diacylglycerol (DAG)), post-translational modification of proteins, or storage in the form of triglycerides.57 Because the heart has a very limited capacity to store triglycerides, and because increased fatty acid supply results in increased fatty acid uptake, the heart is subject to increased susceptibility to “spillover” of toxic lipid byproducts.58
Fatty Acid Metabolites Upstream of β-oxidation
The transgenic overexpression of fatty acyl-CoA synthetase 1 in mice results in dramatic triglyceride accumulation within the cardiomyocyte and systolic dysfunction followed by death.59 In contrast, systolic cardiac function is preserved in mice with the transgenic overexpression of the fatty acid transport protein FATP1 (another model of increased fatty acid uptake), while the mice develop impaired cardiac diastolic function and show a decreased density of the repolarizing voltage-gated K+ current and prolonged corrected Q-T interval on EKG.60 Fatty-acyl CoAs have been demonstrated as modulators of potassium currents mediated by KATP in the heart.61 Thus, lipids are mediators of both systolic and diastolic dysfunction.
Much has been learned about lipotoxicity from studies on the β-cell of the pancreas which show exquisite accumulation of triglycerides and long chain fatty-acyl CoA, β-cell insulin resistance, impaired glucose sensitive insulin secretion and, ultimately, apoptosis. One of the central pathways which mediates this effect is the accumulation of ceramide.62 In the β-cell of the ZDF rat, there is increased expression of the enzyme serine palmitoyltransferase which catalyzes the first step in de novo ceramide biosynthesis. Ceramide can induce ROS generation, as well as apoptosis, and can inhibit insulin signaling at the Akt/PKB complex. Another lipotoxic pathway is the activation of protein kinase C (PKC) by DAG. The isoforms of PKC are thought to confer repression of insulin signaling by serine/threonine phosphorylation of the insulin receptor or the insulin receptor substrates.63 Increased intramyocellular fatty acids and increased PKC activity are present in insulin-resistant skeletal muscle.64,65
Regulation and Dysregulation of Fat Metabolism by Nuclear Receptors
A major mechanism by which the heart adapts to a high fat environment is ligand activation of the peroxisome proliferator-activated receptor alpha (PPARα) transcription factor by fatty acids.66 In a complex “feed-forward” system, PPARα activation enhances the expression of multiple enzymes in the pathways of fatty acid utilization to prevent the accumulation of toxic lipid species. Animals fed a high fat diet usually increase myocardial fatty acid oxidation and maintain near normal cardiac function.54 However, there is also evidence to suggest that inappropriate activation of distinct end-processes of PPARα stimulation in the heart (e.g. fatty acid storage) can be detrimental in the face of specific fatty acid challenges, such as consumption of a diet rich in saturated long chain fatty acids.67 For example, the overexpression of PPARα in the heart mimics features of diabetic cardiomyopathy, 67, 68 while reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in ob/ob mice.48
PPARα is not the only regulator of fatty acid oxidation in the heart. It seems likely that the divergence in the gene cassettes activated to a specific metabolic challenge is mediated through coactivators of the PPAR/retinoic x receptor complex. The PPARγ coactivator-1α (Figure 1) is a master regulator of mitochondrial biogenesis in multiple tissues, including the heart.69 PGC-1α also serves to integrate signaling outcomes of the p38 mitogen activated kinase, β-adrenergic, nitric oxide, AMPkinase and Ca2+-calmodulin kinase signaling pathways to define the energy requirements of the cell.70,71
Figure 1. The Transcriptional Control of Oxidative Metabolism by the Nuclear Receptors.
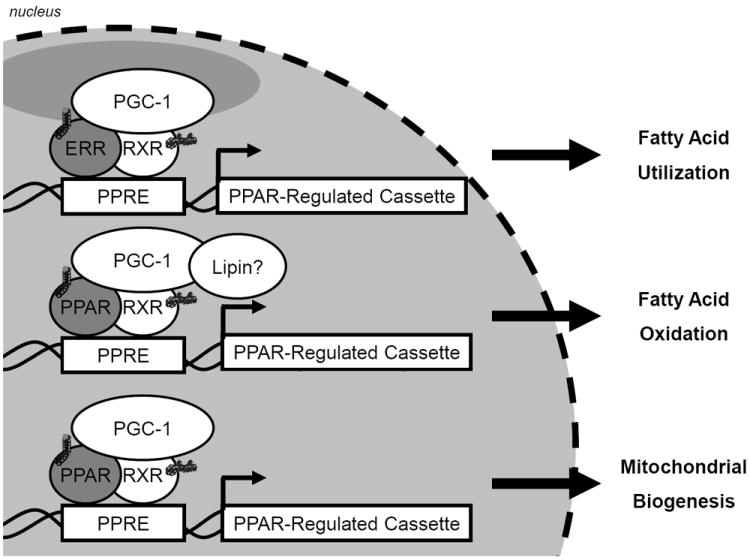
The transcription of distinct cassettes of genes of fatty acid utilization or oxidation are mediated by nuclear receptors (e.g. PPAR and ERR) which require coactivators or corepressors to confer the specificity of response to distinct fatty acid challenges.
Two other PPAR isoforms exist, PPARβ/δ and PPARγ. Like PPARα, PPARβ/δ is also highly expressed in the heart. But, in contrast to mice with cardiac overexpression of PPARα, MHC- PPARβ mice do not develop lipotoxic cardiomyopathy.72 While both nuclear receptors induce expression of genes involved in mitochondrial fatty acid oxidation, PPARβ/δ preferentially induces glucose utilization.72 PPARγ, which is critical for adipocyte differentiation and lipogenesis, is expressed at relatively low levels in the heart and has therefore been ignored for a long time. Although a PPARγ agonist treatment reverses lipotoxic cardiomyopathy, mice overexpressing cardiac PPARγ1 develop a dilated cardiomyopathy associated with increased lipid and glycogen stores.73 In conclusion, the transcriptional regulation of enzymes for fatty acid metabolism is a likely site of dysregulation. When considering ATP- requiring tissues like heart muscle, it is likely that an inadequate mitochondrial activity contributes to the activation of the above mentioned lipotoxic pathways.
Mitochondrial Dysfunction
Mitochondria are the site for β-oxidation of fatty acids. It is known for some time that impaired β-oxidation due to depletion of carnitine induces ‘fatty degeneration’ of the heart.74 More recently, impaired oxidative phosphorylation has been demonstrated in patients with certain forms of genetic cardiomyopathies.75 Today several mouse models of impaired mitochondrial function exist that mimic human disease.76
A potential cause of lipotoxicity is the accumulation of products of incomplete (or inefficient) β-oxidation such as acylcarnitines and ROS.58 Studies by Koves et al. 77 have demonstrated an increase in acylcarnitines in skeletal muscle in a model of diet-induced obesity. The same group suggested the existence of a mitochondria-derived signal that couples incomplete β-oxidation to insulin resistance.78 The authors propose that physical inactivity may be a major contributor of the maladaptive metabolic remodeling of myocytes in response to overnutrition.78 Overexpression of PGC-1α (which promotes increased mitochondrial biogenesis and function) improves the ratio of complete to incomplete oxidation of fatty acids in L6 myoblasts,77 while feeding a high fat diet to mice overexpressing PPARα or activating PPARα in cardiac hypertrophy results in contractile dysfunction.67,79 These results suggest that cardiac maladaptation in obesity may be due to fatty acid-enhanced β-oxidation in the absence of increased energy demand.
An increased reliance on β-oxidation, coupled to a decreased oxidative phosphorylation capacity, is likely to promote the formation of ROS.80 As a consequence of mitochondrial dysfunction, there is excess ROS production and incapacity of mitochondria to reduce superoxide.81 Superoxide is generated from transfer of an electron from the electron transport chain to molecular oxygen (or via NADPH oxidase) and has multiple deleterious consequences to the cell.82 The enzymatic scavenging pathways to reduce superoxide are contained in the mitochondria (and in the cytosol to some extent).82 ROS, in turn, impair Ca2+ homeostasis in isolated cardiomyocytes treated with palmitate.83 The hearts of ob/ob mice exhibit decreased oxidative capacity and decreased protein content of the complexes of the electron transport chain.48 The defects in mitochondria are also present in skeletal muscle of mice with diet-induced obesity.77 Interestingly, oxidative stress is believed to be a major cause of mitochondrial alterations in skeletal muscle of mice fed a high-fat, high-sucrose diet.84 There is, therefore, a noxious feed-forward mechanism in which inadequately enhanced β-oxidation favors the generation of ROS, which will in turn worsen mitochondrial dysfunction.
Uncoupling and Futile Cycles
The heart may have a way to protect itself, at least temporarily, from the deleterious consequences of a high fat supply. We have proposed mitochondrial uncoupling and futile cycling as a major adaptive mechanism of the heart in the setting of high fat feeding.54 One hypothesis is that uncoupling proteins (UCPs) function as a fatty acid anion exporter that preferentially exports those fatty acids that have entered the mitochondrial matrix by a flip-flop mechanism across the inner mitochondrial membrane.85 Because the mitochondrial matrix does not contain ACS isoforms, the negative charge of fatty acid anions would add to the proton gradient (Figure 2). To dissipate this electrical charge, fatty acid anions are exported by UCPs and thus reduce the proton motive force with a net result in the tighter coupling of oxidative phosphorylation with a subsequent decrease in oxidative stress. 85, 86
Figure 2. Putative Role of the Decrease in Fatty Acid-Responsive Genes in the Development of Contractile Dysfunction with Western Diet Compared to High Fat Diet Feeding.
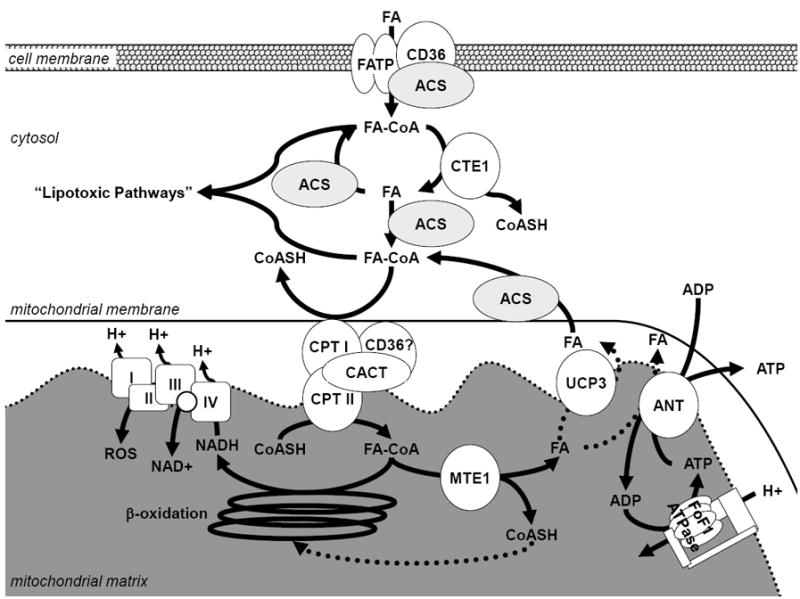
Fatty acid (FA) enters the cell through fatty acid transporters and diffusion, fatty acid transport protein (FATP) and or fatty acid translocase (CD36), and is activated to fatty acyl-CoA (FA-CoA) by an isoform of acyl-CoA synthetase (ACS). The activated Fatty acyl-CoA can undergo hydrolysis (at the indirect expense of ATP) via cytosolic thioesterase 1 (CTE1), which is decreased in the western diet compared to the high fat diet thus decreasing the capacity for fatty acid-mediated futile cycling. After entry of Fatty Acyl-CoAs into the mitochondrion via carnitine palmitoyltransferase (CPT) and carnitine-acylcarnitine translocase (CACT), mitochondrial thioesterase 1 (MTE1) and uncoupling protein 3 (UCP3), or adenine nucleotide translocase (ANT), have the capacity to “uncouple” fat oxidation (indicated with dashed lines), which may result in a increased free CoASH pool for complete β-oxidation and decreased production of reactive oxygen species (ROS) by the electron transport chain. Therefore fatty acyl-CoAs may not accumulate and are not shuttled into “lipotoxic pathways.” Adapted from Wilson et al.54
Similarly, Himms-Hagen and Harper propose that fatty acyl-CoA derivatives are hydrolyzed by mitochondrial thioesterase 1 (MTE-1) in the mitochondrial matrix to release a fatty acid anion and CoASH.87 The CoASH released would be required for other metabolic processes in states of increased oxidation such as reactions needed to maintain fatty acid oxidation (i.e. pyruvate dehydrogenase, ketoglutarate dehydrogenase, and 3-ketoacyl-CoA thiolase)87 (Figure 2). MTE-1 may also promote fatty acid export from cardiac mitochondria. The expression of the MTE-1 protein, as well as its activity, are enhanced in diabetes and by PPARα activation.88, 89 However, the regulation of UCP3 expression by diabetes in these studies is less clear.88, 89 Transcription of ucp3 is fatty acid responsive,90 suggesting fatty acids induce their own futile cycling.
Adenine nucleotide translocase (ANT) has also been proposed to mediate fatty acid efflux from the mitochondrial matrix.91 Fatty acid-induced uncoupling is inhibited by carboxyatractyloside in rat hearts, which correlated with ANT protein content.92 In ANT1-deficient mice, the proton conductance is decreased by up to 50%.86 The dissipation of proton motive force and decrease in cytosolic ATP by palmitate was shown to be regulated by ANT using metabolic control analysis.93 The respective importance of ANT and UCP3 in futile cycling of fatty acids needs to be determined.
Uncoupling and Generation of Reactive Oxygen Species
In line with the above observations we have postulated that the inadequate activation of PPARα responsive genes is a potential mechanism for the development of contractile dysfunction with a Western diet.54 These findings extend previous reports that obese Zucker rat hearts are unable to respond to increased fatty acid availability, showing contractile dysfunction.46 As with deposition of neutral triglyceride in the heart, there is debate whether futile cycling/uncoupling is adaptive or deleterious to cardiac contractile function. Diabetes is known to increase ROS and mitochondrial uncoupling in the heart, and it has been proposed that uncoupling may explain the reduced cardiac efficiency that is measured in diabetes.94 However, contractile function is preserved with a high fat diet where induction of uncoupling components is likely to occur to a greater extent than with a Western diet.54 It is possible that mitochondrial uncoupling is another example of an adaptive mechanism that may, in particular circumstances, become maladaptive.
Conclusion
It is clear that the metabolic dysregulation of obesity is accompanied by adaptive and by maladaptive responses of the heart. The control and regulation of fat or carbohydrate oxidation in the heart is linked to the complex flux of intermediates through distinct pathways that converge at important regulatory complexes or “nodal points” of metabolism. How exactly obesity affects the sites of metabolic regulation in the heart is not completely understood. Likewise, the conditions triggering lipotoxic mechanisms in the heart, as well as the role these mechanisms might have in the development of contractile dysfunction needs better definition. A case in point for adaptation and maladaptation is insulin resistance in the heart. Insulin resistance may be adaptive when it is protecting the heart from excess fuel uptake, or maladaptive when it is associated with ROS formation and activation of signaling pathways of programmed cell death. The present literature reflects an extraordinary complexity of the heart’s metabolic, functional and structural changes in obesity.
Acknowledgments
We thank our reviewers and Dr. Martin E. Young for helpful discussions, as we thank Roxy A. Tate for expert editorial assistance.
Funding
Supported in part by a grant from the National Heart Lung and Blood Institute (RO1-HL073162).
Footnotes
Disclosures
The authors have no conflicts of interest to disclose
Literature Cited
- 1.Virchow R. Cellular pathology as based upon physiological and pathological histology. London: John Churchill; 1860. [DOI] [PubMed] [Google Scholar]
- 2.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc Natl Acad Sci U S A. 2000;97:1784–1789. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Divitiis O, Fazio S, Petitto M, Maddalena G, Contaldo F, Mancini M. Obesity and cardiac function. Circulation. 1981;64:477–482. doi: 10.1161/01.cir.64.3.477. [DOI] [PubMed] [Google Scholar]
- 4.Lauer MS, Anderson KM, Kannel WB, Levy D. The impact of obesity on left ventricular mass and geometry. The framingham heart study. JAMA. 1991;266:231–236. [PubMed] [Google Scholar]
- 5.Alpert MA, Lambert CR, Terry BE, Cohen MV, Mukerji V, Massey CV, Hashimi MW, Panayiotou H. Interrelationship of left ventricular mass, systolic function and diastolic filling in normotensive morbidly obese patients. Int J Obes Relat Metab Disord. 1995;19:550–557. [PubMed] [Google Scholar]
- 6.Peterson LR, Waggoner AD, Schechtman KB, Meyer T, Gropler RJ, Barzilai B, Davila-Roman VG. Alterations in left ventricular structure and function in young healthy obese women: Assessment by echocardiography and tissue doppler imaging. J Am Coll Cardiol. 2004;43:1399–1404. doi: 10.1016/j.jacc.2003.10.062. [DOI] [PubMed] [Google Scholar]
- 7.Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z, Dence C, Klein S, Marsala J, Meyer T, Gropler RJ. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation. 2004;109:2191–2196. doi: 10.1161/01.CIR.0000127959.28627.F8. [DOI] [PubMed] [Google Scholar]
- 8.Leichman JG, Aguilar D, King TM, Vlada A, Reyes M, Taegtmeyer H. Association of plasma free fatty acids and left ventricular diastolic function in patients with clinically severe obesity. Am J Clin Nutr. 2006;84:336–341. doi: 10.1093/ajcn/84.1.336. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe H, Tanabe N, Watanabe T, Darbar D, Roden DM, Sasaki S, Aizawa Y. Metabolic syndrome and risk of development of atrial fibrillation: The niigata preventive medicine study. Circulation. 2008;117:1255–1260. doi: 10.1161/CIRCULATIONAHA.107.744466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Messerli FH, Nunez BD, Ventura HO, Snyder DW. Overweight and sudden death. Increased ventricular ectopy in cardiopathy of obesity. Arch Intern Med. 1987;147:1725–1728. doi: 10.1001/archinte.147.10.1725. [DOI] [PubMed] [Google Scholar]
- 11.Ilercil A, Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Welty TK, Robbins DC, Fabsitz RR, Howard BV, Lee ET. Relationship of impaired glucose tolerance to left ventricular structure and function: The strong heart study. Am Heart J. 2001;141:992–998. doi: 10.1067/mhj.2001.115302. [DOI] [PubMed] [Google Scholar]
- 12.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008;88:389–419. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leichman JG, Lavis VR, Aguilar D, Wilson CR, Taegtmeyer H. The metabolic syndrome and the heart-: A considered opinion. Clin Res Cardiol. 2006;95:i134–i141. doi: 10.1007/s00392-006-1119-7. [DOI] [PubMed] [Google Scholar]
- 14.Avelar E, Cloward TV, Walker JM, Farney RJ, Strong M, Pendleton RC, Segerson N, Adams TD, Gress RE, Hunt SC, Litwin SE. Left ventricular hypertrophy in severe obesity: Interactions among blood pressure, nocturnal hypoxemia, and body mass. Hypertension. 2007;49:34–39. doi: 10.1161/01.HYP.0000251711.92482.14. [DOI] [PubMed] [Google Scholar]
- 15.Lautamaki R, Borra R, Iozzo P, Komu M, Lehtimaki T, Salmi M, Jalkanen S, Airaksinen KE, Knuuti J, Parkkola R, Nuutila P. Liver steatosis coexists with myocardial insulin resistance and coronary dysfunction in patients with type 2 diabetes. Am J Physiol Endocrinol Metab. 2006;291:E282–290. doi: 10.1152/ajpendo.00604.2005. [DOI] [PubMed] [Google Scholar]
- 16.Kankaanpaa M, Lehto HR, Parkka JP, Komu M, Viljanen A, Ferrannini E, Knuuti J, Nuutila P, Parkkola R, Iozzo P. Myocardial triglyceride content and epicardial fat mass in human obesity: Relationship to left ventricular function and serum free fatty acid levels. J Clin Endocrinol Metab. 2006;91:4689–4695. doi: 10.1210/jc.2006-0584. [DOI] [PubMed] [Google Scholar]
- 17.Taegtmeyer H, Wilson CR, Razeghi P, Sharma S. Metabolic energetics and genetics in the heart. Ann N Y Acad Sci. 2005;1047:208–218. doi: 10.1196/annals.1341.019. [DOI] [PubMed] [Google Scholar]
- 18.Perseghin G, Ntali G, De Cobelli F, Lattuada G, Esposito A, Belloni E, Canu T, Costantino F, Ragogna F, Scifo P, Del Maschio A, Luzi L. Abnormal left ventricular energy metabolism in obese men with preserved systolic and diastolic functions is associated with insulin resistance. Diabetes Care. 2007;30:1520–1526. doi: 10.2337/dc06-2429. [DOI] [PubMed] [Google Scholar]
- 19.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. Faseb J. 2004;18:1692–1700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 20.Borisov AB, Ushakov AV, Zagorulko AK, Novikov NY, Selivanova KF, Edwards CA, Russell MW. Intracardiac lipid accumulation, lipoatrophy of muscle cells and expansion of myocardial infarction in type 2 diabetic patients. Micron. 2007 doi: 10.1016/j.micron.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 21.McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, Levine BD, Raskin P, Victor RG, Szczepaniak LS. Cardiac steatosis in diabetes mellitus: A 1h-magnetic resonance spectroscopy study. Circulation. 2007;116:1170–1175. doi: 10.1161/CIRCULATIONAHA.106.645614. [DOI] [PubMed] [Google Scholar]
- 22.Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan R. Obesity and the risk of heart failure. N Engl J Med. 2002;347:305–313. doi: 10.1056/NEJMoa020245. [DOI] [PubMed] [Google Scholar]
- 23.Kenchaiah S, Pocock SJ, Wang D, Finn PV, Zornoff LA, Skali H, Pfeffer MA, Yusuf S, Swedberg K, Michelson EL, Granger CB, McMurray JJ, Solomon SD. Body mass index and prognosis in patients with chronic heart failure: Insights from the candesartan in heart failure: Assessment of reduction in mortality and morbidity (charm) program. Circulation. 2007;116:627–636. doi: 10.1161/CIRCULATIONAHA.106.679779. [DOI] [PubMed] [Google Scholar]
- 24.Davos CH, Doehner W, Rauchhaus M, Cicoira M, Francis DP, Coats AJ, Clark AL, Anker SD. Body mass and survival in patients with chronic heart failure without cachexia: The importance of obesity. J Card Fail. 2003;9:29–35. doi: 10.1054/jcaf.2003.4. [DOI] [PubMed] [Google Scholar]
- 25.Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Woo MA, Tillisch JH. The relationship between obesity and mortality in patients with heart failure. J Am Coll Cardiol. 2001;38:789–795. doi: 10.1016/s0735-1097(01)01448-6. [DOI] [PubMed] [Google Scholar]
- 26.Lavie CJ, Osman AF, Milani RV, Mehra MR. Body composition and prognosis in chronic systolic heart failure: The obesity paradox. Am J Cardiol. 2003;91:891–894. doi: 10.1016/s0002-9149(03)00031-6. [DOI] [PubMed] [Google Scholar]
- 27.Carr DB, Utzschneider KM, Hull RL, Kodama K, Retzlaff BM, Brunzell JD, Shofer JB, Fish BE, Knopp RH, Kahn SE. Intra-abdominal fat is a major determinant of the national cholesterol education program adult treatment panel iii criteria for the metabolic syndrome. Diabetes. 2004;53:2087–2094. doi: 10.2337/diabetes.53.8.2087. [DOI] [PubMed] [Google Scholar]
- 28.Iacobellis G, Sharma AM. Obesity and the heart: Redefinition of the relationship. Obes Rev. 2007;8:35–39. doi: 10.1111/j.1467-789X.2006.00257.x. [DOI] [PubMed] [Google Scholar]
- 29.Poirier P, Martin J, Marceau P, Biron S, Marceau S. Impact of bariatric surgery on cardiac structure, function and clinical manifestations in morbid obesity. Expert Rev Cardiovasc Ther. 2004;2:193–201. doi: 10.1586/14779072.2.2.193. [DOI] [PubMed] [Google Scholar]
- 30.Klein S, Burke LE, Bray GA, Blair S, Allison DB, Pi-Sunyer X, Hong Y, Eckel RH. Clinical implications of obesity with specific focus on cardiovascular disease: A statement for professionals from the american heart association council on nutrition, physical activity, and metabolism: Endorsed by the american college of cardiology foundation. Circulation. 2004;110:2952–2967. doi: 10.1161/01.CIR.0000145546.97738.1E. [DOI] [PubMed] [Google Scholar]
- 31.Mathier MA, Ramanathan RC. Impact of obesity and bariatric surgery on cardiovascular disease. Med Clin North Am. 2007;91:415–431. x–xi. doi: 10.1016/j.mcna.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 32.McCloskey CA, Ramani GV, Mathier MA, Schauer PR, Eid GM, Mattar SG, Courcoulas AP, Ramanathan R. Bariatric surgery improves cardiac function in morbidly obese patients with severe cardiomyopathy. Surg Obes Relat Dis. 2007;3:503–507. doi: 10.1016/j.soard.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 33.Karason K, Wallentin I, Larsson B, Sjostrom L. Effects of obesity and weight loss on left ventricular mass and relative wall thickness: Survey and intervention study. Bmj. 1997;315:912–916. doi: 10.1136/bmj.315.7113.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 35.Unger RH. Minireview: Weapons of lean body mass destruction: The role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144:5159–5165. doi: 10.1210/en.2003-0870. [DOI] [PubMed] [Google Scholar]
- 36.Flier JS. Obesity wars: Molecular progress confronts an expanding epidemic. Cell. 2004;116:337–350. doi: 10.1016/s0092-8674(03)01081-x. [DOI] [PubMed] [Google Scholar]
- 37.Hopkins TA, Ouchi N, Shibata R, Walsh K. Adiponectin actions in the cardiovascular system. Cardiovasc Res. 2007;74:11–18. doi: 10.1016/j.cardiores.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chandran M, Phillips SA, Ciaraldi T, Henry RR. Adiponectin: More than just another fat cell hormone? Diabetes Care. 2003;26:2442–2450. doi: 10.2337/diacare.26.8.2442. [DOI] [PubMed] [Google Scholar]
- 39.Lopaschuk GD, Folmes CD, Stanley WC. Cardiac energy metabolism in obesity. Circ Res. 2007;101:335–347. doi: 10.1161/CIRCRESAHA.107.150417. [DOI] [PubMed] [Google Scholar]
- 40.Graham TE, Yang Q, Bluher M, Hammarstedt A, Ciaraldi TP, Henry RR, Wason CJ, Oberbach A, Jansson PA, Smith U, Kahn BB. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med. 2006;354:2552–2563. doi: 10.1056/NEJMoa054862. [DOI] [PubMed] [Google Scholar]
- 41.Perseghin G, Lattuada G, De Cobelli F, Esposito A, Belloni E, Canu T, Ragogna F, Scifo P, Del Maschio A, Luzi L. Serum retinol-binding protein-4, leptin, and adiponectin concentrations are related to ectopic fat accumulation. J Clin Endocrinol Metab. 2007;92:4883–4888. doi: 10.1210/jc.2007-0325. [DOI] [PubMed] [Google Scholar]
- 42.Steppan CM, Lazar MA. Resistin and obesity-associated insulin resistance. Trends Endocrinol Metab. 2002;13:18–23. doi: 10.1016/s1043-2760(01)00522-7. [DOI] [PubMed] [Google Scholar]
- 43.Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, Watanabe E, Takagi T, Akiyoshi M, Ohtsubo T, Kihara S, Yamashita S, Makishima M, Funahashi T, Yamanaka S, Hiramatsu R, Matsuzawa Y, Shimomura I. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307:426–430. doi: 10.1126/science.1097243. [DOI] [PubMed] [Google Scholar]
- 44.Mathias S, Pena LA, Kolesnick RN. Signal transduction of stress via ceramide. Biochem J. 1998;335(Pt 3):465–480. doi: 10.1042/bj3350465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Unger RH. Hyperleptinemia: Protecting the heart from lipid overload. Hypertension. 2005;45:1031–1034. doi: 10.1161/01.HYP.0000165683.09053.02. [DOI] [PubMed] [Google Scholar]
- 46.Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, Youker KA, Taegtmeyer H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese zucker rat heart. Diabetes. 2002;51:2587–2595. doi: 10.2337/diabetes.51.8.2587. [DOI] [PubMed] [Google Scholar]
- 47.Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 2003;144:3483–3490. doi: 10.1210/en.2003-0242. [DOI] [PubMed] [Google Scholar]
- 48.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 49.Schaffer JE. Lipotoxicity: When tissues overeat. Curr Opin Lipidol. 2003;14:281–287. doi: 10.1097/00041433-200306000-00008. [DOI] [PubMed] [Google Scholar]
- 50.Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK, Dey A, Rothermel B, Kim YB, Kalinowski A, Russell KS, Kim JK. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in c57bl/6 mice. Diabetes. 2005;54:3530–3540. doi: 10.2337/diabetes.54.12.3530. [DOI] [PubMed] [Google Scholar]
- 51.Okere IC, Chandler MP, McElfresh TA, Rennison JH, Kung TA, Hoit BD, Ernsberger P, Young ME, Stanley WC. Carnitine palmitoyl transferase-i inhibition is not associated with cardiac hypertrophy in rats fed a high-fat diet. Clin Exp Pharmacol Physiol. 2007;34:113–119. doi: 10.1111/j.1440-1681.2007.04545.x. [DOI] [PubMed] [Google Scholar]
- 52.Aguila MB, Mandarim-de-Lacerda CA. Heart and blood pressure adaptations in wistar rats fed with different high-fat diets for 18 months. Nutrition. 2003;19:347–352. doi: 10.1016/s0899-9007(02)00934-6. [DOI] [PubMed] [Google Scholar]
- 53.Carroll JF, Zenebe WJ, Strange TB. Cardiovascular function in a rat model of diet-induced obesity. Hypertension. 2006;48:65–72. doi: 10.1161/01.HYP.0000224147.01024.77. [DOI] [PubMed] [Google Scholar]
- 54.Wilson CR, Tran MK, Salazar KL, Young ME, Taegtmeyer H. Western diet, but not high fat diet, causes derangements of fatty acid metabolism and contractile dysfunction in the heart of wistar rats. Biochem J. 2007;406:457–467. doi: 10.1042/BJ20070392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonen A, Chabowski A, Luiken JJ, Glatz JF. Is membrane transport of ffa mediated by lipid, protein, or both? Mechanisms and regulation of protein-mediated cellular fatty acid uptake: Molecular, biochemical, and physiological evidence. Physiology (Bethesda) 2007;22:15–29. doi: 10.1152/physiologyonline.2007.22.1.15. [DOI] [PubMed] [Google Scholar]
- 56.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, Kelly DP. Cd36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100:1208–1217. doi: 10.1161/01.RES.0000264104.25265.b6. [DOI] [PubMed] [Google Scholar]
- 57.Young ME, McNulty P, Taegtmeyer H. Adaptation and maladaptation of the heart in diabetes: Part ii: Potential mechanisms. Circulation. 2002;105:1861–1870. doi: 10.1161/01.cir.0000012467.61045.87. [DOI] [PubMed] [Google Scholar]
- 58.Muoio DM, Newgard CB. Obesity-related derangements in metabolic regulation. Annu Rev Biochem. 2006;75:367–401. doi: 10.1146/annurev.biochem.75.103004.142512. [DOI] [PubMed] [Google Scholar]
- 59.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS, Schaffer JE. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. 2005;96:225–233. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]
- 61.Liu GX, Hanley PJ, Ray J, Daut J. Long-chain acyl-coenzyme a esters and fatty acids directly link metabolism to k(atp) channels in the heart. Circ Res. 2001;88:918–924. doi: 10.1161/hh0901.089881. [DOI] [PubMed] [Google Scholar]
- 62.Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 63.Dey D, Basu D, Roy SS, Bandyopadhyay A, Bhattacharya S. Involvement of novel pkc isoforms in ffa induced defects in insulin signaling. Mol Cell Endocrinol. 2006;246:60–64. doi: 10.1016/j.mce.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 64.Laybutt D, Schmitz-Peiffer C, Saha A, Ruderman N, Biden T, Kraegen E. Muscle lipid accumulation and protein kinase c activation in the insulin-resistant chronically glucose-infused rat. Am J Physiol. 1999;277:E1070–E1076. doi: 10.1152/ajpendo.1999.277.6.E1070. [DOI] [PubMed] [Google Scholar]
- 65.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase c, and ikappab-alpha. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 66.Barger PM, Kelly DP. Ppar signaling in the control of cardiac energy metabolism. Trends Cardiovasc Med. 2000;10:238–245. doi: 10.1016/s1050-1738(00)00077-3. [DOI] [PubMed] [Google Scholar]
- 67.Finck BN, Han X, Courtois M, Aimond F, Nerbonne J, Kovacs A, Gross RW, Kelly DP. A critical role for pparalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: Modulation by dietary fat content. Proc Natl Aad Sci USA. 2003;100:1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by pparalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004;95:568–578. doi: 10.1161/01.RES.0000141774.29937.e3. [DOI] [PubMed] [Google Scholar]
- 71.Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: A question of balance. J Clin Invest. 2005;115:547–555. doi: 10.1172/JCI200524405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM, Shoghi K, Welch MJ, Kelly DP. Nuclear receptors pparbeta/delta and pparalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117:3930–3939. doi: 10.1172/JCI32578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, Homma S, Szabolcs MJ, Huang LS, Goldberg IJ. Cardiomyocyte expression of ppargamma leads to cardiac dysfunction in mice. J Clin Invest. 2007;117:2791–2801. doi: 10.1172/JCI30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wittels B, Bressler R. Biochemical lesion of diphtheria toxin in the heart. J Clin Invest. 1964;43:630. doi: 10.1172/JCI104948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kelly DP, Strauss AW. Inherited cardiomyopathies. N Engl J Med. 1994;330:913–919. doi: 10.1056/NEJM199403313301308. [DOI] [PubMed] [Google Scholar]
- 76.Wallace DC. Mouse models for mitochondrial disease. Am J Med Genet. 2001;106:71–93. doi: 10.1002/ajmg.1393. [DOI] [PubMed] [Google Scholar]
- 77.Koves TR, Li P, An J, Akimoto T, Slentz D, Ilkayeva O, Dohm GL, Yan Z, Newgard CB, Muoio DM. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem. 2005;280:33588–33598. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- 78.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 79.Young ME, Laws FA, Goodwin GW, Taegtmeyer H. Reactivation of peroxisome proliferator-activated receptor alpha is associated with contractile dysfunction in hypertrophied rat heart. J Biol Chem. 2001;276:44390–44395. doi: 10.1074/jbc.M103826200. [DOI] [PubMed] [Google Scholar]
- 80.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 81.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 82.Fridovich I. The biology of oxygen radicals. Science. 1978;201:875–880. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- 83.Fauconnier J, Andersson DC, Zhang SJ, Lanner JT, Wibom R, Katz A, Bruton JD, Westerblad H. Effects of palmitate on ca(2+) handling in adult control and ob/ob cardiomyocytes: Impact of mitochondrial reactive oxygen species. Diabetes. 2007;56:1136–1142. doi: 10.2337/db06-0739. [DOI] [PubMed] [Google Scholar]
- 84.Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, Vidal H, Rieusset J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schrauwen P, Saris WH, Hesselink MK. An alternative function for human uncoupling protein 3: Protection of mitochondria against accumulation of nonesterified fatty acids inside the mitochondrial matrix. Faseb J. 2001;15:2497–2502. doi: 10.1096/fj.01-0400hyp. [DOI] [PubMed] [Google Scholar]
- 86.Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins ucp2 and ucp3. Cell Metab. 2005;2:85–93. doi: 10.1016/j.cmet.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 87.Himms-Hagen J, Harper ME. Physiological role of ucp3 may be export of fatty acids from mitochondria when fatty acid oxidation predominates: An hypothesis. Exp Biol Med (Maywood) 2001;226:78–84. doi: 10.1177/153537020122600204. [DOI] [PubMed] [Google Scholar]
- 88.Gerber LK, Aronow BJ, Matlib MA. Activation of a novel long-chain free fatty acid generation and export system in mitochondria of diabetic rat hearts. Am J Physiol Cell Physiol. 2006;291:C1198–1207. doi: 10.1152/ajpcell.00246.2006. [DOI] [PubMed] [Google Scholar]
- 89.King KL, Young ME, Kerner J, Huang H, O’Shea KM, Alexson SE, Hoppel CL, Stanley WC. Diabetes or peroxisome proliferator-activated receptor alpha agonist increases mitochondrial thioesterase i activity in heart. J Lipid Res. 2007;48:1511–1517. doi: 10.1194/jlr.M600364-JLR200. [DOI] [PubMed] [Google Scholar]
- 90.Young ME, Patil S, Ying J, Depre C, Ahuja HS, Shipley GL, Stepkowski SM, Davies PJ, Taegtmeyer H. Uncoupling protein 3 transcription is regulated by peroxisome proliferator-activated receptor (alpha) in the adult rodent heart. Faseb J. 2001;15:833–845. doi: 10.1096/fj.00-0351com. [DOI] [PubMed] [Google Scholar]
- 91.Skulachev VP. Anion carriers in fatty acid-mediated physiological uncoupling. J Bioenerg Biomembr. 1999;31:431–445. doi: 10.1023/a:1005492205984. [DOI] [PubMed] [Google Scholar]
- 92.Schonfeld P. Does the function of adenine nucleotide translocase in fatty acid uncoupling depend on the type of mitochondria? FEBS Lett. 1990;264:246–248. doi: 10.1016/0014-5793(90)80259-l. [DOI] [PubMed] [Google Scholar]
- 93.Ciapaite J, Bakker SJ, Diamant M, van Eikenhorst G, Heine RJ, Westerhoff HV, Krab K. Metabolic control of mitochondrial properties by adenine nucleotide translocator determines palmitoyl-coa effects. Implications for a mechanism linking obesity and type 2 diabetes. Febs J. 2006;273:5288–5302. doi: 10.1111/j.1742-4658.2006.05523.x. [DOI] [PubMed] [Google Scholar]
- 94.Boudina S, Abel ED. Mitochondrial uncoupling: A key contributor to reduced cardiac efficiency in diabetes. Physiology (Bethesda) 2006;21:250–258. doi: 10.1152/physiol.00008.2006. [DOI] [PubMed] [Google Scholar]