

Abstract
The elderly patients show a significantly elevated mortality rate during sepsis than younger patients, due to their higher propensity to microvascular dysfunction and consequential multiorgan failure. We tested whether aging renders vascular endothelial cells more susceptible to damage induced by inflammatory factors present in the circulation during sepsis. Primary microvascular endothelial cells derived from young (3 months) and aged (24 months) Fischer 344 × Brown Norway rats were treated with sera obtained from sepsis patients and healthy controls. Oxidative stress (MitoSox fluorescence), death receptor activation (caspase 8 activity), and apoptotic cell death (caspase 3 activity) induced by treatment with septic sera were exacerbated in aged endothelial cells as compared with responses obtained in young cells. Induction of heme oxygenase-1 and thrombomodulin in response to treatment with septic sera was impaired in aged endothelial cells. Treatment with septic sera elicited greater increases in tumor necrosis factor-α expression in aged endothelial cells, as compared with young cells, whereas induction of inducible nitric oxide synthase, intercellular adhesion molecule-1, and vascular cell adhesion molecule did not differ between the two groups. Collectively, aging increases sensitivity of microvascular endothelial cells (MVECs) to oxidative stress and cellular damage induced by inflammatory factors present in the circulation during septicemia. We hypothesize that these responses may contribute to the increased vulnerability of elderly patients to multiorgan failure associated with sepsis.
Key Words: Aging, Endothelial cells, Microcirculation, Microvascular injury, Bacteremia, Septicemia, Sepsis, Septic shock.
SEPSIS is a life-threatening systemic inflammatory disorder, which develops in response to an infection. Patients with severe sepsis exhibit refractory hypotension and disseminated intravascular coagulation that lead to multiorgan failure and, often, death. Epidemiological studies show that both the incidence and severity of sepsis significantly increase with age (1–4). Indeed, in developed Western countries, the median age of intensive care unit patients diagnosed with severe sepsis is close to 65 years of age (1,5,6). Importantly, geriatric patients also exhibit a significantly increased mortality of sepsis compared with young patients (1–4). Laboratory studies replicate these findings, demonstrating significant age-related increases in mortality to sepsis in rodents (7–10). The mechanisms underlying increased organismal vulnerability to sepsis in aging remain largely unknown (4).
Recent studies suggest that age-related increase in sepsis mortality, at least in part, is due to the higher propensity to microvascular thrombosis, inflammation, and consequential multiorgan failure in aged animals (10–14). Previous studies have shown that sepsis is associated with significant oxidative–nitrosative stress in the vascular endothelium (15–17). Furthermore, we have recently demonstrated that blood vessels of aged rodents and nonhuman primates exhibit impaired cellular resistance to oxidative injury (18–20). Yet, there are no studies extant investigating age-related alterations in endothelial sensitivity to sepsis-induced oxidative stress and cellular injury.
This study was designed to test the hypothesis that aging increases the vulnerability of MVECs to the deleterious effects of inflammatory factors present in the circulation of patients during sepsis. We assessed the prooxidative, proinflammatory, and proapoptotic effects of treatment with sera derived from sepsis patients and healthy control participants using cultured MVECs isolated from young and aged Fischer 344 × Brown Norway (F344 × BN) rats, as a model system.
Methods
Establishment and Characterization of Primary MVEC Cultures
F344 × BN rats were used as a model of aging, as this strain has a lower incidence of age-specific pathology than other rat strains. Male, 3- and 24-month-old, F344 × BN rats were obtained from the National Institute on Aging. All animals were disease free with no signs of systemic inflammation and/or neoplastic diseases. The rats were housed in an environmentally controlled vivarium under pathogen-free conditions with unlimited access to food and water and a controlled photoperiod (12 hour light; 12 hour dark). All rats were maintained according to National Institutes of Health guidelines, and all animal use protocols were approved by the Institutional Animal Care and Use Committees of the participating institutions. The animals were euthanized with CO2. To establish primary MVECs, the brains were removed aseptically, rinsed in ice-cold phosphate-buffered saline (PBS) and minced into ≈1mm squares. The tissue was washed twice in ice-cold 1× PBS by low-speed centrifugation (50g, 2–3 minutes). The diced tissue was digested in a solution of collagenase (800U/g tissue), hyaluronidase (2.5U/g tissue), and elastase (3U/g tissue) in 1mL PBS/100mg tissue for 45 minutes at 37°C in rotating humid incubator. The digested tissue was passed through a 100 µm cell strainer to remove undigested blocks. The single cell lysate was centrifuged for 2 minutes at 70g. After removing the supernatant carefully, the pellet was washed twice in cold PBS supplemented with 2.5% fetal calf serum (FCS), and the suspension centrifuged at 300g, for 5 minutes at 4°C. To create an endothelial cell–enriched fraction, the cell suspension was gradient centrifuged by using OptiPrep solution (Axi-Shield, PoC, Norway). Briefly, the cell pellet was resuspended in Hanks’ balanced salt solution and mixed with 40% iodixanol thoroughly (final concentration: 17% [w/v] iodixanol solution; ρ = 1.096g/mL). Two millilitres of Hanks’ balanced salt solution was layered on top and centrifuged at 400g for 15 minutes at 20°C. Endothelial cells, which banded at the interface between Hanks’ balanced salt solution and the 17% iodixanol layer, were collected. The endothelial cell–enriched fraction was incubated for 30 minutes at 4°C in dark with anti-CD31/PE (BD Biosciences, San Jose, CA), anti-MCAM/FITC (BD Biosciences). After washing the cells twice with MACS buffer (Milltenyi Biotech, Cambridge, MA), anti-FITC magnetic bead–labeled and anti-PE magnetic bead–labeled secondary antibodies were used for 15 minutes at room temperature. Endothelial cells were collected by magnetic separation using the MACS LD magnetic separation columns according to the manufacturer’s guidelines (Milltenyi Biotech). The endothelial fraction was cultured on fibronectin-coated plates in endothelial growth medium (Cell Application, San Diego, CA) for 10 days. Endothelial cells were phenotypically characterized by flow cytometry (GUAVA 8HT, Merck Millipore, Billerica, MA). Briefly, antibodies against five different endothelial-specific markers were used (anti-CD31-PE, anti-erythropoietin receptor-APC, anti-VEGF R2-PerCP, anti-ICAM-fluorescein, and anti-CD146-PE) and isotype-specific antibody-labeled fractions served as negative controls. Flow cytometric analysis showed that after the third cycle of immunomagnetic selection, there were virtually no CD31-, CD146-, EpoR-, and VEGFR2-cells in the resultant cell populations. All antibodies were purchased from R&D Systems (Minneapolis, MN).
Collection of Sera and Treatment of Endothelial Cells
This study was approved by the ethics committees of the participating institutions. All participants were enrolled after informed consent was obtained. Patients (n = 67) were diagnosed as having sepsis, severe sepsis, or septic shock according to the criteria of the American College of Chest Physicians/Society of Critical Care Medicine consensus conference, as described (21,22). After providing informed consent, patients older than 18 years were enrolled within the first 72 hours of the diagnosis of sepsis or 48 hours after the first organ dysfunction (severe sepsis) or refractory hypotension (septic shock). Patients were excluded from the study if they were known to be infected with human immunodeficiency virus, have any neoplastic disease, had received immunosuppressive agents, or were at risk of imminent death. The epidemiological data of the cohort studied have been previously reported (21,22). In brief, the mean age was 63.1±17.3 years, and 62.7% were men. The primary sources of infection involved the lung (41.8%), abdomen (25.4%), and the urinary tract (13.4%). The mean age of healthy volunteers (n = 32) was 59.6±16.4 years and 62.5% were men. Serum samples obtained from septic patients and healthy volunteers were stored at −80°C.
Primary MVECs were initially cultured in MesoEndo Endothelial Cell Growth Medium (Cell Applications, Inc.) followed by endothelial basal medium supplemented with 10% FCS until the time of serum treatment, as described (23–27). For treatment, FCS was replaced with serum (10%) from sepsis patients or from control participants. Cells cultured in endothelial basal medium supplemented with 10% FCS served as an additional internal control. All reagents used in this study were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated.
Measurement of Mitochondrial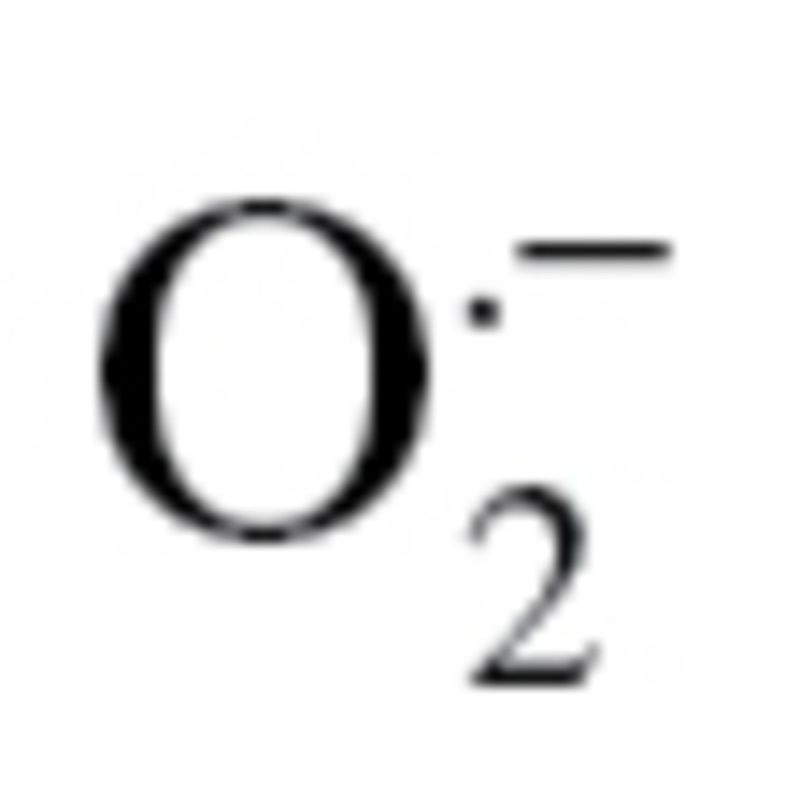 Production
Production
Mitochondrial production in cultured cells from young and aged rats was measured by flow cytometry (Guava 8HT) using MitoSOX Red (Life Technologies), a mitochondrion-specific hydroethidine-derivative fluorescent dye (28,29), as previously reported (23,26,30–32). After 24 hour treatment with human sera (control vs septic), primary endothelial cells were incubated with MitoSox (5 μmol/L at 37°C for 30 minutes), then centrifuged, and washed with PBS. Cell debris (low forward and side scatter) and dead cells (Sytox Green positive) were gated out prior to analysis. The data are presented as mean intensity of MitoSOX fluorescence, normalized to the respective mean fluorescence intensities obtained in FCS-treated rat endothelial cells.
Assessment of Apoptotic Cell Death
Caspase 3/7 and caspase 8 activities in young and aged endothelial cell lysates were measured using Caspase-Glo 3/7 and Caspase-Glo 8 assay kits according to the manufacturer’s guidelines (Promega, Madison, WI) as previously reported (33–35).
Quantitative Real-Time RT-PCR
A quantitative real-time RT-PCR technique was used to analyze mRNA expression of Hmox1, Thbd, Nos2, Icam1, Vcam, Tnfa, and Il6 in sera-treated MVECs, as previously reported (34,36–39). In brief, total RNA was isolated with a Power SYBR Green Cells-to-Ct Kit (Invitrogen) and was reverse transcribed using the same Kit. mRNA expression was analyzed using a Strategen MX3000 platform. Amplification efficiencies were determined using the dilution series of a standard vascular sample. Quantification was performed using the efficiency-corrected ΔΔCq method. The relative quantities of the reference genes Gapdh, Hprt, and Actb were determined, and a normalization factor was calculated based on the geometric mean for internal normalization. Oligonucleotides used for quantitative real-time RT-PCR are listed in Table 1. Fidelity of the PCR reaction was determined by melting temperature analysis and visualization of product on a 2% agarose gel.
Table 1.
Oligonucleotides Used for Real-Time RT-PCR
| mRNA targets | Description | Sense | Antisense |
|---|---|---|---|
| Hmox1 | Heme oxygenase 1 (HO-1) | GGCTGTGAACTCTGTCTC | GGCATCTCCTTCCATTCC |
| Thbd | Thrombomodulin | GGGACTTTGCTTTAATGAA | CCAATTCTTGTGTATAGGC |
| Nos2 | Inducible nitric oxide synthase (iNOS) | CTCTTTCCTTTGCCTCATACTTCC | GCTAAATATTAGAGCAGCGGGATG |
| Icam-1 | Intercellular adhesion molecule-1 | CACAGCCTGGAGTCTC | CCCTTCTAAGTGGTTGGAA |
| Vcam | Vascular cell adhesion molecule 1 | CAGCCTAATTTTATTCCTATGC | GAAACACGTCCCCGTA |
| Tnf-α | Tumor necrosis factor-α | AACCACCAAGCAGAGGAG | CTTGATGGCGGAGAGGAG |
| IL-6 | Interleukin-6 | TACCCCAACTTCCAATGC | GATACCCATCGACAGGAT |
| Hprt | Hypoxanthine phosphoribosyltransferase 1 | AAGACAGCGGCAAGTTGAATC | AAGGGACGCAGCAACAGAC |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | CCAAGGAGTAAGAAACCC | TTGATGGTATTCGAGAGAAGG |
| ACTB | Beta actin | GAAGTGTGACGTTGACAT | ACATCTGCTGGAAGGTG |
Data Analysis
Statistical analyses of data were performed by one-way ANOVA. p < .05 was considered statistically significant. Data are expressed as means ± SEM.
Results
Effect of Treatment with Septic Sera on Programmed Cell Death in Cultured MVECs
Induction of endothelial apoptosis is an important mechanism that contributes to microvascular injury in sepsis. Our results show that MVECs derived from aged rats tend to exhibit higher level of caspase 3/7 and caspase 8 activity, as compared with young cells in the presence of control sera (Figure 1A and 1B). Treatment with septic sera induced apoptosis both in young and aged MVECs, as shown by the increased caspase 3/7 and caspase 8 activities (Figure 1A and 1B). The increase in caspase 3/7 and caspase 8 activities in aged cells upon treatment with septic sera was significantly greater than that in young cells, indicating an age-related exacerbation of endothelial apoptosis under septic conditions (Figure 1A and 1B).
Figure 1.
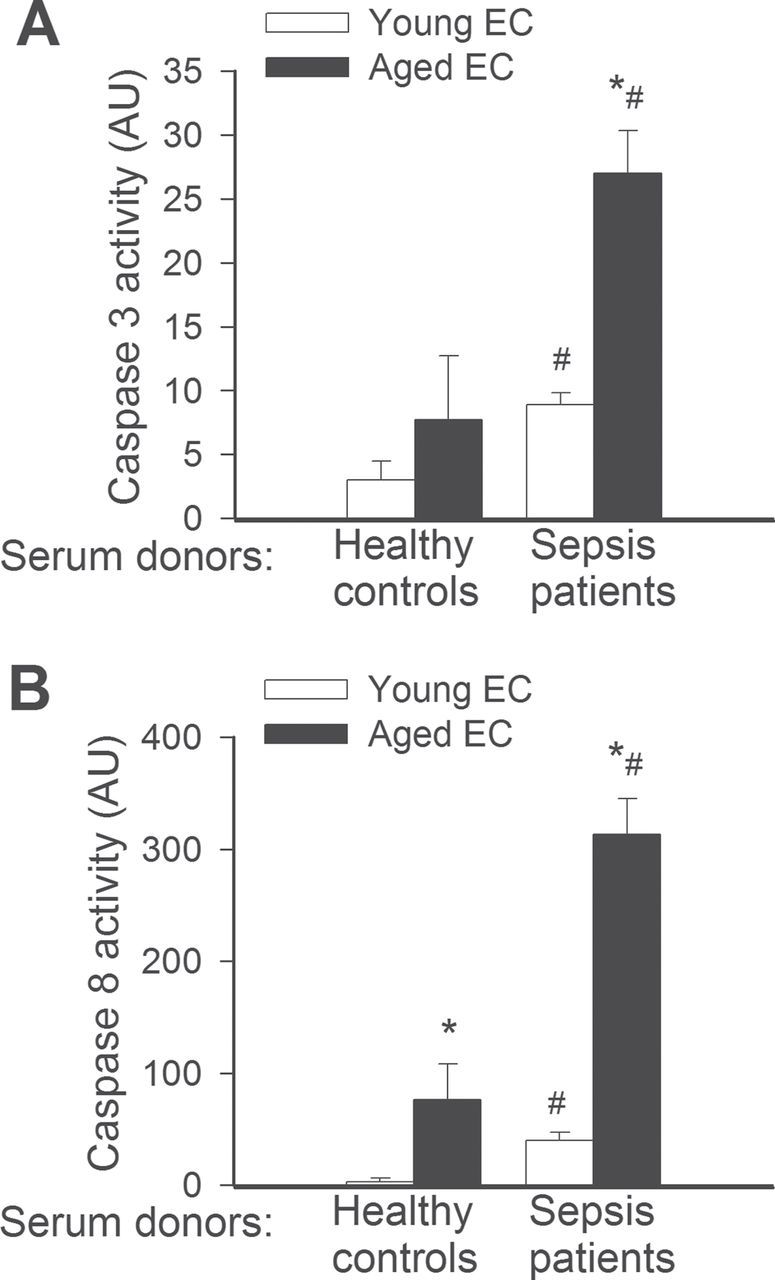
(A) Treatment with sera collected from septic patients elicits significantly greater increases in apoptosis in aged endothelial cells (EC) than in young cells. Apoptotic cell death was assessed by measuring caspase 3 activity in cell lysates. *p < .05 vs young EC, #* < .05 vs control serum treated. Data are mean ± SEM (n = 10–16 for each group). (B) Treatment with sera collected from septic patients elicits significantly greater activation of the death receptor pathway in aged ECs than in young cells. Death receptor activation was assessed by measuring caspase 8 activity in cell lysates. Note that aged cells treated with sera of healthy control paticipants also exhibited more caspase 8 activity than the respective young controls. *p < .05 vs young EC, #*< .05 vs control serum treated. Data are mean ± SEM (n = 10–16).
Effect of Treatment With Septic Sera on Oxidative Stress in Cultured MVECs
On the basis of recent advances in understanding of the apoptotic process, microvascular aging and age-related changes in organ function (18,40–47), we focused on the effects of aging on septic sera-induced mitochondrial oxidative stress. Increased mitochondrial oxidative stress has been shown to impair endothelial function, induce inflammatory gene expression, and promote apoptosis in endothelial cells. MVECs derived from aged rats exhibited higher level of mitochondrial reactive oxygen species (ROS) production as compared with young cells in the presence of control sera (Figure 2). Treatment of both young and aged MVECs with septic sera resulted in a significant increase in mitochondrial oxidative stress. The increase in MitoSox fluorescence in aged cells upon treatment with septic sera was significantly greater than that in young cells, indicating an age-related exacerbation of mitochondrial ROS production under septic conditions (Figure 2).
Figure 2.

Treatment with sera collected from septic patients elicits significantly greater increases in mitochondrial oxidative stress in aged endothelial cells (EC) than in young cells. Mitochondrial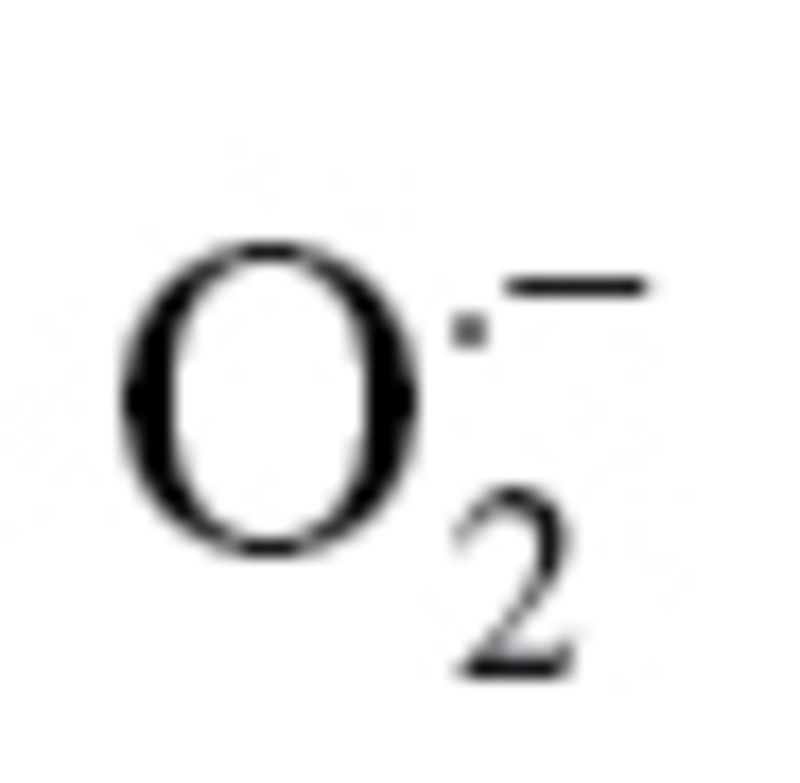 production was assessed using the MitoSox red fluorescence method. *p < .05 vs young ECs, #*< 0.05 vs control serum treated. Data are mean ± SEM (n = 10–16).
production was assessed using the MitoSox red fluorescence method. *p < .05 vs young ECs, #*< 0.05 vs control serum treated. Data are mean ± SEM (n = 10–16).
Effect of Treatment with Septic Sera on Expression of Cytoprotective Factors and Inflammatory Markers in Cultured MVECs
To assess the effect of aging and factors present in the circulation of septic patients on endothelial expression of cytoprotective factors and inflammatory markers, we analyzed mRNA expression of Hmox1, Thbd, Nos2, Icam1, Vcam, Tnfa, and Il6. We found that sera from septic patients upregulated expression of Hmox1 (Figure 3A) and Thbd (Figure 3B) in young endothelial cells, but not in aged cells. The expression levels of Nos2, Icam1, Vcam (Figure 4A–4C), and Il6 (Figure 5B) did not differ between septic sera-treated young and aged MVECs. In contrast, after septic sera treatment, expression of Tnfa was significantly increased in aged MVECs as compared with that in young cells (Figure 5A).
Figure 3.
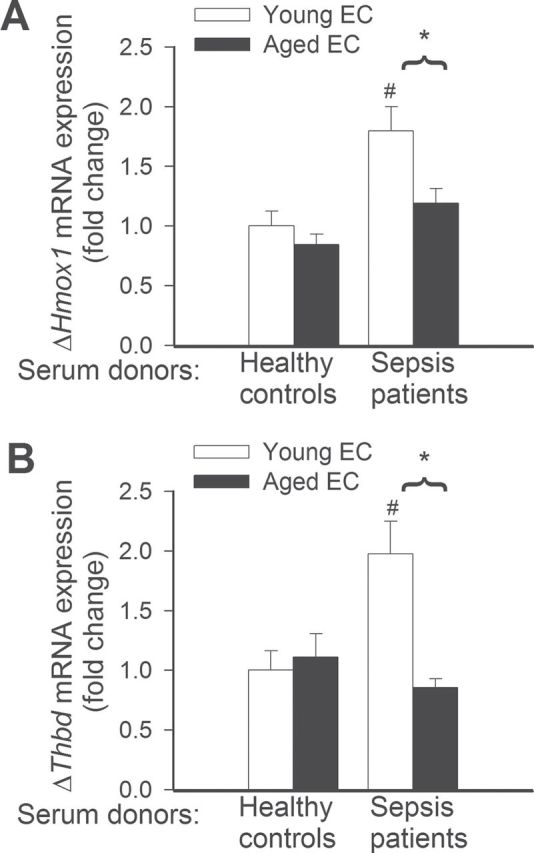
Quantitative real-time RT-PCR data showing that treatment with sera collected from septic patients elicits significantly greater induction of mRNA expression of Hmox1 (A) and Thbd (B) in young endothelial cells (EC) than in aged ECs. *p < .05 vs young ECs, # *< .05 vs control serum treated. Data are mean ± SEM (n = 10–20).
Figure 4.
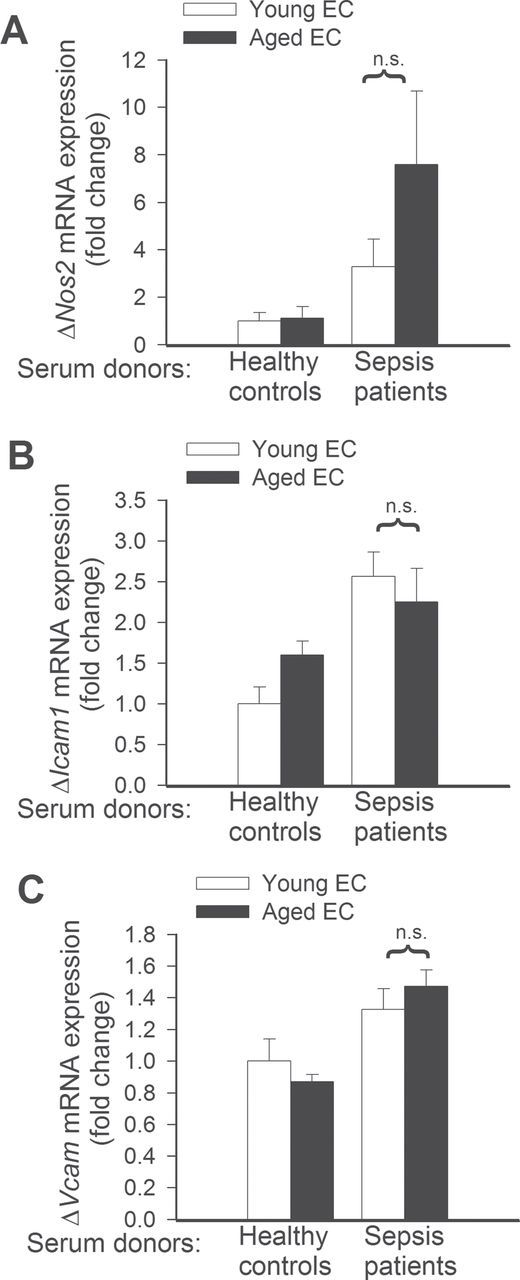
Quantitative real-time RT-PCR data showing that changes in mRNA expression of Nos2 (A), Icam1 (B), and Vcam (C), induced by treatment with sera collected from septic patients, in young and aged endothelial cells (EC) are not significantly different (n.s.). Data are mean ± SEM (n = 10–20).
Figure 5.
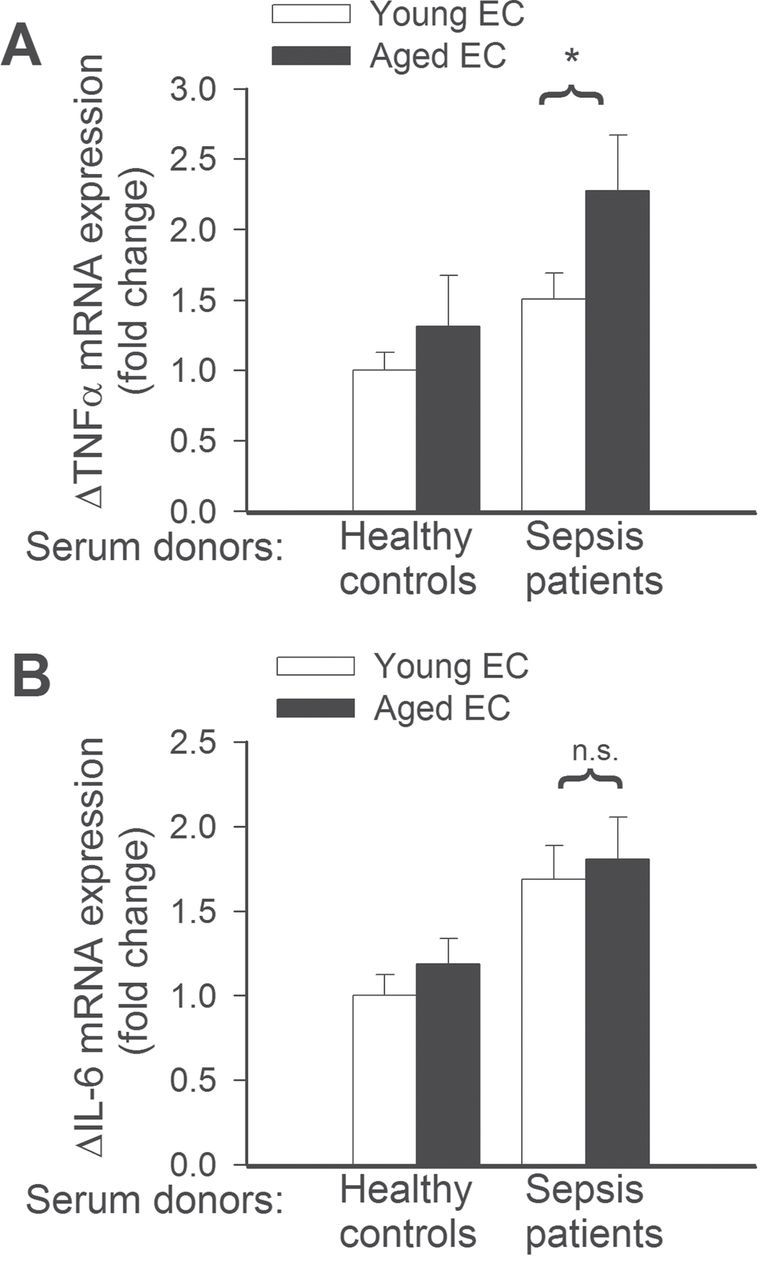
(A) Quantitative real-time RT-PCR data showing that treatment with sera collected from septic patients elicits significantly greater induction of mRNA expression of tumor necrosis factor-α in aged endothelial cells (EC) than in young ECs. *p < .05 vs young ECs. Data are mean ± SEM (n = 10–20). (B) Changes in mRNA expression of interleukin-6 induced by treatment with sera collected from septic patients are not significantly different (n.s.) between young and aged ECs. Data are mean ± SEM (n = 10–20).
Discussion
Serious complications, including oxidative damage to the microvascular endothelium and increased intravascular coagulation, are involved in multiple organ failure, leading to death of septic patients (48). The recent study reported that age-specific incidence of severe sepsis with multiorgan dysfunction increases ~100-fold in older patients (1). There are multiple age-related risk factors that contribute to the increasing incidence of severe sepsis in elderly patients (49), including immunological impairment (e.g., abnormal B and T cell function), comorbidities that require instrumentation (e.g., indwelling urinary catheters, central venous catheters, tracheotomies, peripherally inserted catheters), nutritional problems (malnutrition is commonplace in elderly patients as a result of inactivity, poor mobility, poor diets, chronic disease, dementia, depression, poor dentition, and polypharmacy), oropharyngeal colonization with gram-negative bacilli and institutionalization (in long-term care facilities bacterial flora often demonstrate a level of resistance higher than that seen in the community). Mortality also steadily increases as patients age, with a peak of 38.4% in patients aged older than 85 years (1). The mechanisms underlying age-related alterations in the response of the body to systemic inflammation, resulting in multiple organ failure and increased mortality of severe sepsis in the elderly patients are less well understood.
Recent studies led to the development of the hypothesis that age-related alterations of the microvascular endothelium contribute to increased mortality of sepsis in aged rodents (10–12,14). This study was designed to test a key prediction of this hypothesis, namely, that aging increases the vulnerability of MVECs to oxidative injury induced by inflammatory factors present in the circulation during sepsis. To test this hypothesis, we treated young and aged cultured primary MVECs with sera obtained from sepsis patients and healthy control participants. We focused on the effects of aging on septic sera–induced endothelial apoptosis, oxidative stress, and proinflammatory gene expression.
There is increasing evidence suggesting that endothelial cell apoptosis plays a role in the pathogenesis of sepsis-induced multiorgan failure (48). Here, we demonstrate that systemic factors present in the circulation of septic patients induce apoptosis in MVECs (Figure 1). Our results extend the findings of previous studies demonstrating that treatment of pulmonary MVECs with plasma collected from endotoxemic mice also results in significant increases in the rate of apoptosis (50). It is likely that multiple factors present in the sera of septic patients contribute to activation of programmed cell death in MVECs, including secreted cytokines (eg, tumor necrosis factor-α) and bacterial products (endotoxin, exotoxins of gram-positive bacteria, pore forming toxins, superantigens of gram-positive bacteria, components from the gram-positive bacterial cell wall, including soluble peptidoglycan and lipoteichoic acids) mediated by toll-like receptors (51). Accordingly, abundant in vitro studies suggest that endothelial cell apoptosis can occur in response to administration of tumor necrosis factor-α (52), bacterial lipopolysaccharide and certain pathogenic organisms as well (53,54). Importantly, lipopolysaccharide was recently reported to disrupt ultrastructure of brain MVECs and to induce endothelial apoptosis (55). Interestingly, endothelial apoptosis in response to treatment with septic sera is associated with activation of caspase 8 (Figure 1B). The caspase 8-dependent pathway of apoptosis is activated by ligand binding–induced trimerization of death receptors, which results in recruitment of the Fas-associated death domain (FADD). Activated caspase 8 is known to propagate the apoptotic signal either by directly cleaving and activating downstream caspases or by activating the mitochondrial pathway of apoptosis. It is significant that in vivo delivery of caspase 8-siRNA was reported to decrease mortality in septic mice (56). Yet, it remains to be determined whether the aforementioned protective effect was dependent upon the inhibition of endothelial apoptosis in the animal model used. Endothelial cells undergoing apoptosis express an increasingly procoagulant phenotype, promote leukocyte extravasation, and increase vascular permeability (48), all of which likely play a role in the development of sepsis-induced multiorgan dysfunction. Importantly, we found that aged endothelial cells have an increased propensity to undergo apoptosis in response to treatment with septic sera, likely due to an increased activation of caspase 8-dependent pathways (Figure 1). We propose that increased endothelial apoptosis contributes to the increased vulnerability and mortality of elderly patient with sepsis. Previous studies demonstrated that apoptosis is also an important mechanism of lymphocyte and gastrointestinal epithelial cell death in sepsis (57–60). Thus, further studies are warranted to test whether aging also increases the propensity of the aforementioned cell types to undergo apoptosis in response to factors present in the circulation of septic patients.
Oxidative stress plays a crucial role in endothelial injury during the pathogenesis of sepsis (11). Here, we demonstrate that systemic factors present in the circulation of sepsis patients induce significant oxidative stress in MVECs independent of the effects of sepsis mediated by circulating cells (Figure 2), extending previous findings (61). In patients with sepsis, endothelial oxidative and nitrosative stress is thought to promote endothelial apoptosis and induce endothelial dysfunction impairing microvascular perfusion (16,62). Endothelial oxidative stress also promotes cellular energetic failure and proinflammatory responses by activating the nuclear enzyme poly(ADP-ribose) polymerase-1 (63–67). Here, we provide new evidence that aging exacerbates endothelial oxidative stress induced by inflammatory factors present in the circulation during sepsis (Figure 2), which likely plays a role in induction of apoptosis and predispose aged individuals to microcirculatory failure in sepsis. This finding accords with the results of previous studies showing that during endotoxemia aged mice, compared with young mice, exhibit significantly higher level of oxidative–nitrative stress in the pulmonary vascular endothelium (11).
The mechanisms underlying the increased vulnerability of aged endothelial cells to oxidative stress-mediated injury are not completely understood. Recently, we have shown that in aged vessels increased production of reactive oxygen species fails to activate the transcription factor nuclear factor-erythroid 2–related factor 2 (Nrf2), which has a central role in regulating cellular oxidative stress resistance by controlling the expression of numerous genes for proteins (eg, heme oxygenase-1) that participate in detoxification of ROS during stress conditions (18–20). We demonstrated that the lack of ROS-induced adaptive Nrf2-driven upregulation of antioxidant enzymes contributes to the impaired oxidative stress resistance of the aged vasculature under conditions of metabolic stress (18–20). Nrf2 also confers important anti-inflammatory effects through upregulation of heme oxygenase-1 (68,69). Our findings that sera from septic patients upregulate the Nrf2 target gene heme oxygenase-1 in young endothelial cells, but not in aged cells (Figure 3A), support the idea that age-related vascular Nrf2 dysfunction may contribute to the increased vulnerability of aged animals to sepsis. Further support for this concept is provided by studies showing that genetic disruption of Nrf2 signaling dramatically increases mortality of mice in response to experimentally induced septic shock (70–74).
Thrombomodulin is a glycoprotein expressed on the surface of endothelial cells, which exerts significant vasoprotective and anticoagulant effects in sepsis by serving as a cofactor in the thrombin-induced activation of protein C. Increased shedding of thrombomodulin from the endothelial surface in sepsis associates with the development of multiorgan failure (75,76). Interestingly, we found that following treatment with septic sera expression of thrombomodulin was decreased in aged endothelial cells as compared with that in young cells (Figure 3B). This finding is potentially important as previous studies demonstrated that an age-dependent loss of thrombomodulin in pulmonary microvessels during endotoxemia may contribute to augmented coagulation and increased mortality in aged mice (10). Furthermore, there is clinical evidence that administration of recombinant human thrombomodulin may prevent multiorgan dysfunction in patients with sepsis-induced DIC (77). The factor(s) that regulate endothelial thrombomodulin expression and release under septic conditions may include heme oxygenase-1-dependent pathways (78) and inflammatory cytokines (eg, tumor necrosis factor-α [75]).
Increased ROS production was shown to promote endothelial activation and vascular inflammation by activating redox-sensitive transcription factors including nuclear factor kappa-B (NF-κB [79–81]). As predicted, in septic sera–treated endothelial cells increased oxidative stress tended to be associated with an increased expression of inducible nitric oxide synthase, adhesion molecules, and inflammatory cytokines. Interestingly, contrary to our prediction (82) after septic sera treatment only expression of tumor necrosis factor-α was significantly increased in aged endothelial cells as compared with that in young cells. Previous studies demonstrate that the pattern of endotoxemia-induced inflammatory gene expression shows considerable organ specificity in aged mice (12,13). Thus, further studies are warranted to characterize the expression pattern of a wide set of inflammatory markers in young and aged endothelial cells treated with septic sera.
Taken together, aging increases sensitivity of MVECs to oxidative stress and cellular damage induced by inflammatory factors present in the circulation during septicemia. These results can partly explain the age-associated increase in susceptibility to microvascular dysfunction and multiorgan failure associated with sepsis.
Funding
This work was supported by grants from the American Heart Association (to Z.U., P.T., and A.C.), American Federation for Aging Research (to A.C.), the Oklahoma Center for the Advancement of Science and Technology (to A.C. and Z.U.), the National Institutes of Health (AG031085 to A.C.; AT006526 to Z.U.; AG038747, NS056218, and P01 AG11370 to W.E.S.), the Ellison Medical Foundation and the Arkansas Claude Pepper Older Americans Independence Center at University of Arkansas Medical Center (to A.C.), and the American Diabetes Association (to C.S.). The authors would like to express their gratitude for the support of the Donald W. Reynolds Foundation, which funds aging research at the University of Oklahoma Health Sciences Center under its Aging and Quality of Life Program.
References
- 1. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001; 29:1303–1310 [DOI] [PubMed] [Google Scholar]
- 2. Girard TD, Opal SM, Ely EW. Insights into severe sepsis in older patients: from epidemiology to evidence-based management. Clin Infect Dis. 2005; 40:719–727 [DOI] [PubMed] [Google Scholar]
- 3. Stanley M. Sepsis in the elderly. Crit Care Nurs Clin North Am. 1996; 8:1–6 [PubMed] [Google Scholar]
- 4. Martin GS, Mannino DM, Moss M. The effect of age on the development and outcome of adult sepsis. Crit Care Med. 2006; 34:15–21 [DOI] [PubMed] [Google Scholar]
- 5. Padkin A, Goldfrad C, Brady AR, Young D, Black N, Rowan K. Epidemiology of severe sepsis occurring in the first 24 hrs in intensive care units in England, Wales, and Northern Ireland. Crit Care Med. 2003; 31:2332–2338 [DOI] [PubMed] [Google Scholar]
- 6. Brun-Buisson C, Meshaka P, Pinton P, Vallet B. EPISEPSIS Study Group EPISEPSIS: a reappraisal of the epidemiology and outcome of severe sepsis in French intensive care units. Intensive Care Med. 2004; 30:580–588 [DOI] [PubMed] [Google Scholar]
- 7. Tateda K, Matsumoto T, Miyazaki S, Yamaguchi K. Lipopolysaccharide-induced lethality and cytokine production in aged mice. Infect Immun. 1996; 64:769–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saito H, Sherwood ER, Varma TK, Evers BM. Effects of aging on mortality, hypothermia, and cytokine induction in mice with endotoxemia or sepsis. Mech Ageing Dev. 2003; 124:1047–1058 [DOI] [PubMed] [Google Scholar]
- 9. Chorinchath BB, Kong LY, Mao L, McCallum RE. Age-associated differences in TNF-alpha and nitric oxide production in endotoxic mice. J Immunol. 1996; 156:1525–1530 [PubMed] [Google Scholar]
- 10. Starr ME, Ueda J, Takahashi H, et al. Age-dependent vulnerability to endotoxemia is associated with reduction of anticoagulant factors activated protein C and thrombomodulin. Blood. 2010; 115:4886–4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Starr ME, Ueda J, Yamamoto S, Evers BM, Saito H. The effects of aging on pulmonary oxidative damage, protein nitration, and extracellular superoxide dismutase down-regulation during systemic inflammation. Free Radic Biol Med. 2011; 50:371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saito H, Papaconstantinou J. Age-associated differences in cardiovascular inflammatory gene induction during endotoxic stress. J Biol Chem. 2001; 276:29307–29312 [DOI] [PubMed] [Google Scholar]
- 13. Starr ME, Evers BM, Saito H. Age-associated increase in cytokine production during systemic inflammation: adipose tissue as a major source of IL-6. J Gerontol A Biol Sci Med Sci. 2009; 64:723–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamamoto K, Shimokawa T, Yi H, et al. Aging accelerates endotoxin-induced thrombosis: increased responses of plasminogen activator inhibitor-1 and lipopolysaccharide signaling with aging. Am J Pathol. 2002; 161:1805–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Szabó C, Cuzzocrea S, Zingarelli B, O’Connor M, Salzman AL. Endothelial dysfunction in a rat model of endotoxic shock. Importance of the activation of poly (ADP-ribose) synthetase by peroxynitrite. J Clin Invest. 1997; 100:723–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szabo C, Goldstein B. Endothelial dysfunction as predictor of mortality in sepsis. Crit Care Med. 2011; 39:878–879 [DOI] [PubMed] [Google Scholar]
- 17. Szabó C, Módis K. Pathophysiological roles of peroxynitrite in circulatory shock. Shock. 2010; 34(Suppl 1):4–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Csiszar A, Sosnowska D, Wang M, Lakatta EG, Sonntag WE, Ungvari Z. Age-associated proinflammatory secretory phenotype in vascular smooth muscle cells from the non-human primate macaca mulatta: reversal by resveratrol treatment. J Gerontol A Biol Sci Med Sci. 2012; 67:811–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ungvari Z, Bailey-Downs L, Gautam T, et al. Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-{kappa}B activation in the nonhuman primate Macaca mulatta. J Gerontol A Biol Sci Med Sci. 2011; 66:866–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ungvari Z, Bailey-Downs L, Sosnowska D, et al. Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am J Physiol Heart Circ Physiol. 2011; 301:H363–H372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brunialti MK, Santos MC, Rigato O, Machado FR, Silva E, Salomao R. Increased percentages of T helper cells producing IL-17 and monocytes expressing markers of alternative activation in patients with sepsis. PLoS ONE. 2012; 7:e37393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Santos SS, Brunialti MK, Rigato O, Machado FR, Silva E, Salomao R. Generation of nitric oxide and reactive oxygen species by neutrophils and monocytes from septic patients and association with outcomes. Shock. 2012; 38:18–23 [DOI] [PubMed] [Google Scholar]
- 23. Ungvari Z, Labinskyy N, Mukhopadhyay P, et al. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol. 2009; 297:H1876–H1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ungvari Z, Bagi Z, Feher A, et al. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am J Physiol Heart Circ Physiol. 2010; 299:H18–H24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ungvari Z, Bailey-Downs L, Gautam T, et al. Adaptive induction of NF-E2-related factor-2-driven antioxidant genes in endothelial cells in response to hyperglycemia. Am J Physiol Heart Circ Physiol. 2011; 300:H1133–H1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Csiszar A, Labinskyy N, Perez V, et al. Endothelial function and vascular oxidative stress in long-lived GH/IGF-deficient Ames dwarf mice. Am J Physiol Heart Circ Physiol. 2008; 295:H1882–H1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Csiszar A, Sosnowska D, Tucsek Z, et al. Circulating factors induced by caloric restriction in the non-human primate Macaca mulatta activate angiogenic processes in endothelial cells. J Gerontol A Biol Sci Med Sci. 2012. 10.1093/gerona/gls158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mukhopadhyay P, Rajesh M, Haskó G, Hawkins BJ, Madesh M, Pacher P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc. 2007; 2:2295–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mukhopadhyay P, Rajesh M, Yoshihiro K, Haskó G, Pacher P. Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem Biophys Res Commun. 2007; 358:203–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Csiszar A, Podlutsky A, Podlutskaya N, et al. Testing the oxidative stress hypothesis of aging in primate fibroblasts: is there a correlation between species longevity and cellular ROS production? J Gerontol A Biol Sci Med Sci. 2012; 67:841–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Labinskyy N, Mukhopadhyay P, Toth J, et al. Longevity is associated with increased vascular resistance to high glucose-induced oxidative stress and inflammatory gene expression in Peromyscus leucopus. Am J Physiol Heart Circ Physiol. 2009; 296:H946–H956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ungvari Z, Bailey-Downs L, Gautam T, et al. Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-{kappa}B activation in the nonhuman primate Macaca mulatta. J Gerontol A Biol Sci Med Sci. 2011; 66:866–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ungvari Z, Bagi Z, Feher A, et al. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am J Physiol Heart Circ Physiol. 2010; 299:H18–H24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z, Ungvari Z. Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am J Pathol. 2007; 170:388–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ungvari Z, Orosz Z, Rivera A, et al. Resveratrol increases vascular oxidative stress resistance. Am J Physiol Heart Circ Physiol. 2007; 292:H2417–H2424 [DOI] [PubMed] [Google Scholar]
- 36. Csiszar A, Ungvari Z, Edwards JG, et al. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002; 90:1159–1166 [DOI] [PubMed] [Google Scholar]
- 37. Ungvari Z, Labinskyy N, Gupte S, Chander PN, Edwards JG, Csiszar A. Dysregulation of mitochondrial biogenesis in vascular endothelial and smooth muscle cells of aged rats. Am J Physiol Heart Circ Physiol. 2008; 294:H2121–H2128 [DOI] [PubMed] [Google Scholar]
- 38. Ungvari Z, Orosz Z, Labinskyy N, et al. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am J Physiol Heart Circ Physiol. 2007; 293:H37–H47 [DOI] [PubMed] [Google Scholar]
- 39. Csiszar A, Smith K, Labinskyy N, Orosz Z, Rivera A, Ungvari Z. Resveratrol attenuates TNF-alpha-induced activation of coronary arterial endothelial cells: role of NF-kappaB inhibition. Am J Physiol Heart Circ Physiol. 2006; 291:H1694–H1699 [DOI] [PubMed] [Google Scholar]
- 40. Jane-Wit D, Chun HJ. Mechanisms of dysfunction in senescent pulmonary endothelium. J Gerontol A Biol Sci Med Sci. 2012; 67:236–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci. 2010; 65:1028–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bailey-Downs LC, Mitschelen M, Sosnowska D, et al. Liver-specific knockdown of igf-1 decreases vascular oxidative stress resistance by impairing the nrf2-dependent antioxidant response: A novel model of vascular aging. J Gerontol A Biol Sci Med Sci. 2012; 67:313–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bailey-Downs LC, Sosnowska D, Toth P, et al. Growth hormone and IGF-1 deficiency exacerbate high-fat diet-induced endothelial impairment in obese lewis dwarf rats: implications for vascular aging. J Gerontol A Biol Sci Med Sci. 2011; 67:553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ungvari Z, Sosnowska D, Podlutsky A, Koncz P, Sonntag WE, Csiszar A. Free radical production, antioxidant capacity, and oxidative stress response signatures in fibroblasts from lewis dwarf rats: effects of life span-extending peripubertal GH treatment. J Gerontol A Biol Sci Med Sci. 2011; 66:501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Valcarcel-Ares MN, Gautam T, Warrington JP, et al. Disruption of Nrf2 signaling impairs angiogenic capacity of endothelial cells: implications for microvascular aging. J Gerontol A Biol Sci Med Sci. 2012; 67:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vaz Fragoso CA, Lee PJ. The aging lung. J Gerontol A Biol Sci Med Sci. 2012; 67:233–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Horan MP, Pichaud N, Ballard JW. Quantifying mitochondrial dysfunction in complex diseases of aging. J Gerontol A Biol Sci Med Sci. 2012; 67:1022–1035 [DOI] [PubMed] [Google Scholar]
- 48. Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003; 101:3765–3777 [DOI] [PubMed] [Google Scholar]
- 49. Destarac LA, Ely EW. Sepsis in older patients: an emerging concern in critical care. Advances in Sepsis. 2002; 2:15–22 [Google Scholar]
- 50. Hu H, Li X, Li Y, et al. Calpain-1 induces apoptosis in pulmonary microvascular endothelial cells under septic conditions. Microvasc Res. 2009; 78:33–39 [DOI] [PubMed] [Google Scholar]
- 51. Peters K, Unger RE, Brunner J, Kirkpatrick CJ. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res. 2003; 60:49–57 [DOI] [PubMed] [Google Scholar]
- 52. Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol Genomics. 2004; 17:21–30 [DOI] [PubMed] [Google Scholar]
- 53. Hotchkiss RS, Tinsley KW, Swanson PE, Karl IE. Endothelial cell apoptosis in sepsis. Crit Care Med. 2002; 30(5 Suppl):S225–S228 [DOI] [PubMed] [Google Scholar]
- 54. Londoño D, Carvajal J, Strle K, Kim KS, Cadavid D. IL-10 Prevents apoptosis of brain endothelium during bacteremia. J Immunol. 2011; 186:7176–7186 [DOI] [PubMed] [Google Scholar]
- 55. Cardoso FL, Kittel A, Veszelka S, et al. Exposure to lipopolysaccharide and/or unconjugated bilirubin impair the integrity and function of brain microvascular endothelial cells. PLoS ONE. 2012; 7:e35919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wesche-Soldato DE, Chung CS, Lomas-Neira J, Doughty LA, Gregory SH, Ayala A. In vivo delivery of caspase-8 or Fas siRNA improves the survival of septic mice. Blood. 2005; 106:2295–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Coopersmith CM, Chang KC, Swanson PE, et al. Overexpression of Bcl-2 in the intestinal epithelium improves survival in septic mice. Crit Care Med. 2002; 30:195–201 [DOI] [PubMed] [Google Scholar]
- 58. Bommhardt U, Chang KC, Swanson PE, et al. Akt decreases lymphocyte apoptosis and improves survival in sepsis. J Immunol. 2004; 172:7583–7591 [DOI] [PubMed] [Google Scholar]
- 59. Efron PA, Tinsley K, Minnich DJ, et al. Increased lymphoid tissue apoptosis in baboons with bacteremic shock. Shock. 2004; 21:566–571 [DOI] [PubMed] [Google Scholar]
- 60. Hotchkiss RS, Chang KC, Swanson PE, et al. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol. 2000; 1:496–501 [DOI] [PubMed] [Google Scholar]
- 61. Huet O, Obata R, Aubron C, et al. Plasma-induced endothelial oxidative stress is related to the severity of septic shock. Crit Care Med. 2007; 35:821–826 [DOI] [PubMed] [Google Scholar]
- 62. Zingarelli B, Day BJ, Crapo JD, Salzman AL, Szabó C. The potential role of peroxynitrite in the vascular contractile and cellular energetic failure in endotoxic shock. Br J Pharmacol. 1997; 120:259–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Goldfarb RD, Marton A, Szabó E, et al. Protective effect of a novel, potent inhibitor of poly(adenosine 5’-diphosphate-ribose) synthetase in a porcine model of severe bacterial sepsis. Crit Care Med. 2002; 30:974–980 [DOI] [PubMed] [Google Scholar]
- 64. Pacher P, Mabley JG, Soriano FG, Liaudet L, Komjáti K, Szabó C. Endothelial dysfunction in aging animals: the role of poly(ADP-ribose) polymerase activation. Br J Pharmacol. 2002; 135:1347–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pacher P, Vaslin A, Benko R, et al. A new, potent poly(ADP-ribose) polymerase inhibitor improves cardiac and vascular dysfunction associated with advanced aging. J Pharmacol Exp Ther. 2004; 311:485–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007; 87:315–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Csiszar A, Pacher P, Kaley G, Ungvari Z. Role of oxidative and nitrosative stress, longevity genes and poly(ADP-ribose) polymerase in cardiovascular dysfunction associated with aging. Curr Vasc Pharmacol. 2005; 3:285–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ha YM, Ham SA, Kim YM, et al. β1-adrenergic receptor-mediated HO-1 induction, via PI3K and p38 MAPK, by isoproterenol in RAW 264.7 cells leads to inhibition of HMGB1 release in LPS-activated RAW 264.7 cells and increases in survival rate of CLP-induced septic mice. Biochem Pharmacol. 2011; 82:769–777 [DOI] [PubMed] [Google Scholar]
- 69. Piantadosi CA, Withers CM, Bartz RR, et al. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J Biol Chem. 2011; 286:16374–16385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Thimmulappa RK, Lee H, Rangasamy T, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006; 116:984–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nagai N, Thimmulappa RK, Cano M, et al. Nrf2 is a critical modulator of the innate immune response in a model of uveitis. Free Radic Biol Med. 2009; 47:300–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kong X, Thimmulappa R, Craciun F, et al. Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am J Respir Crit Care Med. 2011; 184:928–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Thimmulappa RK, Scollick C, Traore K, et al. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem Biophys Res Commun. 2006; 351:883–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Suh JH, Shenvi SV, Dixon BM, et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci USA. 2004; 101:3381–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Redl H, Schlag G, Schiesser A, Davies J. Thrombomodulin release in baboon sepsis: its dependence on the dose of Escherichia coli and the presence of tumor necrosis factor. J Infect Dis. 1995; 171:1522–1527 [DOI] [PubMed] [Google Scholar]
- 76. Iba T, Yagi Y, Kidokoro A, Fukunaga M, Fukunaga T. Increased plasma levels of soluble thrombomodulin in patients with sepsis and organ failure. Surg Today. 1995; 25:585–590 [DOI] [PubMed] [Google Scholar]
- 77. Yamakawa K, Fujimi S, Mohri T, et al. Treatment effects of recombinant human soluble thrombomodulin in patients with severe sepsis: a historical control study. Crit Care. 2011; 15:R123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fei D, Meng X, Kang K, et al. Heme oxygenase-1 modulates thrombomodulin and activated protein C levels to attenuate lung injury in cecal ligation and puncture-induced acute lung injury mice. Exp Lung Res. 2011; 38:173–182 [DOI] [PubMed] [Google Scholar]
- 79. Kim YH, Choi KH, Park JW, Kwon TK. LY294002 inhibits LPS-induced NO production through a inhibition of NF-kappaB activation: independent mechanism of phosphatidylinositol 3-kinase. Immunol Lett. 2005; 99:45–50 [DOI] [PubMed] [Google Scholar]
- 80. Bozinovski S, Jones JE, Vlahos R, Hamilton JA, Anderson GP. Granulocyte/macrophage-colony-stimulating factor (GM-CSF) regulates lung innate immunity to lipopolysaccharide through Akt/Erk activation of NFkappa B and AP-1 in vivo. J Biol Chem. 2002; 277:42808–42814 [DOI] [PubMed] [Google Scholar]
- 81. Paterson RL, Galley HF, Dhillon JK, Webster NR. Increased nuclear factor kappa B activation in critically ill patients who die. Crit Care Med. 2000; 28:1047–1051 [DOI] [PubMed] [Google Scholar]
- 82. Belmin J, Bernard C, Corman B, Merval R, Esposito B, Tedgui A. Increased production of tumor necrosis factor and interleukin-6 by arterial wall of aged rats. Am J Physiol. 1995; 268(6 Pt 2):H2288–H2293 [DOI] [PubMed] [Google Scholar]