

Abstract
PTEN plays an important role not only in tumorigenesis but also in the normal development of central nervous system. PTEN loss in neural progenitor cells during embryogenesis disrupts migration and proper formation of the brain laminar structure. We generated a conditional PTEN knockout mouse by crossing mice that express Cre recombinase driven by the human GFAP promoter to a floxed PTEN gene to investigate the role of astroglial PTEN signaling pathway in neuronal patterning and lamination. We found PTEN loss not only in astroglial cells, but also in radial glia-derived neurons in hGFAP-Cre+/−/PTENloxp/loxp transgenic mice. Homozygous hGFAP-Cre+/−/PTENloxp/loxp transgenic mice showed progressive brain enlargement with cellular disorganization that occurred predominantly in hippocampus and cerebellum and died by postnatal day 20. Confocal images show that nestin-positive radial glial cells were observed in the hippocampus, cortex, and cerebellum at postnatal day 0 in homozygous hGFAP-Cre+/−/PTENloxp/loxp, but not in heterozygous hGFAP-Cre+/−/PTENloxp/− and hGFAP-Cre−/−/PTENloxp/loxp mice. Homozygous hGFAP-Cre+/−/PTENloxp/loxp transgenic mouse eyes, which lack radial glial lineage, were able to develop normal architectonics after birth. In addition, we also found that neuronal progenitor migration was defected at postnatal day 0 in homozygous hGFAP-Cre+/−/PTENloxp/loxp mice. These results suggest that PTEN has a critical role in regulating radial glial differentiation, proliferation, maturation, and eventually neuronal patterning in central nervous system in a spatio-temporal dependent manner.
Keywords: PTEN, radial glia, neurogenesis, neuronal lamination
Phosphatase and tensin homolog (PTEN) is a lipid phosphatase, and functions as a tumor suppressor [1, 2]. It is ubiquitous and highly conserved among many species. Previous studies show that PTEN has a fundamental role with many signaling cascades in numerous functions, such as cell growth, survival, proliferation, migration and metabolism [3, 4]. In addition, PTEN may have phosphatase-independent activities as well as other functions [5]. Mutations in PTEN are associated with a group of inherited neurological disorders, known as the PTEN hamartoma tumor syndromes (PHTS) [6, 7]. Inherited mutation of PTEN is associated with a wide range of phenotypes with variable penetrance including hamartomas in multiple tissues, and a high risk of breast, thyroid and endometrial cancer, as well as multiple neurological features [8, 9].
Neuronal patterning in the central nervous system is controlled by a variety of signaling pathways that guide neuronal migration to form laminae. Directed migration of neuronal precursor cells or immature neurons to the proper positions is critical during the lamination. During early development, newborn neuroblasts undergo extensive migration to establish the predetermined organization and patterning in the central nervous systems. Such migration terminates upon arrival at the targeted region, followed by maturation and differentiation. Neuronal migration and patterning process are regulated by many specific extracellular factors and intrinsic electrical activity in a spatiotemporally specific manner, including calcium, growth factors, and adhesion molecules. Radial glial cells are a pivotal cell type supporting such neuronal patterning and migration, and may function as precursors during neurogenesis [10–12]. Studies have suggested that PTEN might be involved in neuronal patterning and lamination. In the nervous system, conditional deletion of PTEN from mouse GFAP promoter directed cre-loxP system causes seizures, ataxia and defects in soma [13, 14]. On the other hand, PTEN deletion in Bergmann glia leads to premature differentiation and affects cerebellar laminar organization [15].
In the current report, we generated mice with astroglial specific (human GFAP promoter directed Cre-loxP system) PTEN deletion. We observed that astroglial PTEN deficiency altered radial glia proliferation, extension and scaffold formation in cortex, hippocampus, and cerebellum. These results provided in vivo evidence that PTEN functions as an inhibitory factor in the early stage of neurogenesis and gliogenesis through regulation of radial glia at the brain. PTEN deletion induced accelerated and prolonged proliferation of radial glia, dysregulated neuronal migration in late stage developments, and subsequently caused neuronal lamination defects in selective brain regions that involve early radial glia based neurogenesis. These results suggest that PTEN has a critical role in regulating radial glial differentiation, maturation, and eventually neuronal patterning in central nervous system in a temporal, spatial, and cell type-specific manner.
MATERIALS AND METHODS
Animals and Reagents
To generate astroglial specific PTEN deletion mice, the hGFAP-Cre line [16] was crossed to PTENloxp/loxp mice [17]. Both strains were obtained from Jax Mice (Bar Harbor, ME). The double transgenic hGFAP-Cre+/−/PTENloxp were backcrossed to PTENloxp/loxp mice. Homozygous hGFAP-Cre+/−/PTENloxp/loxp (gKO) and homozygous hGFAP-Cre−/−/PTENloxp/loxp (control) were used in all the experiments except indicated, where hGFAP-Cre+/−/PTENloxp−/− were examined together with control and gKO. All mice were housed with controlled temperature (22–25°C) and humidity (55%). A 12-hour light-dark cycle was maintained, with lights on between 7 a.m. and 7 p.m. All housing and procedures were performed in accordance with the guidelines of the Institutional Care and Use Committee of the National Research Council, and were approved by the University of North Texas Health Science Center Animal Care and Use Committee.
Immunohistochemistry and Histological Analysis
Immunohistochemical analysis was performed on 4% paraformaldehyde-fixed paraffin embedded sections (8–12μm). Sections were then immunostained with Picture Plus immunohistochemistry kits (Invitrogen, Carlsbad, CA). 3,3′-diaminobenzidine tetrachloride (DAB) was used to visualize the sections. The primary antibodies used include mouse anti-PTEN (1:100), rabbit anti-GFAP (1:100), goat anti-DCX (Santa Cruz, Santa Cruz CA),and rabbit anti-NeuN (1:100; Millipore Billerica, MA). The procedure followed the instruction of the manufacture and previous publications [18, 19]. For control slides, primary antibodies were omitted.
Double or Multiple Immunostaining
For double or multiple immunostaining or immunofluorescence, the primary antibodies used include goat anti-DCX (1:200), goat anti-MCM2 (1:100; Santa Cruz Santa Cruz, CA), and rabbit anti-GFAP (1:1,000; Sigma, St. Louis, MO), The secondary antibodies were Alexa Fluor 488-, 594-, or 647-conjugated donkey anti-goat, or anti-rabbit IgG (1:200 to 500; Invitrogen, CA,). Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI) using prolong Gold antifade reagent (Invitrogen, CA). Fluorescence signals were detected using an LSM 510 NLO Confocal Scanning System mounted on an Axiovert 200 inverted microscope (Carl Zeiss Ltd) equipped with a two-photon Chameleon laser (Coherent Inc.), and images were acquired using LSM 510 Imaging Software (Carl Zeiss Ltd). Two-, three-, or four-color images were scanned using argon, 543 HeNe, 633 HeNe, and Chameleon (750 to 780nm for DAPI) lasers. GFAP and NeuN whole brain staining were obtained with low magnification, and consistent exposure throughout the brain, and were then collaged with Adobe Photoshop CS3.
Western Blot Analysis
For Western blot, we followed standard procedures according to our previous publication [20]. Mice were sacrificed and the brains were immediately removed, dissected, and kept on dry ice, or snap frozen for further analysis. Hemibrains were weighed and homogenized without thawing in 1× PBS buffer containing 1 mM EDTA, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, 10 μl/ml protease inhibitor mix (EMD bioscience). The samples were then diluted in equal volume of 2X loading buffer (125 mM Tris-HCl pH 6.8, 20% glycerol, 10% 2-mercaptoethanol, 4% SDS, 2 mM EGTA, 2 mM EDTA, 2 mM PMSF, 2 mM Na3VO4, 2 mM NaF, 20μl/ml protease inhibitor mix) and incubated at 85 °C for 10 minutes, and sonicated for 5 seconds to break the DNA. Depending on the primary antibody used, 10–50 μg of protein was analyzed following electrophoresis on SDS-PAGE and transferred onto 0.2 μm nitrocellulose membranes (Whatman, Florham Park, NJ). Membranes were blocked, incubated with appropriate primary and secondary antibody dilutions (Jackson ImmunoResearch, West Grove, PA), and treated using chemiluminescent developing agent (Thermo). The membranes were probed with antibodies specific for mouse anti-actin, mouse anti-PTEN, rabbit anti-GFAP (Santa Cruz Biotech), rabbit anti-p308AKT, p473AKT, AKT, ERK1/2, and pERK1/2 (all were from Cell Signaling Technologies), rabbit anti-PSD95, NeuN, and NR2B (Millipore). The data were quantified with Vision Works LS Image Acquisition and Analysis Software (UVP), and expressed as percent of mean control optical density from the same blots.
Statistical Analysis
All statistical analyses were performed with Prism (Graphpad Software, San Diego, CA), using unpaired Student’s t test between two groups, and one-way ANOVA among multiple groups. Data are shown in mean ± SEM. Groups were considered significantly different when p < 0.05 (*p < 0.05, **p < 0.01, and ***p < 0.001).
RESULTS
hGFAP promoter directed PTEN deletion in the central nervous system
To investigate the effects of astroglial specific PTEN deficiency, we generated compound mice by crossing PTENloxp/loxp mice [17] with transgenic mice bearing an hGFAP-Cre [16] (Fig 1A). To determine the efficiency of PTEN deletion, we examined PTEN expression with immunohistochemistry in mouse brain at postnatal day 15. In control mice, PTEN expression was mainly observed in cortex and hippocampus. In the cortex, most of the PTEN immunoreactivity was observed in neuronal soma and axons, projecting from pial surface into the cortex. In the hippocampus, PTEN was localized in the axons of neuronal cells, such as mossy fiber regions and the projecting axons in the CA1 region (Fig 1B). However, in gKO mice, PTEN-positive cells were barely detectable in astroglia and radial glial-derived neuronal cells. Interestingly, a large number of microglia shaped PTEN-positive cells were observed. These observations are consistent with previous studies with hGFAP derived PTEN expression [16, 21, 22]. To further confirm PTEN loss, we examined PTEN and the downstream signaling pathway activation by PTEN loss with immunoblotting analysis. At P15, we observed substantial loss of PTEN in the whole brain. Consistently, AKT and ERK pathways were constitutively activated, demonstrated by the increased phosphorylation of Akt (p308 and p473) and ERK1/2 (Thr202/Tyr204) in the gKO mice, which are well documented hallmarks of PTEN loss. We found no significant difference in Akt activation in the heterozygous mouse brain, but a slightly increase in ERK1/2 activation. The protein levels of AKT and ERK1/2 were reduced, due to constitutive activation (Fig. 1C).
Figure 1.
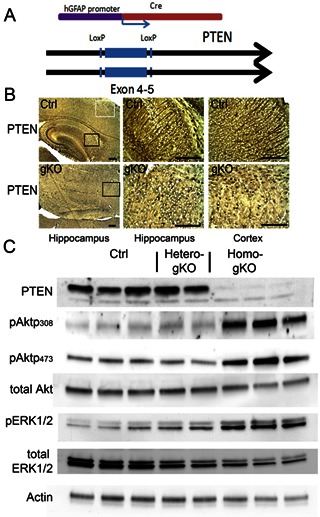
hGFAP targeted astroglial specific PTEN loss in the brain. A. Schematic illustration for homo-gKO mouse genotypes. B. Immunohistochemistry of PTEN in hippocampus (left and middle panel) and cortex (right panel) from control mice (Ctrl) and homo-gKO mice (gKO). C. Representative immunoblotting of PTEN, pAkt 308, pAkt 473, AKT, pERK1/2, ERK1/2, and actin from control, hetero-gKO and homo-gKO mice at postnatal day 15. All scale bars are equal to 200 μm.
Astroglial PTEN loss results in severe macrocephaly and cellular disorganizations in hippocampus and cerebellum
The homozygous GFAP-Cre/ PTENloxp/loxp (gKO) mice were viable and showed slight macrocephaly at birth. However, gKO mouse brain size progressively increased and died between 13 to 20 days after birth. At the age of 15 days, all of gKO mice developed severe motor function deficiency, such as ataxia, tremor, and spasms. The wet brain mass were approximately two and half folds of the PTENloxp/loxp mice (control), while the body weight was much smaller than control mice (Fig 2A, B).
Figure 2.
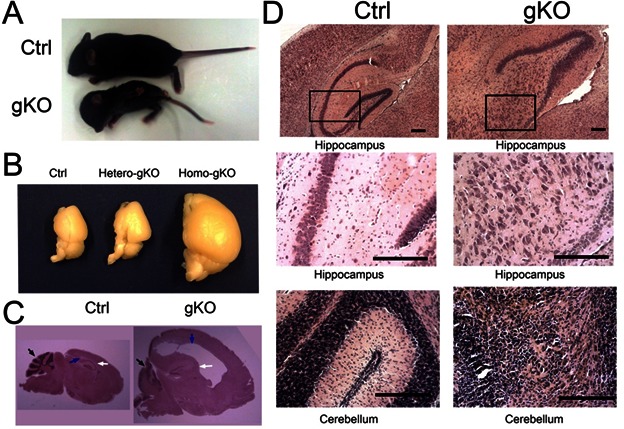
Astroglial PTEN loss induced macrocephaly, severe gliosis in hippocampus and cerebellum. A. The representative image of control and gKO mice whole body. gKO mice showed lower body weight and larger brain size compared to the control mice. B. Dissected hemi-brain of control, hetero-gKO, and homo-gKO. The hetero-gKO did not show significant difference in brain size, compared with control mice. C. H&E histological analysis on the sagittal section of control and gKO mice. Defect cerebellar neuronal formation (black arrow) and severe macrocephaly (blue arrow) are indicated. D. H&E histological analysis revealed neuronal layering defect in the hippocampus (upper and middle panels) and cerebellum (lower panels). All scale bars are equal to 200 μm.
Histological analysis at postnatal day 15 shows many pathological features, including but not limited to gliosis, brain hypertrophy, hyperplasia, and hydrocephalus, a similar phenotype as previously reported using a mouse GFAP-Cre directed PTEN inactivation [13, 14]. However, we found that much more severe phenotypes and earlier occurrence in gKO, along with the abnormal ventricular regions as shown in Fig. 2C.
The cortical neuronal layer in gKO mice remained similar to the control mice, but the hippocampal neuronal pattern was completely disrupted, where the whole CA1, CA2 and CA3 regions no longer maintained the compact formation as that in the control mice. All gKO mice showed striking scattered dispersion of pyramidal neurons in the hippocampus, together with undulation of the granule-cell layer of the dentate gyrus. Intercellular spaces between neuronal cell bodies were filled with thick neuronal dendrites and astrocytes, compared with the tightly packed lamination noted in the hippocampal formation (Fig 2D). In the cerebellum, gKO mice had increased cerebellar volume, together with almost complete loss of neuronal organization and layering in all mice examined. In control mice, three layers of the cerebellar cortex (molecular, Purkinje, and granular layers) had been formed and all lobes were well structured by postnatal day 15. In gKO mice, the Purkinje cells and the granule neurons were still present in the cerebellum, but the layering formation was almost completely disorganized in which Purkinje cells were mixed with granule neurons and high density astrocytes (Fig 2D).
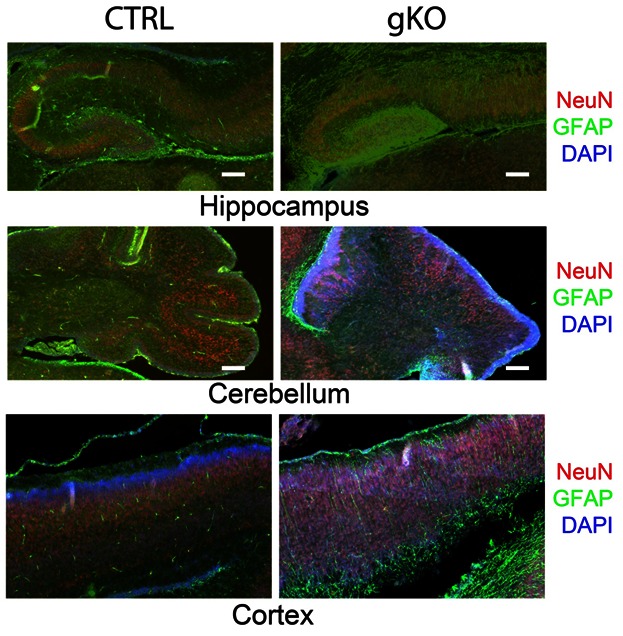
Further double immunofluorescence labeling of GFAP and NeuN of the entire brain were examined with a collaged image of the whole sagittal section. Severe defects of neuronal patterning and laminar organization at hippocampus and cerebellum were observed (Fig 3). In control mice, GFAP were primarily localized in the corpus callosum, hippocampus, cerebellum, and amygdale. In gKO mice, hypertrophic astrocytes with enhanced GFAP immunoreactivity were widespread. The overall pattern of GFAP immunoreactivity was similar, but, in a much more diffusive manner. Other than the lateral ventricle, several brain regions as hippocampus, amygdala and cerebellum were considerably enlarged. Further immunohistological analysis of GFAP (astrocytes) and NeuN (Neurons) at hippocampus (Fig 4A) and cerebellum (Fig 4B) shows highly organized radial glia interlaced with correct neuronal layer formation in control mice, but prominent astrogliosis and high density GFAP immunostaining in gKO mice. A complete loss of radial glia and disorganizations of neuronal lays were observed in both hippocampus and cerebellum (Fig 4A, B).
Figure 3:
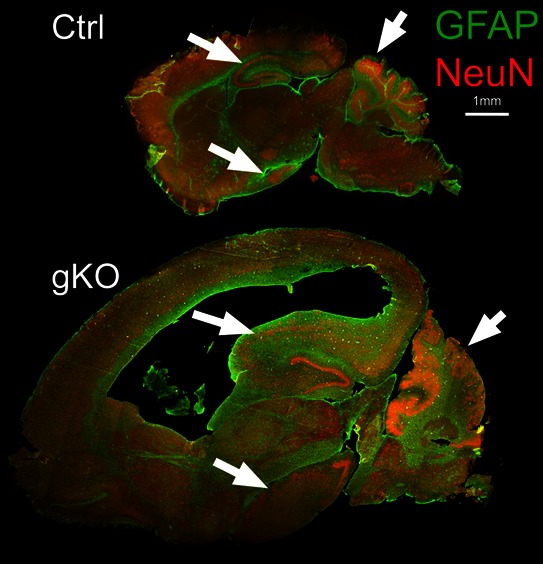
Astroglial PTEN loss induces severe defects of neuronal patterning and laminar organization. Double immunofluorescence labeling of neurons (NeuN) and astrocytes (GFAP) in the whole brain sagittal section from control and gKO mice. The arrows indicate hippocampus, cerebellum, and amygdala in control and gKO mice.
Figure 4:
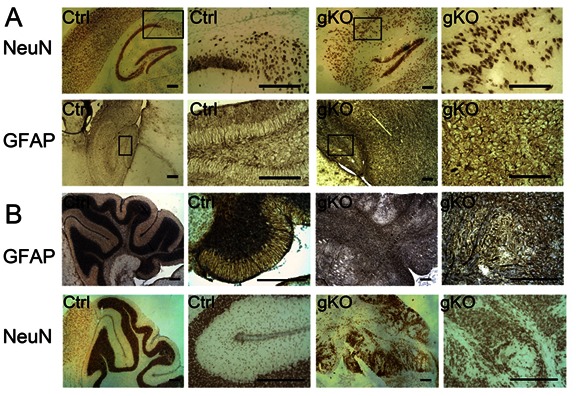
Neuronal and astrocyte patterning defect in the hippocampus and cerebellum of astroglial PTEN knockout mice. Representative immunohistochemistry of neurons (NeuN) and astrocytes (GFAP) in the control and gKO mice from hippocampus (A) and cerebellum (B). All scale bars are equal to 200 μm.
Dysregulation of neuronal progenitor cell migration in gKO mice
In gKO mice, we observed more than four-fold increase in GFAP levels and more than two-fold increase in a neuronal progenitor cell marker DCX levels compared to control mice. While, post-synaptic marker PSD95 and pre-synaptic marker NMDA receptor 2B (NR2B) both significantly were decreased in the gKO mice (Fig 5A,B). Such reduction may not necessarily indicate neurodegeneration, but a reduced proportion of neural components in the overall brain. We found no significant difference in control and heterogeneous transgenic mice in the expression levels of these proteins.
Figure 5:
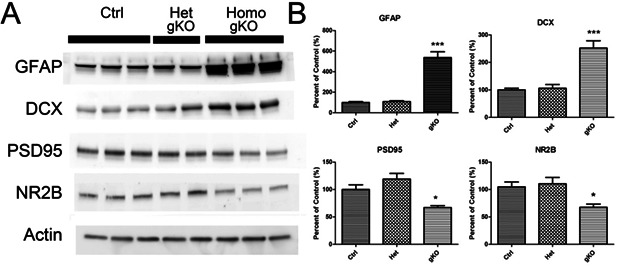
Astroglial PTEN loss induces GFAP and DCX over expression. A. Western blot of astrocyte marker (GFAP), double cortin (DCX), post-synaptic neuronal marker (PSD95), and presynaptic neuronal marker (NR2B) in brain of control, hetero and homozygous astroglial PTEN null mice. B. Quantitative immune blot analysis. * (P<0.05) and *** (P<0.001) shows significant difference between the indicated group and the control group.
Immunofluorescence of DCX confirms the increased DCX at both hippocampal regions and subventricular zone in the gKO mice as compared with control mice. In the gKO mice, DCX–positive cells in dentate gyrus displayed dysregulating migration pattern, compared with normal mice. The DCX positive neuronal progenitor cells show a migrating pattern toward the pial surface in the gKO mice, while they appeared completely “anchored” in the control mice (Fig 6A). In order to characterize these neuronal progenitor cells, we examined the co-localizations of DCX with GFAP and another proliferation marker, the DNA replication licensing factor MCM2. In the dentate gyrus, DCX-positive cells did not co-localize with GFAP. In gKO mice, GFAP immunoreactivity was observed mostly in a highly scattered pattern, without colocalization with DCX positive neuronal progenitor cells. MCM2 was decreased in these regions in gKO mice (Fig 6B).
Figure 6:
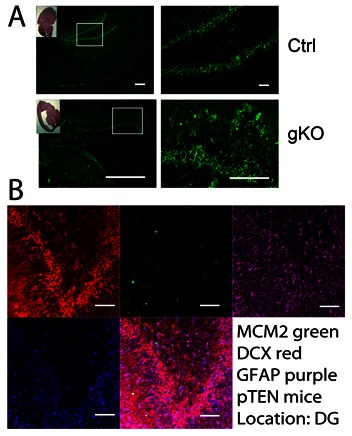
Astroglial PTEN loss leads to increased neuronal progenitor cell population that does not colocalize with GFAP. A. immunofluorescence of DCX in the control and gKO mice from dentate gyrus. B. representative multiple labeling of MCM2, DCX, PTEN and DAPI in the dentate gyrus. We found no co-localization of GFAP and DCX. All scale bars are equal to 200 μm in A, and equal to 20 μm in B.
Abnormal radial glial patterning in astroglial PTEN null mice
During development of mouse brain, neuronal precursors use the radial glial cells as a scaffold to establish the neuronal laminae. We determined whether abnormal neuronal migration and laminar formation described above was due to defects of radial glial-guided neuronal migration. We examined the expression profile of radial glia in the control mice and gKO mice. Chronological immunohistochemistry analysis suggests that at postnatal day 0, the overall anatomical structures between control mice and gKO mice were quite similar in both cortex and hippocampus. However, GFAP-immunoreactive radial glia was much more intense in gKO mice than control mice (Fig 7). In control mice, radial shaped glia cells were barely detectable in the hippocampus and cortex. However, in the gKO mice, a great number of the radial shaped GFAP positive glia were detected, extending from the apical surface, into the CA1 and CA2 regions, or to the pial surface. These astroglial structures were totally absent in the adjacent hypothalamus regions. At this stage in the neocortex, neuronal migration was almost complete in the control mice. Numerous radial glia were found projecting from the apical surface of the ventricle lumen to the pial surface in the gKO mice. Dramatic layering defect was detectable at the end of the first week in gKO mice, and progressed very rapidly to almost complete disorganization of hippocampus and cerebellum by postnatal day 15. A double immunofluorescence of GFAP and nestin, an early proliferation marker, showed that many of the GFAP-positive radial glia expressed nestin in gKO mice, but not in control mice, suggesting that these were immature radial glia and able to divide (Fig 8).
Figure 7.
Double immunofluorescence labeling of GFAP and NeuN in hippocampus, cerebellum, and cortex of astroglial PTEN knockout mouse and littermate control at postnatal day 0. Sections were counter-stained with DAPI, and represented as merged images. All scale bars are equal to 200 μm.
Figure 8.
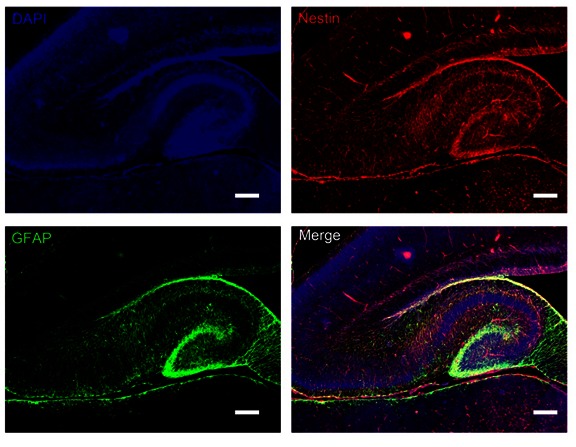
Double immunofluorescence labeling of GFAP and nestin in the hippocampus from gKO mice. We observed substantial co-localization between GFAP and Nestin in the dentate gyrus and hippocampus. All scale bars are equal to 200 μm.
In order to further characterize the role of PTEN loss in the brain development, we examined the retina in control and gKO mice. Retina lacks radial glial lineage at the early development stage. We found similar PTEN expression in both control and gKO mice. In gKO mice, retina was able to develop normal architectonics at post natal day 15 (Fig. 9).
Figure 9.
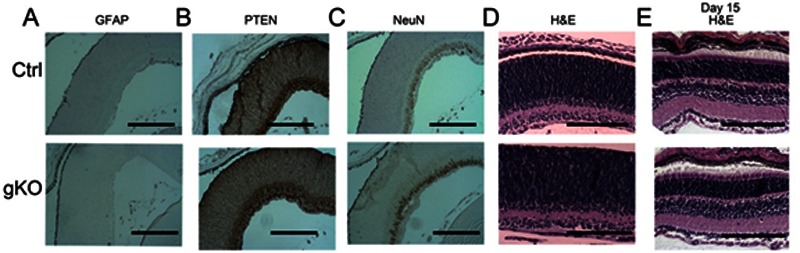
Histological and immunohistochemical analysis of retinal structure in control and gKO mice. Eyes from postnatal day 0 (A–D) were immune labeled with GFAP, PTEN, and NeuN. We observed no difference between control mice and gKO mice. At postnatal day 15, we found no observable structural difference between control and gKO mice (E). All scale bars are equal to 200 μm.
DISCUSSION
In this study, we characterized the functions of astroglial specific PTEN deficiency during central nervous system development with conditional knockout mice. We showed that hGFAP-directed PTEN deletion resulted in dramatic neuronal layering and patterning defect. PTEN deficiency in astroglial system induced dysregulated radial glial proliferation and scaffold formation in the hippocampus, cortex, and cerebellum, leading to misguided neuronal proliferation, differentiation, and migration. These results demonstrate the critical role of PTEN in regulating radial glial functions during development. These data support the recent concept that radial glial functions as fundamental neuronal progenitor cells during the development, that asymmetrically divide for neuronal progenitor genesis, proliferation, and recruitment and forming a scaffold that guides and facilitates the neuronal migration.
Both human and mouse GFAP promoter-directed PTEN loss has been investigated previously. Two groups [13, 14] used a mouse GFAP-CRE that restricted PTEN deletion in postmitotic neurons and demonstrated an increase in neuronal soma size upon PTEN loss. Another mouse GFAP-CRE line induced PTEN deletion in a subpopulation of adult neural stem cells in the subependymal zone [23]. On the other hand, the human GFAP-CRE line induced PTEN loss in both astroglia and a subset of neuronal population which is consistent with the present study [22]. PTEN loss does not affect neuronal migration in in vitro experiments, but its deletion in Bergmann glia was shown to cause premature differentiation and affects laminar organization in the cerebellum [15]. Another group used En2-CRE and L7- CRE lines to inactivate PTEN in all neuronal and glial cell populations at cerebellum and observed that PTEN is required for the cerebellar architecture and for proper neuronal and glial migration, while, not for cell differentiation [24]. Our data further extend the previous findings and suggest that PTEN deletion in radial glia induced accelerated and prolonged proliferation of radial glia, and dramatic cellular disorganization of the hippocampus, cortex, and cerebellum.
In the traditional concept, generation of neurons only occurred during embryonic and perinatal development. However, adult neurogenesis has been now accepted as a common feature of vertebrate brains in the last two decades. The stem cell lineage has also been thoroughly investigated. Historically, it was believed that neuroepithelial cells (NE) produced two separate pools of committed glial and neuronal progenitors. Recent data support a different model, in which NE cells either produce or transform into radial glia, which can self replicate, or divide asymmetrically to produce neuronal progenitor cells, neurons and glia. Radial glial cells are defined as cells with a radial morphology and astroglial characteristics. Similar to NE, they typically stretch from the apical surface from the basement membrane to the pial surface. Radial glial cells have cellular and molecular characteristics of astroglia and long radial processes. Radial glia have similar ultrastructural characteristics of astrocytes, and typically contain S100β and GFAP, which are absent in NE. Radial glia functions both as neuronal progenitors during development and in mature adult brain and as scaffolds that direct migration of newborn neurons. In the late stages of cortical development, radial glial cells may divide asymmetrically in the ventricular zone to generate radial glial cells, postmitotic neurons, or intermediate progenitor cells, with various lineages [25, 26]. In mammals, most radial cells disappear perinatally, then, they either went into apoptosis or transformed into post-mitotic astrocytes. In the adult subventricular zone, some astrocytes may continue to produce neurons, glia and perhaps other cell types. In several brain regions, specialized radial glia (Bergmann glia, Muller cell) can be induced or persist into adulthood, which arguably continue to produce new neurons under certain pathological conditions [27]. In the current study, Cre-floxed recombinant PTEN deletion driven by the human GFAP promoter not only induces loss of PTEN in astrocytes but also in neurons. The nascent neuronal progenitor cells, labeled by DCX are GFAP negative but PTEN deficient, suggesting that these progenitor cells carry an astroglial lineage, the radial glia. This clarifies the radial glia based neuronal progenitor linage in different brain regions. In our observation, almost all hippocampal and most of the cortical neurons carry an astroglial linage, represented by neuronal PTEN loss. This finding provides firm in vivo evidence that radial glia asymmetrically generate neurons [28, 29], and supports the hypothesis [30, 31] that cortical and hippocampal neuronal progenitor cells are derived from radial glia, which are ultimately derived from neuroepithelial cells. These results are consistent with previous publications that radial glia in the neonatal ventricular wall produces multiple classes of brain cells: astrocytes, oligodendrocytes, ependymal cells, and neurons [32, 33]. Indeed, radial glial cells divide throughout neurogenesis and give rise to the majority of projection neurons in the cerebral cortex and hippocampus [34].
The present study also provides in vivo evidence, regarding the role of radial glial in neuronal migration. Our mice clearly presented the evidence that astroglial PTEN loss leads to the defect of neuronal migration at cortex, hippocampus and cerebellum. Initially, proliferating multipotent precursor cells generated from radial glia form a thick layer around the ventricles. The proper migration of the neuroblasts depends on radial glia for guidance to the targeted compartments. The soma of radial glia is located in the apical surface, and its processes stretch vertically to the pial surface, forming a long scaffold of cellular processes which provides the cue for neurons to migrate to the pial surface. Migrating neurons are guided by many factors, such as calcium, adhesion molecules, growth factors that interact with the receptors or ligands in the radial glial fibers that guide the migration [35, 36]. The neurons that form the permanent cortical plate migrate in an inside out pattern (from apical to pial surface) to form the cortex and hippocampus. Evidently, PTEN has a critical role in this process, as loss of astroglial PTEN induces macrocephaly and severe hydrocephalus. The migrating neurons lack anchoring target with unrestricted outgrowth. The current study demonstrates that PTEN has a critical regulatory role in radial glial scaffold formation during CNS development, and interfering with the PTEN/PI3 kinase system results in severe migration and layering defect in various brain regions as cortex, hippocampus and cerebellum (Fig 10).
Figure 10.
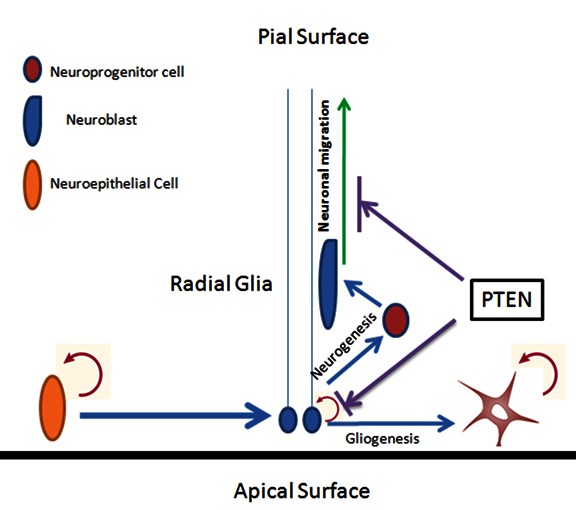
Schematic illustration of PTEN negatively regulating the radial glial proliferation and formation of scaffold that guides the neuronal migration and proper layering in the hippocampus and cortex.
In summary, we generated conditional knockout mice with astroglial specific PTEN deficiency. Our study demonstrated that PTEN loss in radial glia resulted in enhanced radial glia proliferation and defect of neuronal migration in multiple brain regions. The corresponding increased neuronal progenitor population, including but not limited to cortex, hippocampus, cerebellum, and amygdala in early stage development caused several severe phenotypes. This provides solid evidence on the critical role of PTEN in gliogenesis, neurogenesis and neuronal migration.
Acknowledgments
This work was partly supported by National Institutes of Health grants R01NS054687 (SY), R01NS054651 (SY).
Reference
- [1].Bostrom J, Cobbers JM, Wolter M, Tabatabai G, Weber RG, Lichter P, Collins VP, Reifenberger G. Mutation of the PTEN (MMAC1) tumor suppressor gene in a subset of glioblastomas but not in meningiomas with loss of chromosome arm 10q. Cancer research. 1998;58:29–33. [PubMed] [Google Scholar]
- [2].Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer research. 1997;57:2124–2129. [PubMed] [Google Scholar]
- [3].Chen X, Thakkar H, Tyan F, Gim S, Robinson H, Lee C, Pandey SK, Nwokorie C, Onwudiwe N, Srivastava RK. Constitutively active Akt is an important regulator of TRAIL sensitivity in prostate cancer. Oncogene. 2001;20:6073–6083. doi: 10.1038/sj.onc.1204736. [DOI] [PubMed] [Google Scholar]
- [4].Endersby R, Baker SJ. PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene. 2008;27:5416–5430. doi: 10.1038/onc.2008.239. [DOI] [PubMed] [Google Scholar]
- [5].Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD, Liu X, Wu H. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer cell. 2003;3:117–130. doi: 10.1016/s1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- [6].Zhou XP, Marsh DJ, Morrison CD, Chaudhury AR, Maxwell M, Reifenberger G, Eng C. Germline inactivation of PTEN and dysregulation of the phosphoinositol-3-kinase/Akt pathway cause human Lhermitte-Duclos disease in adults. American journal of human genetics. 2003;73:1191–1198. doi: 10.1086/379382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Eng C. PTEN: one gene, many syndromes. Human mutation. 2003;22:183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- [8].Hobert JA, Eng C. PTEN hamartoma tumor syndrome: an overview. Genet Med. 2009;11:687–694. doi: 10.1097/GIM.0b013e3181ac9aea. [DOI] [PubMed] [Google Scholar]
- [9].Wallace JA, Li F, Leone G, Ostrowski MC. Pten in the breast tumor microenvironment: modeling tumor-stroma coevolution. Cancer research. 71:1203–1207. doi: 10.1158/0008-5472.CAN-10-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Alvarez-Buylla A, Garcia-Verdugo JM, Tramontin AD. A unified hypothesis on the lineage of neural stem cells. Nat Rev Neurosci. 2001;2:287–293. doi: 10.1038/35067582. [DOI] [PubMed] [Google Scholar]
- [11].Farkas LM, Huttner WB. The cell biology of neural stem and progenitor cells and its significance for their proliferation versus differentiation during mammalian brain development. Current opinion in cell biology. 2008;20:707–715. doi: 10.1016/j.ceb.2008.09.008. [DOI] [PubMed] [Google Scholar]
- [12].Merkle FT, Alvarez-Buylla A. Neural stem cells in mammalian development. Current opinion in cell biology. 2006;18:704–709. doi: 10.1016/j.ceb.2006.09.008. [DOI] [PubMed] [Google Scholar]
- [13].Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nature genetics. 2001;29:404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- [14].Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nature genetics. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- [15].Yue Q, Groszer M, Gil JS, Berk AJ, Messing A, Wu H, Liu X. PTEN deletion in Bergmann glia leads to premature differentiation and affects laminar organization. Development (Cambridge, England) 2005;132:3281–3291. doi: 10.1242/dev.01891. [DOI] [PubMed] [Google Scholar]
- [16].Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- [17].Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, Wu H. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32:148–149. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- [18].Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan LJ, Ju X, Liu R, Qian H, Marvin MA, Goldberg MS, She H, Mao Z, Simpkins JW, Yang SH. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. The Journal of biological chemistry. 286:16504–16515. doi: 10.1074/jbc.M110.208447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wen Y, Yu WH, Maloney B, Bailey J, Ma J, Marie I, Maurin T, Wang L, Figueroa H, Herman M, Krishnamurthy P, Liu L, Planel E, Lau LF, Lahiri DK, Duff K. Transcriptional regulation of beta-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron. 2008;57:680–690. doi: 10.1016/j.neuron.2008.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wen Y, Planel E, Herman M, Figueroa HY, Wang L, Liu L, Lau LF, Yu WH, Duff KE. Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J Neurosci. 2008;28:2624–2632. doi: 10.1523/JNEUROSCI.5245-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- [22].Fraser MM, Zhu X, Kwon CH, Uhlmann EJ, Gutmann DH, Baker SJ. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer research. 2004;64:7773–7779. doi: 10.1158/0008-5472.CAN-04-2487. [DOI] [PubMed] [Google Scholar]
- [23].Gregorian C, Nakashima J, Le Belle J, Ohab J, Kim R, Liu A, Smith KB, Groszer M, Garcia AD, Sofroniew MV, Carmichael ST, Kornblum HI, Liu X, Wu H. Pten deletion in adult neural stem/progenitor cells enhances constitutive neurogenesis. J Neurosci. 2009;29:1874–1886. doi: 10.1523/JNEUROSCI.3095-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Marino S, Krimpenfort P, Leung C, van der Korput HA, Trapman J, Camenisch I, Berns A, Brandner S. PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development (Cambridge, England) 2002;129:3513–3522. doi: 10.1242/dev.129.14.3513. [DOI] [PubMed] [Google Scholar]
- [25].Marin-Padilla M. [The development of the human cerebral cortex. A cytoarchitectonic theory] Revista de neurologia. 1999;29:208–216. [PubMed] [Google Scholar]
- [26].Casanova MF, Trippe J., 2nd Regulatory mechanisms of cortical laminar development. Brain research reviews. 2006;51:72–84. doi: 10.1016/j.brainresrev.2005.10.002. [DOI] [PubMed] [Google Scholar]
- [27].Lorber B, Berry M, Douglas MR, Nakazawa T, Logan A. Activated retinal glia promote neurite outgrowth of retinal ganglion cells via apolipoprotein E. Journal of neuroscience research. 2009;87:2645–2652. doi: 10.1002/jnr.22095. [DOI] [PubMed] [Google Scholar]
- [28].Alvarez-Buylla A, Theelen M, Nottebohm F. Proliferation “hot spots” in adult avian ventricular zone reveal radial cell division. Neuron. 1990;5:101–109. doi: 10.1016/0896-6273(90)90038-h. [DOI] [PubMed] [Google Scholar]
- [29].Malatesta P, Hartfuss E, Gotz M. Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development (Cambridge, England) 2000;127:5253–5263. doi: 10.1242/dev.127.24.5253. [DOI] [PubMed] [Google Scholar]
- [30].Imura T, Kornblum HI, Sofroniew MV. The predominant neural stem cell isolated from postnatal and adult forebrain but not early embryonic forebrain expresses GFAP. J Neurosci. 2003;23:2824–2832. doi: 10.1523/JNEUROSCI.23-07-02824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kriegstein AR, Gotz M. Radial glia diversity: a matter of cell fate. Glia. 2003;43:37–43. doi: 10.1002/glia.10250. [DOI] [PubMed] [Google Scholar]
- [32].Merkle FT, Tramontin AD, Garcia-Verdugo JM, Alvarez-Buylla A. Radial glia give rise to adult neural stem cells in the subventricular zone. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:17528–17532. doi: 10.1073/pnas.0407893101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gubert F, Zaverucha-do-Valle C, Pimentel-Coelho PM, Mendez-Otero R, Santiago MF. Radial glia-like cells persist in the adult rat brain. Brain research. 2009;1258:43–52. doi: 10.1016/j.brainres.2008.12.021. [DOI] [PubMed] [Google Scholar]
- [34].Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annual review of neuroscience. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chedotal A, Rijli FM. Transcriptional regulation of tangential neuronal migration in the developing forebrain. Current opinion in neurobiology. 2009;19:139–145. doi: 10.1016/j.conb.2009.04.005. [DOI] [PubMed] [Google Scholar]
- [36].Zupanc GK, Clint SC. Potential role of radial glia in adult neurogenesis of teleost fish. Glia. 2003;43:77–86. doi: 10.1002/glia.10236. [DOI] [PubMed] [Google Scholar]