

Abstract
Alzheimer’s disease (AD) develops for a yet unknown period of time and can progress undiagnosed for years before its first clinical manifestation consisting of characteristic cognitive impairments. Current AD treatments offer only a small symptomatic benefit, likely because AD is diagnosed when the pathology is already well advanced, whereas treatments may be most efficient in the early phases of pathology. An accurate, early marker of AD is therefore needed to help diagnose AD earlier. It is now well documented that AD patients and animal models of AD exhibit reorganization of hippocampal and cortical networks. This reorganization is initiated by an early imbalance between excitation and inhibition, leading to altered network activity. The mechanisms underlying these changes are unknown but recent evidence suggests that either soluble amyloid-beta (Aß) or fibrillar forms of Aß are central to various network alterations observed in AD. However, recent evidence also suggests that Aβ over-production in animal models is not systematically linked to network over-excitation. We hypothesize here that early changes in the excitation-inhibition balance within the hippocampus occurs much earlier than currently believed and initially produces only slight changes in overall hippocampal activity. In this review, we introduce the concept according to which the subtle changes in theta and gamma rhythms might occur during the very first stages of AD and thus could be used as a possible predictor for the disease.
Keywords: Oscillatory activity, theta rhythm, gamma oscillations, Aβ, hippocampus
Alzheimer’s disease (AD) is the most common form of dementia. Generally, it is diagnosed in people over 65 years of age, although the less prevalent early-onset AD can occur much earlier. As of September 2010, this number is reported to be 35.6 million worldwide, increasing to 65.7 million by 2030 and 115.4 million by 2050. Its prevalence (5% over 65 years, 20% of those over 80 years) and the social and economic burden it imposes on society (70% of beds in hospitals, long stays) represent a major public health problem in all industrialized countries. The diagnosis of the disease depends mainly on neuropsychological tests and the detection of cortical atrophy affecting the medial temporal lobe including the hippocampus. Early symptoms consist of memory loss (amnesia) manifested initially by minor distractions. These memory failures increase with the progression of the disease, while long-term memory remains relatively preserved. A recent French epidemiological study that focused on 3000 subjects followed for 20 years, had retrospectively demonstrated signs of memory impairments up to 12 years before diagnosis, reinforcing the necessity to identify new early markers of AD [1].
In high-income countries, only 20–50% of dementia cases are recognized and treated in the context of primary care. Such “treatment gap” is certainly much greater in low- and middle-income countries. This is clearly a major concern, given that the world’s population is growing older, new cases of dementia and Alzheimer’s disease are increasing, and earlier diagnosis and early intervention are important mechanisms by which the “treatment gap” can be filled.
The World Alzheimer Report 2011 has pointed-out that lack of detection is a significant barrier to improving life quality of AD and other dementia patients. Indeed, there is no cure for AD. Moreover, all available treatments offer symptomatic benefit only in early phases of the disease, and are only “disease modifying”. Thus, these treatments can only delay the progression of the disease. Finally, and as stated by the American Alzheimer Association, “Disclosing the diagnosis early in the disease process allows the individual to maximize quality of life and play an active role in planning for the future. If you disclose the diagnosis after the dementia has advanced, it may no longer be meaningful to the affected individual.” (http://www.alz.org/professionals_and_researchers_diagnostic_disclosure.asp). Therefore, an early and easily accessible marker of AD is desperately needed.
Currently, the cause of AD is not known. Genetic studies [2] have identified mutations in the amyloid precursor protein (APP), presenilin 1 and 2 (PS1, PS2) that cause rare, dominantly inherited familial AD (FAD). Proteolytic processing of APP by BACE (β-site APP cleaving enzyme) followed by the PS-containing γ-secretase complex generates amyloid-β (Aβ) peptides that deposit in amyloid plaques. Genetic and cell biological studies show increased production of amyloidogenic Aβ peptides with FAD-linked mutations, providing strong support for the amyloid hypothesis [3], which posits that Aβ peptides play a pivotal role in AD pathogenesis. In addition, AD is characterized by the intracellular accumulation of hyperphosphorylated tau protein (neurofibrillary tangles), which together with amyloid plaques remain the major histological hallmarks of AD. A very recent longitudinal study of 128 individuals bearing FAD mutations has suggested that the diagnosis of clinical dementia is made late in the course of AD pathogenesis [4]. The authors therefore suggest that the targeting of Aβ earlier in the course of the disease (the increase in Aβ being seen 20 years before onset of memory deficits) may provide better clinical outcomes than the treatment of mild to moderate dementia, reinforcing the need for an early, easily accessible biomarker of AD.
Altered network activity in AD
AD patients also present an increased incidence of seizures [5, 6]. Until recently, it was believed that this increase in seizure susceptibility was the result of neuronal cell loss. Multiple studies indeed indicate that hippocampal inhibitory GABAergic transmission might be altered. At the cellular level, inhibitory GABAergic interneurons of the hippocampus form a very heterogenous population (there is 21 different types of interneurons) expressing different neurochemical markers and projections[7]. A number of recent studies have focused on phenotype identification of hippocampal interneurons that are lost in AD but there is yet no consensus on this issue [8–11]. However, a common finding in all these studies is the clear decrease in interneuron number within the hippocampal formation, which is detectable even during early phases of the disease. As a corollary, it was postulated that the increased incidence of seizures seen in AD results from the loss of hippocampal interneurons. However, recent evidence using the transgenic hAPPFAD mouse, have challenged this concept. Indeed, in a seminal paper that rejuvenated the field, Palop and colleagues showed that high levels of Aβ are sufficient to elicit epileptiform activity and seizures, even at early stages of the disease process and in the absence of overt neuronal loss [12]. Electroencephalographic epileptiform activity or increases in epileptic susceptibility have been subsequently identified independently in a number of additional transgenic AD mouse models (see [13] for a specific review on this topic), clearly indicating that the imbalance between excitation and inhibition within the hippocampus is a robust hallmark of early stage AD. These observations strongly suggest 1) that the increased incidence of epileptic seizures is not the consequence of neuronal cell loss, but rather its cause, 2) that this increased incidence of seizures in both hippocampus and cortex indicate that there is an imbalance between network inhibition and excitation and 3) that neuronal over-excitation in the hippocampus is linked to Aβ overproduction. Finally, this study raised the possibility that at least some cognitive decline seen in animal AD mouse models might be in part related to seizure activity. Indeed, a recent study has shown that chronic treatment with Levetiracetam (an FDA-approved antiepileptic drug) drastically reduced seizure activity in hAPPJ20 mice. This reduced seizure activity was correlated with a clear improvement of hippocampus-dependent learning and memory [14].
Regarding human pathology, elevated hippocampal activation is observed in conditions that confer risk for AD, including amnestic mild cognitive impairment (aMCI). In a recent study, Bakker and colleagues tested the counterintuitive idea that elevated hippocampal activation might in fact have deleterious effects on cognitive performance. Chronic treatment with Levetiracetam indeed normalized hippocampal activation in aMCI patients and improved memory performance [15], again suggesting that hippocampal over-excitation might be responsible, at least partly, for cognitive decline observed in prodromal AD. Most importantly, this hippocampal hyperactivation is correlated with cortical thinning (a sensitive and specific marker of AD neurodegeneration) within MCI patients [16], indicating that hippocampal hyperactivation might be viewed as a biomarker for AD.
Since Palop’s study [12], multiple groups have attempted to define the link between Aβ overproduction and hippocampal network dysfunction. An important step forward has been achieved recently by linking the altered network activity to a specific type of interneurons within the hippocampus, namely the parvalbumin (PV)-positives interneurons [17]. More specifically, Verret and colleagues have shown that restoring normal expression of a specific voltage-gated sodium channel (Nav1.1) reduced abnormal network synchronization and memory deficits in hAPPJ20 mice. However, hAPP mice over-expressing Nav1.1 still present similar levels of Aβ compared to control hAPP mice, suggesting that Aβ alone is not responsible for network dysfunction.
Indeed, despite the evidence obtained in genetic and cell biology studies, which support the amyloid hypothesis, it is becoming clear that AD etiology is complex and that Aβ alone cannot account for all aspects of AD [18, 19]. For example, recent neuroimaging studies have confirmed previous autopsy findings indicating that amyloid deposits are present in cognitively normal individuals, whereas some AD patients show no amyloid deposits in positron emission tomography (PET) scans [20, 21]. Therefore, mutations in APP and presenilins may be able to contribute to AD pathology by amyloid-independent mechanisms. In agreement, the recent data suggests that Aβ over-production is not systematically linked to network alterations [22] and acute injection of Aβ in either hippocampus or medial septum induces changes in hippocampal oscillatory activity but do not trigger epileptic activity [23, 24]. Furthermore, lowering tau levels in different AD mouse models reduces cognitive deficits and normalizes hippocampal activity despite an elevated Aβ level [25, 26]. In addition, because full-length APP is expressed in many mouse models of AD, it is difficult to distinguish the contribution of APP, from that of the individual peptides generated from APP. Relevantly, emerging evidence points to non-amyloid factors, such as β-CTF or the APP-intracellular domain (AICD) as possible culprits in AD [27].
Altered hippocampal oscillatory activity as an early marker of AD?
Since epileptiform activity likely results from drastic alterations at the network level [28], we and others have hypothesized that early changes in the excitation-inhibition balance within the hippocampus will produce slight changes in overall hippocampal activity. Indeed, among the first symptoms of AD are alterations in episodic memory. Information processing and storage by brain networks requires a highly synchronized operation of multiple neuronal groups. One possible mechanism of such synchronization is through the coordinated, rhythmic activity of neuronal populations, which give rise to oscillations in local field potentials. Among the brain rhythms generated by these oscillations, the hippocampal theta and gamma oscillations have been described as critical players in memory consolidation [29] and rely extensively on the integrity of hippocampal networks. Theta rhythm in the hippocampus has generated substantial interest since 1950’s because of its association with arousal and attention [30]. Theta oscillations occur at frequencies in the range of 3–12 Hz and are the most prominent extracellular signal recorded in the mammalian brain [31]. This rhythmic activity is present during exploration of a novel environment, is elicited by sensory stimuli, present during REM sleep [29, 32] and arises from the coordinated activity of multiple brain areas [29] (including the medial entorhinal cortex and the medial septum) in coordination with intra-hippocampal oscillators [33]. Because the hippocampus is widely known for its critical role in learning and memory, many studies have examined how theta contributes to these cognitive phenomena. Two seminal studies published in the late 1970’s have demonstrated that theta prevalence was significantly correlated with learning [34] while eliminating hippocampal theta oscillations abolished spatial memory [35]. Since then, many studies have investigated how theta activity participates in memory formation. Particularly, theta oscillations play a key role in terms of setting, not only the right conditions of excitability for the functional expression of plasticity, but also the direction of plasticity [36–40]. There is also convincing evidence that theta oscillations plays an important role in pacing place cells [41, 42]. These so called “place cells” are pyramidal neurons that increase firing when an animal enters a particular place or area of a given environment. Hippocampal pyramidal cells undergo a phenomena known as “phase precession” when an animal enters the cell’s place field. During phase precession, the cell fires at a progressively earlier phase of the network theta [41]. Therefore, place cells exploit both rate and phase coding mechanisms to increase the spatial information contained in a given spike train [43].
In addition to theta, hippocampus also generates two types of gamma oscillations: the slow gamma and the fast gamma activities [44]. Both forms of gamma oscillations are nested within the theta oscillations and this co-modulation has been proposed as a mechanism by which particular cell assemblies are recruited throughout the theta cycle [45]. Phase-amplitude coupling between theta and gamma oscillations has been reported across species including mice, rats, and humans, and there is evidence to support the idea that this type of coupling has an important functional role in the execution of cognitive functions [46, 47]. Thus, gamma oscillations, in conjunction with theta rhythm, play a critical role in hippocampus-dependent memory processes and therefore might be altered in the early stages of AD [17].
Conclusion
In conclusion, recent evidence indicates that changes in theta and gamma rhythms could be used as a possible predictor for AD [48]. To our knowledge, only one study has attempted to characterize hippocampal oscillatory activity in young animal model of AD. In these experiments, hippocampal theta oscillations were recorded in young and old anaesthetized APP/PS1 mice following brainstem stimulation and the first alterations of elicited theta rhythm were found at 4 months of age, concomitant with the increase in Aβ level and appearance of plaques burden [49]. However, it is now clear that the first impairments in learning and memory may arise before Aβ plaques become fully apparent [50], reinforcing the necessity of assessing hippocampal oscillatory activity in young freely-moving animal models of AD. Understanding the mechanisms behind the original alterations in neuronal network activities may provide the earliest AD markers. Defining such markers is in turn the obvious prerequisite in establishment of diagnostic criteria for the initial stages of the pathophysiological process of AD. Moreover, discovery of the key players underlying the early neuronal network impairments will also provide new therapeutic targets. As a corollary, the efficiency of the newly developed treatments will become testable by monitoring the delay or prevention in appearance of the earliest AD markers and thus the clinical onset of AD.
Figure 1.
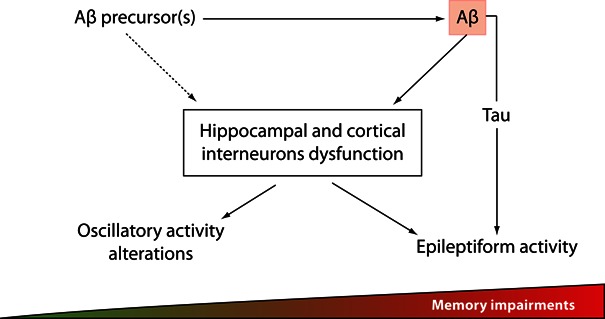
Changes in hippocampal and/or cortical oscillatory activity might precede Aβ induced-epileptiform activity and correlate with the first alterations in memory processes in early stages of AD
Acknowledgments
This work was supported by the University of Strasbourg, the Centre National de la Recherche Scientifique (CNRS) and the Institut National de la Santé et de la Recherche Médicale (INSERM). RG is supported by grants from the Fondation Fyssen, the European Research Executive Agency and the NARSAD. We are grateful to Chelsea Cavanagh for comments that greatly improved the clarity of this manuscript.
References
- [1].Amieva H, Le Goff M, Millet X, Orgogozo JM, Peres K, Barberger-Gateau P, Jacqmin-Gadda H, Dartigues JF. Prodromal Alzheimer’s disease: successive emergence of the clinical symptoms. Ann Neurol. 2008;64(5):492–498. doi: 10.1002/ana.21509. [DOI] [PubMed] [Google Scholar]
- [2].Price DL, Tanzi RE, Borchelt DR, Sisodia SS. Alzheimer’s disease: genetic studies and transgenic models. Annu Rev Genet. 1998;32:461–493. doi: 10.1146/annurev.genet.32.1.461. [DOI] [PubMed] [Google Scholar]
- [3].Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. (New York, N.Y. [DOI] [PubMed] [Google Scholar]
- [4].Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. The New England journal of medicine. 2012;367(9):795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia. 2006;47(5):867–872. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- [6].Hauser WA, Morris ML, Heston LL, Anderson VE. Seizures and myoclonus in patients with Alzheimer’s disease. Neurology. 1986;36(9):1226–1230. doi: 10.1212/wnl.36.9.1226. [DOI] [PubMed] [Google Scholar]
- [7].Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321(5885):53–57. doi: 10.1126/science.1149381. (New York, N.Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ramos B, Baglietto-Vargas D, del Rio JC, Moreno-Gonzalez I, Santa-Maria C, Jimenez S, Caballero C, Lopez-Tellez JF, Khan ZU, Ruano D, Gutierrez A, Vitorica J. Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenic model of Alzheimer’s disease. Neurobiology of aging. 2006;27(11):1658–1672. doi: 10.1016/j.neurobiolaging.2005.09.022. [DOI] [PubMed] [Google Scholar]
- [9].Loreth D, Ozmen L, Revel FG, Knoflach F, Wetzel P, Frotscher M, Metzger F, Kretz O. Selective degeneration of septal and hippocampal GABAergic neurons in a mouse model of amyloidosis and tauopathy. Neurobiol Dis. 2012;47(1):1–12. doi: 10.1016/j.nbd.2012.03.011. [DOI] [PubMed] [Google Scholar]
- [10].Baglietto-Vargas D, Moreno-Gonzalez I, Sanchez-Varo R, Jimenez S, Trujillo-Estrada L, Sanchez-Mejias E, Torres M, Romero-Acebal M, Ruano D, Vizuete M, Vitorica J, Gutierrez A. Calretinin interneurons are early targets of extracellular amyloid-beta pathology in PS1/AbetaPP Alzheimer mice hippocampus. Journal of Alzheimer’s disease : JAD. 2010;21(1):119–132. doi: 10.3233/JAD-2010-100066. [DOI] [PubMed] [Google Scholar]
- [11].Krantic S, Isorce N, Mechawar N, Davoli MA, Vignault E, Albuquerque M, Chabot JG, Moyse E, Chauvin JP, Aubert I, McLaurin J, Quirion R. Hippocampal GABAergic neurons are susceptible to amyloid-beta toxicity in vitro and are decreased in number in the Alzheimer’s disease TgCRND8 mouse model. Journal of Alzheimer’s disease : JAD. 2012;29(2):293–308. doi: 10.3233/JAD-2011-110830. [DOI] [PubMed] [Google Scholar]
- [12].Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55(5):697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66(4):435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, Mucke L. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(42):E2895–2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74(3):467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Putcha D, Brickhouse M, O’Keefe K, Sullivan C, Rentz D, Marshall G, Dickerson B, Sperling R. Hippocampal hyperactivation associated with cortical thinning in Alzheimer’s disease signature regions in non-demented elderly adults. J Neurosci. 2011;31(48):17680–17688. doi: 10.1523/JNEUROSCI.4740-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149(3):708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pimplikar SW. Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. Int J Biochem Cell Biol. 2009;41(6):1261–1268. doi: 10.1016/j.biocel.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Golde TE, Schneider LS, Koo EH. Anti-Abeta Therapeutics in Alzheimer’s Disease: The Need for a Paradigm Shift. Neuron. 2011;69(2):203–213. doi: 10.1016/j.neuron.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, Rossor M, Brooks DJ. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68(7):501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- [21].Li Y, Rinne JO, Mosconi L, Pirraglia E, Rusinek H, DeSanti S, Kemppainen N, Nagren K, Kim BC, Tsui W, de Leon MJ. Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2008;35(12):2169–2181. doi: 10.1007/s00259-008-0833-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vogt DL, Thomas D, Galvan V, Bredesen DE, Lamb BT, Pimplikar SW. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiology of aging. 2011;32(9):1725–1729. doi: 10.1016/j.neurobiolaging.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Colom LV, Castaneda MT, Banuelos C, Puras G, Garcia-Hernandez A, Hernandez S, Mounsey S, Benavidez J, Lehker C. Medial septal beta-amyloid 1–40 injections alter septo-hippocampal anatomy and function. Neurobiology of aging. 2010;31(1):46–57. doi: 10.1016/j.neurobiolaging.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Villette V, Poindessous-Jazat F, Simon A, Lena C, Roullot E, Bellessort B, Epelbaum J, Dutar P, Stephan A. Decreased rhythmic GABAergic septal activity and memory-associated theta oscillations after hippocampal amyloid-beta pathology in the rat. J Neurosci. 2010;30(33):10991–11003. doi: 10.1523/JNEUROSCI.6284-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011;31(2):700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–754. doi: 10.1126/science.1141736. (New York, N.Y. [DOI] [PubMed] [Google Scholar]
- [27].Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH. Amyloid-Independent Mechanisms in Alzheimer’s Disease Pathogenesis. J Neurosci. 2010;30(45):14946–14954. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(16):9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33(3):325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- [30].Green JD, Arduini AA. Hippocampal electrical activity in arousal. Journal of neurophysiology. 1954;17(6):533–557. doi: 10.1152/jn.1954.17.6.533. [DOI] [PubMed] [Google Scholar]
- [31].Vertes RP, Hoover WB, Viana Di Prisco G. Theta rhythm of the hippocampus: subcortical control and functional significance. Behavioral and cognitive neuroscience reviews. 2004;3(3):173–200. doi: 10.1177/1534582304273594. [DOI] [PubMed] [Google Scholar]
- [32].Vanderwolf CH. Hippocampal electrical activity and voluntary movement in the rat. Electroencephalography and clinical neurophysiology. 1969;26(4):407–418. doi: 10.1016/0013-4694(69)90092-3. [DOI] [PubMed] [Google Scholar]
- [33].Goutagny R, Jackson J, Williams S. Self-generated theta oscillations in the hippocampus. Nat Neurosci. 2009;12(12):1491–1493. doi: 10.1038/nn.2440. [DOI] [PubMed] [Google Scholar]
- [34].Berry SD, Thompson RF. Prediction of learning rate from the hippocampal electroencephalogram. Science. 1978;200(4347):1298–1300. doi: 10.1126/science.663612. (New York, N.Y. [DOI] [PubMed] [Google Scholar]
- [35].Winson J. Loss of hippocampal theta rhythm results in spatial memory deficit in the rat. Science. 1978;201(4351):160–163. doi: 10.1126/science.663646. (New York, N.Y. [DOI] [PubMed] [Google Scholar]
- [36].Greenstein YJ, Pavlides C, Winson J. Long-term potentiation in the dentate gyrus is preferentially induced at theta rhythm periodicity. Brain research. 1988;438(1–2):331–334. doi: 10.1016/0006-8993(88)91358-3. [DOI] [PubMed] [Google Scholar]
- [37].Larson J, Lynch G. Induction of synaptic potentiation in hippocampus by patterned stimulation involves two events. Science. 1986;232(4753):985–988. doi: 10.1126/science.3704635. (New York, N.Y. [DOI] [PubMed] [Google Scholar]
- [38].Pavlides C, Greenstein YJ, Grudman M, Winson J. Long-term potentiation in the dentate gyrus is induced preferentially on the positive phase of theta-rhythm. Brain research. 1988;439(1–2):383–387. doi: 10.1016/0006-8993(88)91499-0. [DOI] [PubMed] [Google Scholar]
- [39].Holscher C, Anwyl R, Rowan MJ. Stimulation on the positive phase of hippocampal theta rhythm induces long-term potentiation that can Be depotentiated by stimulation on the negative phase in area CA1 in vivo. J Neurosci. 1997;17(16):6470–6477. doi: 10.1523/JNEUROSCI.17-16-06470.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hyman JM, Wyble BP, Goyal V, Rossi CA, Hasselmo ME. Stimulation in hippocampal region CA1 in behaving rats yields long-term potentiation when delivered to the peak of theta and long-term depression when delivered to the trough. J Neurosci. 2003;23(37):11725–11731. doi: 10.1523/JNEUROSCI.23-37-11725.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].O’Keefe J, Recce ML. Phase relationship between hippocampal place units and the EEG theta rhythm. Hippocampus. 1993;3(3):317–330. doi: 10.1002/hipo.450030307. [DOI] [PubMed] [Google Scholar]
- [42].Skaggs WE, McNaughton BL, Wilson MA, Barnes CA. Theta phase precession in hippocampal neuronal populations and the compression of temporal sequences. Hippocampus. 1996;6(2):149–172. doi: 10.1002/(SICI)1098-1063(1996)6:2<149::AID-HIPO6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- [43].Huxter J, Burgess N, O’Keefe J. Independent rate and temporal coding in hippocampal pyramidal cells. Nature. 2003;425(6960):828–832. doi: 10.1038/nature02058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Colgin LL, Denninger T, Fyhn M, Hafting T, Bonnevie T, Jensen O, Moser MB, Moser EI. Frequency of gamma oscillations routes flow of information in the hippocampus. Nature. 2009;462(7271):353–357. doi: 10.1038/nature08573. [DOI] [PubMed] [Google Scholar]
- [45].Lisman J, Redish AD. Prediction, sequences and the hippocampus. Philos Trans R Soc Lond B Biol Sci. 2009;364(1521):1193–1201. doi: 10.1098/rstb.2008.0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Canolty RT, Knight RT. The functional role of cross-frequency coupling. Trends Cogn Sci. 2010;14(11):506–515. doi: 10.1016/j.tics.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fell J, Axmacher N. The role of phase synchronization in memory processes. Nature reviews. 2011;12(2):105–118. doi: 10.1038/nrn2979. [DOI] [PubMed] [Google Scholar]
- [48].Jelic V, Johansson SE, Almkvist O, Shigeta M, Julin P, Nordberg A, Winblad B, Wahlund LO. Quantitative electroencephalography in mild cognitive impairment: longitudinal changes and possible prediction of Alzheimer’s disease. Neurobiology of aging. 2000;21(4):533–540. doi: 10.1016/s0197-4580(00)00153-6. [DOI] [PubMed] [Google Scholar]
- [49].Scott L, Feng J, Kiss T, Needle E, Atchison K, Kawabe TT, Milici AJ, Hajos-Korcsok E, Riddell D, Hajos M. Age-dependent disruption in hippocampal theta oscillation in amyloid-beta overproducing transgenic mice. Neurobiology of aging. 2012;33(7):1481, e1413–1423. doi: 10.1016/j.neurobiolaging.2011.12.010. [DOI] [PubMed] [Google Scholar]
- [50].Francis BM, Kim J, Barakat ME, Fraenkl S, Yucel YH, Peng S, Michalski B, Fahnestock M, McLaurin J, Mount HT. Object recognition memory and BDNF expression are reduced in young TgCRND8 mice. Neurobiology of aging. 2012;33(3):555–563. doi: 10.1016/j.neurobiolaging.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]