

Abstract
Sarcomas are cancers arising from the mesenchymal layer that affect children, adolescents, young adults, and adults. Although most sarcomas are localized, many display a remarkable predilection for metastasis to the lungs, liver, bones, subcutaneous tissue, and lymph nodes. Additionally, many sarcoma patients presenting initially with localized disease may relapse at metastatic sites. While localized sarcomas can often be cured through surgery and often radiation, controversies exist over optimal management of patients with metastatic sarcoma. Combinations of chemotherapy are the most effective in many settings, and many promising new agents are under active investigation or are being explored in preclinical models. Metastatic sarcomas are excellent candidates for novel approaches with additional agents as they have demonstrated chemosensitivity and affect a portion of the population that is motivated toward curative therapy. In this paper, we provide an overview on the common sarcomas of childhood (rhabdomyosarcoma), adolescence, and young adults (osteosarcoma, Ewing sarcoma, synovial sarcoma, and malignant peripheral nerve sheath tumor) and older adults (leiomyosarcoma, liposarcoma, and undifferentiated high grade sarcoma) in terms of the epidemiology, current therapy, promising therapeutic directions and outcome with a focus on metastatic disease. Potential advances in terms of promising therapy and biologic insights may lead to more effective and safer therapies; however, more clinical trials and research are needed for patients with metastatic sarcoma.
Keywords: chemotherapy, pediatric sarcoma, rhabdomyosarcoma, osteosarcoma, Ewing sarcoma, synovial sarcoma
Introduction
Sarcomas, cancers of tissues derived from the mesenchymal layer, represent 1% of all cancers in adults, 10% of cancers in children, and 8% of cancer in adolescents and young adults. This rarity and the diversity across ages render diagnosis and treatment difficult. In 2012, 2890 new cases of bone and joint cancer and 11,280 new cases of soft tissue cancer were estimated in the USA.1 In this same year, 1410 and 3900 deaths due to bone and soft tissue cancers, respectively, were also estimated. Despite being rare, sarcomas contribute to a substantial loss of years of life compared to other cancers because of the many children, adolescents, and young adults diagnosed with sarcoma.
Sarcomas are broadly classified as either soft tissue or bone neoplasms. There is substantial diversity in the more than 50 histologic soft tissue sarcoma (STS) subtypes.2 Peak incidence differs according to the histologic subtype with rhabdomyosarcoma being the most common type in early childhood, bone sarcomas predominating in adolescence, and multiple histologic types of soft tissue sarcomas predominating in young adulthood (<40 year old) and in older adults. From 2005–2009 approximately 29% of bone and joint cancer cases were diagnosed in patients under 20 years and 15% were diagnosed in 20–34 year olds. Nine percent of soft tissue cancer cases were diagnosed at <20 years or 20–34 years.3
At present, it should be noted that metastatic sarcomas are defined by the presence of disease to any metastatic site. This definition of metastasis may change over time with the advent of more sensitive measures for detecting metastatic disease. Currently, micrometastases are frequently below the detection limit of modern scans. Perhaps biomarkers such as circulating tumor cells, tumor-specific DNA markers such as translocations, tumor-specific antigens, or microRNAs (miRNAs) may eventually be incorporated into the definition of metastases.4–9
In this report, we will review the epidemiology, current therapy and promising therapeutic directions, and outcomes of patients with metastatic sarcoma. This review will include the common sarcomas of childhood (rhabdomyosarcoma), adolescence and young adults (osteosarcoma, Ewing sarcoma, synovial sarcoma, and malignant peripheral nerve sheath tumor), and older adults (leiomyosarcoma, liposarcoma, and undifferentiated sarcoma). We will also discuss patients who present with localized disease but unfortunately relapse at metastatic sites.
Epidemiology
Incidence and survival
Incidence and survival statistics were obtained from Surveillance, Epidemiology, and End Results (SEER) Fast Stats, an interactive tool that allows access to SEER and US cancer statistics.10 In 2009, STS accounted for 85.6% of all sarcomas diagnosed in the USA, whereas bone and joint sarcoma accounted for 14.4%. The incidence of STS is highest in individuals aged 65+ years and lowest in those <20 years (Figure 1). The 10-year relative survival rate for STS among patients > 20 years old was 70%, but approximately 50% among patients > 65 years old (Figure 2). The incidence of bone and joint cancer is highest in individuals aged 65+ years and lowest in those aged 20–49 years (Figure 3). The 10-year relative survival rate for patients diagnosed at 20–49 years with bone and joint sarcoma is 70% and about 40% for patients diagnosed > 65 years (Figure 4). SEER does not distinguish between metastatic and lower stage sarcomas and, as detailed throughout this report, patients with metastatic sarcoma have a worse prognosis than those listed in the figures.
Figure 1.
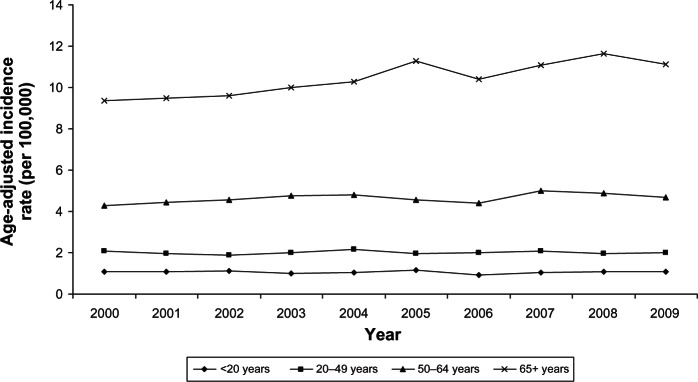
Age-adjusted incidence rates of soft tissue sarcoma by age at diagnosis (SEER18 areas).
Abbreviation: SEER, Surveillance, Epidemiology, and End Results.
Figure 2.

Relative survival of soft tissue sarcoma for different age groups, 1988–2008 (SEER 9 areas).
Abbreviation: SEER, Surveillance, Epidemiology, and End Results.
Figure 3.

Age-adjusted incidence rates of bone and joint cancer for different ages at diagnosis (SEER 18 areas).
Abbreviation: SEER, Surveillance, Epidemiology, and End Results.
Figure 4.
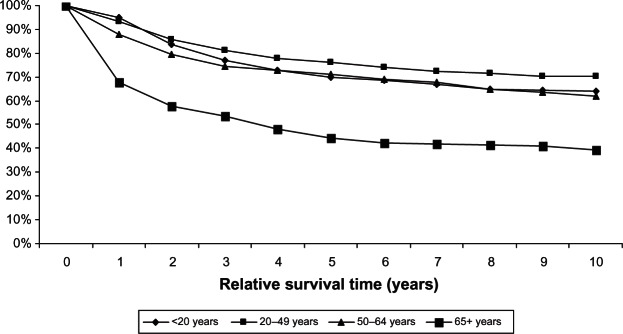
Relative survival of bone and joint cancer for different age groups, 1988–2008 (SEER 9 areas).
Abbreviation: SEER, Surveillance, Epidemiology, and End Results.
Risk factors
Sarcoma most typically presents spontaneously without a demonstrable cause. However, several risk factors have been associated with its development, including exposure to radiation and chemotherapeutic agents, viral infections, occupational factors, hereditary syndromes, certain diseases, and hormones.
Radiation and chemotherapeutic agents
Ionizing radiation has been consistently reported to be associated with increased risk of bone and STS of up to 1%. In children, studies have shown increased risk after high-dose fractionated radiation exposure (>10 Gy), which increases approximately linearly in dose until about 40+ Gy.11 Interestingly, this risk declines after 40+ Gy, possibly due to high levels of cell killing, which may include precursors of sarcoma.12 These findings are controversial as the majority of the study participants had retinoblastoma, a condition that increases the risk for sarcoma development. Regarding radiotherapy, limited data have suggested an increased risk after high-dose exposure in adult sarcoma patients.11 In a study of 100,000 women with breast cancer in Sweden, dose of radiotherapy predicted sarcoma development, except angiosarcoma.13 In addition, recent findings from studies on Japanese atomic bomb survivors have suggested that the risk of sarcomas is elevated by acute lower doses of radiation (<5 Gy) at any age.11 Osteosarcoma is the most common type of post- radiation sarcoma, followed by malignant fibrous histiocytoma and fibrosarcoma.14 The good absorption of radiation by bone may account for the high incidence of bone sarcoma relative to other sites.
In addition to radiation, alkylating agents such as cyclophosphamide have been reported to be an independent risk factor for bone sarcoma. Tamoxifen, a commonly used agent for the treatment of estrogen receptor-positive breast cancer, has been linked to the development of uterine sarcomas.15
Viral infection
Epstein–Barr virus (EBV) and Kaposi’s sarcoma-associated herpes virus (KSHV), also known as human herpes virus 8 (HHV-8), play direct roles in carcinogenesis-encoding oncoproteins that are able to promote cellular transformation. Both of these human gamma herpes viruses are known for their oncogenic properties, for the viral products that mimic or interfere with the functions of critical cellular proteins, and for their ability to escape immune responses.16 Greater than 90% of the world population is infected with EBV, and EBV has been implicated in the development of leiomyosarcomas in HIV-infected and transplant patients.17 Associations between EBV and HHV-8 have been noted to occur in the setting of chronic immunosuppression (ie, untreated HIV patients or patients with a history of organ transplantation).
Occupational factors
Associations between job type and sarcoma risk have been reported, but with inconsistent results. For example, the association between agriculture-based occupations and risk of sarcoma has been positive in some analyses and negative in others.18–20 These inconsistencies may potentially be due to the small number of cases in some of the studies. Other occupations, such as blacksmiths, toolmakers, machine-tool operators, carpenters, health-related occupations (such as radiologists), and individuals working in the manufacturing of wood, cork products, and straw, have been reported to have an increased risk for bone tumor.21,22 Exposures to several chemical agents, including dioxin, chlorophenols, and other solvents have also been associated with the risk of STS development;23–25 other studies did not observe these associations, however.26–28
Hereditary syndromes
The risk of sarcoma increases with different inherited genetic syndromes, including Li–Fraumeni syndrome, retinoblastoma, Werner’s syndrome, Rothmund-Thompson syndrome, neurofibromatosis, and enchondromatosis. Li–Fraumeni syndrome results from germline mutations in the TP53 suppressor gene. Sarcoma patients are more likely to have TP53 germline mutations, and sarcoma represents 25% of tumors in TP53 mutation carriers.29 Retinoblastoma develops through germline mutations in the RB1 tumor suppressor gene, and retinoblastoma survivors have shown an increased risk of sarcoma compared to the general population.30 Werner syndrome is an autosomal inherited disease caused by a mutation in the DNA helicase gene, WRN. Werner syndrome patients have an increased propensity to develop sarcomas31. Neurofibromatosis patients have a 10% cumulative lifetime risk of developing sarcoma.
Disease
Paget’s disease of bone is a focal disorder of bone metabolism mediated by abnormal osteoclast function. This disease has been associated with about 1% of osteosarcomas. These cancers occur mostly in patients with long-standing polyostotic disease who are older than 65 years of age.32,33 A molecular basis for the association of osteosarcoma with Paget’s disease is unclear.
Diamond Black fan anemia is an inherited red blood disorder where 40% of patients have mutations in genes important in ribosomal function.34 This condition is associated with an increased risk of osteosarcoma,35 suggesting a potential role of ribosomal proteins in osteosarcoma development, although data in this area are very limited.
Hormones
The differential incidence of histologic subtype-specific sarcomas between the genders has suggested that hormones may be involved in tumorigenesis of sarcoma, including leiomyosarcoma, but data on their potential role are limited. In a case control study of 104 STS patients and 505 controls, no significant associations between female hormone-related factors, including parity, age at menopause, and menstrual cycle patterns, were shown.36 However, a suggestive association was observed in women who were first pregnant at age > 29 years. In addition, use of oral contraceptives has been reported to increase the risk of chondrosarcoma.37
Other risk factors
Various studies have reported on other risk factors associated with the development of sarcoma in children, including the use of antibiotics38 or medications for vomiting and nausea during pregnancy39 or the use of antibiotics in children soon after birth. Some studies have suggested an association between pediatric sarcoma and birth weight,40 gestational age,41 birth order and maternal age,42,43 and occupation.21
However, results have been inconsistent, and further studies are needed to confirm these observations. Available data on height suggest that taller individuals are more likely to develop osteosarcoma.40 Additional information on the epidemiology of sarcoma has been summarized nicely in a recent review article.44
Rhabdomyosarcoma
Rhabdomyosarcoma, a malignancy of primitive muscle, predominates in pediatric patients but can be seen at any age.45,46 In 2009, rhabdomyosarcomas comprised 3.2% of STS diagnosed in the USA. It typically presents as a painless mass that interferes with organ function, with symptoms depending on location. There are multiple histologic subtypes, with the alveolar and embryonal subtypes being the most common forms in childhood.45 Many clinical features contribute to rhabdomyosarcoma staging and risk grouping, including tumor size, location of primary tumor, degree of surgical resection, histologic subtype, and presence of nodal or metastatic disease.
Current therapy
Roughly 16% of newly diagnosed rhabdomyosarcoma patients have metastatic disease.47–49 The most common metastatic sites include lung (18%–39%), lymph node (30%–49%), bone marrow (32%–37%), and bone (27%–33%).50–52 Chemotherapy is standard for all pediatric high-risk and relapsed patients. Surgery, radiation, or a combination is employed for local control of primary tumors. Patients with lymph node-only metastases and the alveolar subtype have a prognosis similar to patients with hematogenous metastasis. On the other hand, embryonal rhabdomyosarcoma patients who are young (<10 years old) with lymph node-only metastasis have a prognosis similar to patients with localized disease.53 Patients with metastases are more likely to be older, have alveolar subtype, have larger primary tumors, and present with an extremity or other unfavorable site primary tumor.51,52 Although outcomes have been stagnant over many years, a recently completed clinical trial (ARST0431) demonstrated improved short-term survival, especially for those with embryonal rhabdomyosarcoma, using a combination of past-tested agents, including vincristine, dactinomycin, cyclophosphamide, ifosfamide, etoposide, doxorubicin, and irinotecan; follow-up for this regimen is ongoing.54
Emerging therapies
Another ongoing study, through the Children’s Oncology Group (COG), has added an insulin growth factor receptor 1 antibody (IMC-A12) to the ARST0431 regimen (National Clinical Trial [NCT] #01055314), with aims of improving early disease control and determining the feasibility of the chemotherapy combination with biologic correlates. Bevacizumab and temsirolimus are also being evaluated in relapsed and refractory patients in a randomized fashion when added to vinorelbine and cyclophosphamide to estimate and compare event-free survival between groups (NCT#01222715). The role of higher dose chemotherapy with stem cell rescue has thus far not been validated.55,56 The pleomorphic and undifferentiated histologic subtypes of rhabdomyosarcoma, which are more common in older adults, are more resistant to chemotherapy and may present more commonly with metastatic disease. These are not treated the same as childhood rhabdomyosarcoma and are included in the leiomyosarcoma section.
Osteosarcoma
Osteosarcoma is the most common bone sarcoma in children and adolescents. Osteosarcoma accounts for approximately 400 new cancer diagnoses per year in children less than 18 years old and for a total of about 1000 new diagnoses per year. Although it affects all ages, the clear peak incidence is the period of puberty and adolescence followed by another peak in the 7th decade,34 with the majority of cases in the adult population. However, more is known about pediatric osteosarcoma through carefully conducted, randomized, single institution, and cooperative group trials.57 When treated by surgical resection alone, only 16% of patients have long-term survival, suggesting that micrometastasis is present in an overwhelming majority of newly diagnosed patients.58,59 In a large collaborative pediatric series, osteosarcoma presented with detectable metastatic disease in 11% of patients.60 The importance of complete surgical resection, burden, and location of disease for the prognosis of metastatic patients has been established.61
Metastatic osteosarcoma at diagnosis and relapsed disease with metastases both portend a poor prognosis. With primarily diagnosed metastatic osteosarcoma, metastasis location and burden correlate with outcome, with unresectable disease generally being incurable. Patients with multifocal bone osteosarcoma have high rates of Li–Fraumeni syndrome.62
Current therapy
Osteosarcomas require surgical control for cure, but a clear benefit of cytotoxic chemotherapy was established in the early 1980s. Patients with metastatic and unresectable disease have a dismal prognosis with very few long-term survivors. Additional agents such as MTP-PE (muramyl tripeptide phosphatidylethanolamine) have not improved cure rates in patients with osteosarcoma.63,64 In adolescent and young adult patients aged 18–40, less information is known, and very little has been reported in metastatic patients over 40. One report of young adults enrolled in cooperative group studies shows an inferior outcome for young adults due to increased distant relapse.65 Retrospective reports differ regarding the outcome of older patients versus pediatric patients.66,67 Improving outcomes was the aim of two recent COG studies: one studied the addition of trastuzumab and the other studied the addition of zoledronic acid to a chemotherapeutic backbone of doxorubicin, cisplatin, high-dose methotrexate, ifosfamide, and etoposide. Although trastuzumab plus doxorubicin was tolerated with acceptable cardiotoxicity, no clear overall benefit was shown, including in patients with high Human Epidermal Growth Factor Receptor 2 (HER2) levels by immunohistochemistry.68 It is too early for results on the zoledronic acid study.
Roughly 30% of localized osteosarcoma patients and 80% of patients presenting with metastatic disease will ultimately relapse.69 Many reviews have evaluated prognostic factors for distant relapsed patients. Relapses are frequently in the lung (60%–85% of patients), are local (10%–20%), or in bone (10%–20%).70,71 In the largest reported series (576 patients) median time from diagnosis to relapse was 1.6 years, with 469 patients (81%) with lung metastasis and 90 (16%) with bone metastases.70 Metastasectomy is clearly necessary for long-term disease-free interval or for potential cure, as there were no 5-year survivors in the unresected group (229 patients), whereas resected patients had a 5-year survival of 39%.70 Long interval to relapse, single site or side of pulmonary metastasis, and use of chemotherapy were among the positive prognostic variables.70 Short time to progression to lung metastases, central location, increased number of lesions, and bilateral disease are adverse prognostic factors, and patients with bilateral relapse have almost universally poor outcomes.72 Second and beyond relapses are very likely to be pulmonary and have a poor overall outcome.73 Bone metastatic disease either synchronous or metachronous is associated with a very poor but not uniformly fatal prognosis and, similar to patients with local and lung metastatic disease, requires surgery for a chance of cure.62,70,74
Emerging therapies
Two of the most well studied cell-cycle related tumor suppressors are RB1 and p53. The genes, or their pathways are mutated in the majority of osteosarcomas, but targeting these aberrations has proved to be difficult.33,75 No other clear, recurrent, genetic changes have thus far been observed in osteosarcoma, although putative genes and pathways have been explored, including HER2.68 An evaluation of 98 models or tumor samples of osteosarcoma showed very few point mutations of oncogenes and tumor suppressor genes commonly mutated in carcinomas; the PI3K pathway did emerge as a recurrently disrupted pathway in a subset of the patients.76 Emerging deep sequencing data have supported the idea that a process known as chromothripsis may be important in osteosarcoma development.77 This process involves numerous translocations acquired as a single event from chromosome shattering and re-annealing. A potential therapeutic target has thus emerged, with microtubule inhibitors demonstrating preclinical activity in osteosarcoma models.78
There are two ongoing studies in patients with relapsed metastatic osteosarcoma to lung: one with inhaled liposomal cisplatin (NCT01650090) and another with the SRC inhibitor saracatinib in the adjuvant setting following lung only recurrence (NCT00752206). Other compounds that may be translated into upcoming trials include mammalian target of rapamycin (mTOR) inhibitors, PI3K pathway inhibitors, anti-microtubular agents, cell cycle protein inhibitors, disruptors of osteoclast activity (receptor activator of nuclear factor-κB ligand inhibitors and bisphosphonates), immune strategies, and other targeted agents.
Ewing sarcoma
There are roughly 250 new cases of Ewing sarcoma per year in the USA, with 20%–30% of these patients presenting with metastases.79 More than 90% of patients display rearrangement of the EWS gene, most often to FLI1. Although results are mixed, in general, adult patients have a poorer prognosis than children and adolescents. It has long been known that metastasis is the poorest clinical prognostic marker for patients with Ewing sarcoma;80 however, there have been improvements in survival for both localized and metastatic patients over time.81
Current therapy
Patients with metastatic or unresectable Ewing sarcoma have a dismal prognosis, especially those with a large metastatic tumor burden or bone marrow disease. Vincristine, doxorubicin, and cyclophosphamide, alone or alternating with ifosfamide and etoposide, are typically used for patients with metastatic Ewing sarcoma. Metastatic presentation is most often in the lungs, although it can occur in bones, bone marrow, and other soft tissues.82 Unfortunately, systemic chemotherapy trials have not been able to improve dramatically upon the durable remission rates for patients with metastatic Ewing sarcoma. Patients without marrow disease may benefit from intensive chemotherapy followed by autologous hematologic stem cell rescue, but new strategies are actively sought in patients with metastatic disease, including targeted agents.82 An example includes the addition of agents targeted to the insulin growth factor receptor though support for these agents for a rare disease is waning among drug companies.
Emerging therapies
Although the pathophysiology of Ewing sarcoma is thought to be tightly related to transcriptional alterations in the context of the t(11;22) EWS/FLI1 translocation, therapies related to this altered fusion protein have not yet been incorporated into practice. There has been a steady identification of biological targets in Ewing sarcoma over the past few years, including clinical trials focused on the insulin growth actor receptor 1 (IGFR-1), a receptor tyrosine kinase overexpressed in Ewing sarcoma cells. Several trials completed in unselected populations of Ewing sarcoma patients have unfortunately not shown these dramatic responses (temporary responses limited to 5%–15% of patients on the order of a few months).83–88 Although this strategy is still being explored in the metastatic setting, IGFR-1 inhibition in patients with localized disease has lost the support of the drug industry, and to date no salient predictive biomarker has been identified. Another recent strategy, which added an mTOR inhibitor (temsirolimus) to further downregulate the pathway, was attempted with IGFR-1 immunohistochemistry (IHC) used to stratify patients in all sarcomas, including those with Ewing sarcoma. Overall, there was no clear predictive value to IGFR-1 IHC for response (personal communication).
Other promising therapeutic strategies include poly(ADP-ribose) polymerase (PARP) inhibitors, mithramycin, and a small molecule inhibitor of a protein–protein interaction, which includes EWS/FLI1. Although elevated levels of PARP in Ewing sarcoma cells were noted 20 years ago, only recently has this strategy attracted strong interest.89,90 An ongoing trial is investigating what proportion of unselected Ewing sarcoma patients would benefit from this strategy (NCT01583543).
Two other small molecules are being explored with a mechanism of action of disrupting the translocation protein itself. Mithramycin (ongoing study NCT01610570) is an older antibiotic identified in a drug screen, and YK-4-279 disrupts the EWS/FLI1 interaction with a binding partner, RNA helicase A.91–96 Presumably, these agents would not face the same barriers that PARP inhibitors or the IGFR-1 antibody have faced, as the fusion transcript is thought to be more central to the pathophysiology of Ewing sarcoma.
Soft tissue sarcomas
Soft tissue sarcomas most often present as a painless mass and the diagnosis is made by a combination of history and physical exam, radiologic features, and tissue biopsy. Most STS are localized, and histologic grade, tumor size, patient age, and tumor subtype are the major determinants of the therapeutic approach. Many STS display a remarkable predilection for metastasizing through circulation to the lungs. Other sites of metastasis include, but are not limited to, the liver, bones, and subcutaneous tissue with wide variation in the likelihood of metastasis to these areas depending on subtype. A minority of subtypes (synovial sarcoma, rhabdomyosarcoma, epithelioid sarcoma, clear cell sarcoma, and angiosarcoma) may metastasize to lymph nodes and, even more rarely, to other sites of the body.97,98 Histologic grade is an independent predictor of metastasis development for the main histologic types of adult STS.99
Adolescent and young adults (AYA) (18–40 years old) typically fare worse than both the younger and older cohorts of patients for a given histologic diagnosis.100–102 This is thought to arise from a multitude of factors including location of care, patient education, poor clinical trial participation, and a lack of a care system focused on the needs of this patient group.103–107 In addition to osteosarcoma and Ewing sarcoma, sarcomas with a peak incidence in the AYA population include synovial sarcoma and Malignant Peripheral Nerve Sheath Tumor (MPNST).
Current therapy
Excepting gastrointestinal stromal tumor, which is treated with targeted agents primarily, adults with metastatic soft tissue sarcomas have a poor overall survival of roughly 20%–25% at 2 years and a short median survival of 12–18 months, correlated with the grade of tumor and burden of disease. The overall survival has improved over time on patients enrolled prospectively in the early 2000s compared with historical controls in a French database.108 Patients presenting with poor performance status have significantly shorter survival, with an increased risk of early death within the first 90 days.109
The primary therapy for the treatment of localized soft tissue sarcoma is surgery with radiation being used for difficult margins or high grade tumors. Adjuvant chemotherapy is utilized by certain sarcoma centers, but is considered controversial primarily due to potential long-term toxicities and impact upon survival. Intermediate or high-grade soft tissue sarcomas may be treated with neoadjuvant chemotherapy to determine chemosensitivity, induce cytoreduction, and presumably eradicate micrometastasis. Reports on response rates vary across studies due to variations in regimens, dosing schedules, and histologic subtype selection.110
Resectable metastatic disease does confer a more favorable prognosis than unresectable, based largely on retrospective, single institution reviews.111–113 In a series of 97 patients at Massachusetts General Hospital, an increased number of pulmonary lesions, shorter time to progression, bilateral disease, and larger size of disease had inferior survival.111 Patients with multiple operations lived longer than patients who had undergone only one operation, perhaps secondary to being better candidates for surgery for the aforementioned reasons.111 Similar factors were found in another review and overall survival was near 50% in both series.111,113
Combination therapy for metastatic or unresectable disease, mainly with ifosfamide, has shown benefit in response rate. However, there have been increased toxicities and no clear corresponding benefit in overall survival.114–118 Response rates to frontline chemotherapy vary considerably in published reports both with single agents and with combinations of chemotherapy with ranges of 10%–46%.117–127 Confounding this even further are a spectrum of response rates between histologies, whereby synovial cell sarcoma and liposarcoma are more chemosensitive, while leiomyosarcoma and epithelioid sarcoma are less chemosensitive.124,128 Age also confounds analyses as there are better responses in younger patients.129–133
Beyond first-line therapy, gemcitabine and docetaxel are commonly used based on a Sarcoma Alliance for Research through Collaboration (SARC) trial which demonstrated improved activity of the combination of these agents over gemcitabine alone.124 Response rates were modest by size criteria at 16% overall, and the median progression-free survival (PFS) was just over 6 months. A recently completed trial of the tyrosine kinase inhibitor, pazopanib, demonstrated an improvement in PFS to 4.6 months versus 1.6 months in the placebo arm.134 This study included previously treated soft tissue sarcomas and excluded liposarcoma patients. Based on these study results, pazopanib was approved by the US Food and Drug Association (FDA) for the treatment of soft tissue sarcoma.
Synovial sarcoma
Based on SEER data and a large, single institution retrospective review, synovial sarcoma accounts for 6%–10% of new sarcoma diagnoses, with 70% of these diagnoses occurring in patients under 40 years old with a peak incidence in the third decade of life.135 In 2005–2009, synovial sarcoma affected 602 people in the United States, accounting for 5% of STS during that period136. Although lung metastases are present in about 6% of newly diagnosed patients, synovial sarcoma can metastasize through the lymph nodes with clinically detectable disease in 15%–20% of newly diagnosed patients.137,138 Synovial sarcomas are associated with a high risk of recurrence, estimated to be 12% locally and 39% at distant sites at 5 years,139 with a median survival of 22 months from onset of metastatic disease.140 Synovial sarcoma is characterized by the presence of a translocation between SYT on the X chromosome and SSX1, SSX2, or SSX4 on chromosome 18.141 Fusion type has been explored in terms of prognosis, but size of the primary tumor and presence of metastases at diagnosis are the most significant prognostic variables for synovial sarcoma.142,143
Current therapy
Current therapy for synovial sarcoma is similar to that for most adult type STS in the first-line setting, with surgery and radiation for all resectable localized tumors, and chemotherapy being given for larger tumors or metastatic disease. Synovial sarcoma demonstrates better response rates to conventional chemotherapy than other STS, with approximately half of patients responding.138 Among the STS, synovial sarcoma is particularly sensitive to high doses of ifosfamide, which can be given alone in the second-line setting.144
Emerging therapies
Multiple strategies for treating synovial sarcoma have demonstrated some promise. Pazopanib, a multi-tyrosine kinase inhibitor with antiangiogenic activity, has recently been approved by the FDA for sarcomas and has demonstrated increased progression-free interval, compared with placebo.134,145 Immunotherapy with T cells targeting NY-ESO, a tumor antigen, has also demonstrated promise in an ongoing trial (NCT01343043).146 Data from Phase I trials suggest promise for the use of proapoptotic modes of therapy and angiogenesis inhibitors in synovial sarcoma.147,148
MPNST
In 2005–2009, MPNST affected 220 people per year in the USA, accounting for 1.8% of all STS.136 Distant metastasis has been shown in 35 (42%) of 84 patients with pathologically confirmed MPNST from 1999 to 2011.149 MPNST presents in the context of neurofibromatosis type 1 (NF1) in about half of patients and occurs spontaneously in the other half. NF1 typically is identified either by family history or characteristic skin findings such as café au lait macules, axillary freckling, and/or multiple cutaneous neurofibromas. NF1 affects 1 in 3500 people and is characterized by a mutation in NF1, a tumor suppressor gene whose protein product, neurofibromin, is an RAS-GTPase activating protein, which negatively regulates RAS.150–153 MPNST occurs with a 10% incidence in patients with NF1 and derives from a known precursor lesion, the deep neurofibroma.154
Current therapy
Therapy for MPNST is similar to that of most adult type STS in the first-line setting, although it tends to be one of the more chemoresistant histologies and has a poorer prognosis than other histologies.155,156 An active clinical trial is incorporating etoposide into frontline therapy to establish the response rate and natural history of this malignancy (NCT00304083). Because NF1 patients often have more than one mass and because there are no reliable characteristics that distinguish benign neurofibroma from MPNST by computerized tomography or magnetic resonance imaging, positron emission tomography is particularly useful in distinguishing benign from malignant disease.157
Emerging therapies
MPNST associated with NF1 may represent a targetable malignancy with known RAS pathway activation. Although additional molecular changes are also necessary for oncogenesis, an interesting anecdote supporting the homogeneity MPNST development is a case of monozygous twins with remarkably similar phenotypes.158 Constitutive activation of the RAS/RAF/MEK/ERK pathway through biallelic loss of function of neurofibromin is a uniform and critical event in MPNST pathogenesis.159,160 mTOR inhibitors have been studied alone in clinical trials for plexiform neurofibromas and in combination for MPNST (NCT01412892). No targeted agent has thus far demonstrated activity to replace the standard of care, although a combination of the mTOR inhibitor everolimus and the anti-angiogenic agent bevacizumab are being explored (NCT01661283). Based on the current knowledge of neurofibromatosis and MPNST, it could be predicted that inhibiting pathways downstream of RAS such as MEK/ERK and PI3K/AKT/mTOR may be promising therapeutic strategies.161–166
Leiomyosarcoma
Leiomyosarcomas are sarcomas of smooth muscle origin that generally occur in older patients and account for 10% of all soft tissue sarcomas and 30% of all uterine sarcomas.167 They can arise in the retroperitoneum, uterus, extremities, blood vessels, and dermis. Unlike sarcomas associated with unbalanced translocation events or activating mutations in a tyrosine kinase receptor, these tumors exhibit complex cytogenetics. Mutations in the TP53 gene, inactivation of the PTEN gene, and mTOR activation have been observed in a subset of tumors.168
Current therapy
For patients with extra-uterine leiomyosarcomas, unresectable, metastatic disease is often treated with doxorubicin or doxorubicin-based combinations. Gemcitabine and docetaxel are often considered either in the first or second line setting in leiomyosarcomas with particular activity in uterine leiomyosarcoma.124,169 Gemcitabine and docetaxel demonstrated a response rate of 53% and PFS was 5.6 months in a single institution series of 35 patients.121
Emerging therapies
Unlike other sarcomas with a definable target, leiomyosarcomas appear to be a disease involving a series of aberrations that collectively contribute to its pathogenesis. Loss of cell cycle regulation is inferred by the frequency of p53 inactivation and alterations in RB1 function. Cellular proliferation is commonly impaired in leiomyosarcomas as evidenced by the inactivation of PTEN and upregulation of AKT and mTOR.170,171 Agents that target the AKT/mTOR pathway are of clear interest for this disease; unfortunately, no large-scale study specific to leiomyosarcomas has been performed.168 A phase I/IIa trial with ridaforolimus demonstrated a clinical benefit rate, defined by response or stable disease for at least 4 months in a quarter of the patients.172 Another Phase II trial demonstrated a medium PFS of 15.3 weeks and a clinical benefit rate of 28.8% for all sarcomas, with this rate being 33% for leiomyosarcomas, the highest subset.173
Despite the interest in targeted molecular therapeutics in leiomyosarcomas, a very promising agent is trabectedin, a marine-derived compound that inhibits transcription by forming covalent bonds with the minor groove of DNA. Activity leading to its approval in Europe has been demonstrated in liposarcoma and leiomyosarcoma.174 Trabectedin also has activity in uterine leiomyosarcoma, as shown in a retrospective series demonstrating a 3-month PFS of 53% and an impressive 6-month PFS of 33%, with mostly stable disease rather than objective responses seen.175 A Phase II study of 20 patients demonstrated similar findings.176 Another agent which is being actively explored in a Phase III study is the halichondrin B analog, eribulin, which inhibits microtubules in a novel manner. This agent demonstrated a reasonable progression free rate of 31.6% at 12 weeks in leiomyosarcomas and is being actively studied in comparison to dacarbazine in an ongoing Phase III trial (NCT01327885).177
Liposarcoma
Liposarcomas are soft tissue neoplasms of adipocytic lineage, which comprise the single largest group of sarcomas. They affect adults in the overwhelming majority of cases, and are most commonly identified in individuals in their 5th decade of life.178 The most common site involves the retroperitoneum, but they can also occur in the extremities. The most common form of it is the adipocytic neoplasm, a well-differentiated liposarcoma followed by dedifferentiated liposarcoma, myxoid/round cell liposarcoma, and pleomorphic liposarcoma, which is least common. Well-differentiated liposarcoma and dedifferentiated liposarcoma are considered to be a disease spectrum with low histologic features favoring the well-differentiated disease while the areas of dedifferentiation may closely resemble a high-grade, spindle cell-shaped sarcoma. While both ends of this spectrum are associated with a low-risk (10%) distant metastasis, these diseases can be devastating to local structures, especially in the abdomen. Myxoid liposarcoma, which is characterized by a t(12;16) chromosome translocation between the CHOP and FUS genes, is a less common entity that typically arises in the extremities and has a predilection for distant metastasis to areas such as the chest wall, paraspinal musculature, retroperitoneum, and the lungs.179,180
Current therapy
Liposarcomas are treated by surgical resection. Unfortunately, these tumors are often quite large at diagnosis and provide a significant challenge to the surgical team. Neoadjuvant therapies may be utilized for large, high-grade disease. Chemosensitivity is variable, with well-differentiated and dedifferentiated liposarcomas generally considered insensitive to chemotherapy;178,181 however, myxoid and pleomorphic liposarcomas may benefit from neoadjuvant chemotherapy. For patients with unresectable/metastatic disease, the primary therapeutic option is chemotherapy. Front-line therapy consists of doxorubicin or doxorubicin compounds, and second-line therapies generally consist of gemcitabine/docetaxel.
Emerging therapies
Each liposarcoma subtype exhibits unique molecular features. Liposarcomas treated as a group by a novel microtubule inhibitor, eribulin, demonstrated nearly stable disease at 3 months in roughly half of patients.177 Well- differentiated and dedifferentiated liposarcomas frequently exhibit abnormalities in chromosome 12, and approximately 95% of dedifferentiated liposarcomas display up-regulation of HDM2 and CDK4. Recent early-phase studies involving inhibitors of HDM2 and CDK4 have yielded modest results in terms of response and survivorship, although these Phase I studies, which aim to determine an adequate dose, are not powered for response analysis.
As with leiomyosarcomas, trabectedin has shown promising activity, with stable disease shown in a majority of liposarcoma patients and response rates approaching 50% by RECIST (Response Criteria in Solid Tumors).182–184 Retrospective analyses of responders have shown that trabectedin induces fat maturation similar to that seen with doxorubicin-based regimens and radiation. Liposarcomas were not included in the pazopanib study. Data from a single-institution phase II with sunitinib did show promising 3-month PFS rates for liposarcomas of 75%, suggesting activity.185
Conclusion
There is considerable diversity in pathophysiology, current and emerging therapies, and outcome for pediatric and young adult patients with metastatic sarcoma. Some subtypes, such as alveolar rhabdomyosarcoma, Ewing sarcoma, and synovial sarcoma, are well defined in regard to translocations in a majority of cases; however, events that lead to osteosarcoma and embryonal rhabdomyosarcoma are less well understood. Many patients with MPNST have a uniform first hit of a neurofibromin mutation.
Overall, the burden of disease correlates with prognosis in general. When local control is not possible, patients often have a poor prognosis. Patients who can have all areas of disease addressed by surgery or radiation to visible disease often have the best outcomes. Because of years of collaborative studies on these rare tumors, there is a well-established tumor-specific systemic therapy regimen for each histologic type. These tumors are excellent candidates for novel approaches with additional agents as they affect a portion of the population that is motivated toward curative therapy and have demonstrated that they are chemosensitive. Combinations of chemotherapy are the most effective in most settings, and many promising new agents are under active investigation or are being explored in preclinical models. There is a plethora of opportunities to explore these therapies. Ideally, clinical trials will explore multiple promising agents, be studied in a context where historical outcome is known, and will be used for a specific clinical context for a single histology.
Acknowledgment
We thank Rasa Hamilton (Moffitt Cancer Center) for editorial assistance.
Footnotes
Disclosure
Ernest Amankwah is supported by a cancer prevention fellowship from the National Cancer Institute (grant R25T CA147832). Anthony Conley serves as a consultant for Novartis Pharmaceuticals. Damon Reed receives support from the Pediatric Cancer Foundation, the Gonzmart Family Foundation, Swim Across America, and the Moffitt Cancer Center Sarcoma Foundation. No significant relationship exists between the authors and the companies/organizations whose products or services may be referenced in this article. The authors report no other conflicts of interest in this work.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Fletcher CDM, Krishnan Unni K, Mertens F, editors. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. Vol. 11. Lyon: IRAC Press; 2002. p. 298. [Google Scholar]
- 3.Howlader N, Noone AM, Krapcho M.SEER Cancer Statistics Review, 1975–2009 (Vintage 2009 Populations) Bethesda, MD: National Cancer Institute; 2012Available from: http://seer.cancer.gov/csr/1975_2009_pops09/Accessed Dec 11, 2012 [Google Scholar]
- 4.Bruland OS, Hoifodt H, Hall KS, Smeland S, Fodstad O. Bone marrow micrometastases studied by an immunomagnetic isolation procedure in extremity localized non-metastatic osteosarcoma patients. Cancer Treat Res. 2009;152:509–515. doi: 10.1007/978-1-4419-0284-9_30. [DOI] [PubMed] [Google Scholar]
- 5.Dubois SG, Epling CL, Teague J, Matthay KK, Sinclair E. Flow cytometric detection of Ewing sarcoma cells in peripheral blood and bone marrow. Pediatr Blood Cancer. 2010;54(1):13–18. doi: 10.1002/pbc.22245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DuBois SG, Stempak D, Wu B, et al. Circulating endothelial cells and circulating endothelial precursor cells in patients with osteosarcoma. Pediatr Blood Cancer. 2012;58(2):181–184. doi: 10.1002/pbc.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallego S, Llort A, Roma J, Sabado C, Gros L, de Toledo JS. Detection of bone marrow micrometastasis and microcirculating disease in rhabdomyosarcoma by a real-time RT-PCR assay. J Cancer Res Clin Oncol. 2006;132(6):356–362. doi: 10.1007/s00432-006-0083-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoshino M, Ogose A, Kawashima H, et al. Molecular analyses of cell origin and detection of circulating tumor cells in the peripheral blood in alveolar soft part sarcoma. Cancer Genet Cytogenet. 2009;190(2):75–80. doi: 10.1016/j.cancergencyto.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 9.Pennacchioli E, Tosti G, Barberis M, et al. Sarcoma spreads primarily through the vascular system: are there biomarkers associated with vascular spread? Clin Exp Metastasis. 2012;29(7):757–773. doi: 10.1007/s10585-012-9502-4. [DOI] [PubMed] [Google Scholar]
- 10.Surveillance Research Program NCI Fast Stats: An Interactive Tool for Access to SEER Cancer Statistics Available from: http://profiles.wizfolio.com/LayYiFei/publications/26760/178397/Accessed April 8, 2013
- 11.Berrington de Gonzalez A, Kutsenko A, Rajaraman P. Sarcoma risk after radiation exposure. Clin Sarcoma Res. 2012;2(1):18. doi: 10.1186/2045-3329-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hawkins MM, Wilson LM, Burton HS, et al. Radiotherapy, alkylating agents, and risk of bone cancer after childhood cancer. J Natl Cancer Inst. 1996;88(5):270–278. doi: 10.1093/jnci/88.5.270. [DOI] [PubMed] [Google Scholar]
- 13.Karlsson P, Holmberg E, Samuelsson A, Johansson KA, Wallgren A. Soft tissue sarcoma after treatment for breast cancer – a Swedish population-based study. Eur J Cancer. 1998;34(13):2068–2075. doi: 10.1016/s0959-8049(98)00319-0. [DOI] [PubMed] [Google Scholar]
- 14.Mavrogenis AF, Pala E, Guerra G, Ruggieri P. Post-radiation sarcomas. Clinical outcome of 52 Patients. Journal of surgical oncology. 2012;105(6):570–576. doi: 10.1002/jso.22122. [DOI] [PubMed] [Google Scholar]
- 15.Wickerham DL, Fisher B, Wolmark N, et al. Association of tamoxifen and uterine sarcoma. J Clin Oncol. 2002;20(11):2758–2760. doi: 10.1200/JCO.2002.20.11.2758. [DOI] [PubMed] [Google Scholar]
- 16.da Silva SR, de Oliveira DE. HIV, EBV and KSHV: viral cooperation in the pathogenesis of human malignancies. Cancer Lett. 2011;305(2):175–185. doi: 10.1016/j.canlet.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 17.Bhatia K, Shiels MS, Berg A, Engels EA. Sarcomas other than Kaposi sarcoma occurring in immunodeficiency: interpretations from a systematic literature review. Curr Opin Oncol. 2012;24(5):537–546. doi: 10.1097/CCO.0b013e328355e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balarajan R, Acheson ED. Soft tissue sarcomas in agriculture and forestry workers. J Epidemiol Community Health. 1984;38(2):113–116. doi: 10.1136/jech.38.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serraino D, Franceschi S, La Vecchia C, Carbone A. Occupation and soft-tissue sarcoma in northeastern Italy. Cancer Causes Control. 1992;3(1):25–30. doi: 10.1007/BF00051908. [DOI] [PubMed] [Google Scholar]
- 20.Wingren G, Fredrikson M, Brage HN, Nordenskjold B, Axelson O. Soft tissue sarcoma and occupational exposures. Cancer. 1990;66(4):806–811. doi: 10.1002/1097-0142(19900815)66:4<806::aid-cncr2820660435>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 21.Merletti F, Richiardi L, Bertoni F, et al. Occupational factors and risk of adult bone sarcomas: a multicentric case-control study in Europe. Int J Cancer. 2006;118(3):721–727. doi: 10.1002/ijc.21388. [DOI] [PubMed] [Google Scholar]
- 22.Pukkala E, Martinsen JI, Lynge E, et al. Occupation and cancer – follow-up of 15 million people in five Nordic countries. Acta Oncol. 2009;48(5):646–790. doi: 10.1080/02841860902913546. [DOI] [PubMed] [Google Scholar]
- 23.Eriksson M, Hardell L, Adami HO. Exposure to dioxins as a risk factor for soft tissue sarcoma: a population-based case-control study. J Natl Cancer Inst. 1990;82(6):486–490. doi: 10.1093/jnci/82.6.486. [DOI] [PubMed] [Google Scholar]
- 24.Hoppin JA, Tolbert PE, Herrick RF, et al. Occupational chlorophenol exposure and soft tissue sarcoma risk among men aged 30–60 years. Am J Epidemiol. 1998;148(7):693–703. doi: 10.1093/aje/148.7.693. [DOI] [PubMed] [Google Scholar]
- 25.Kogevinas M, Kauppinen T, Winkelmann R, et al. Soft tissue sarcoma and non-Hodgkin’s lymphoma in workers exposed to phenoxy herbicides, chlorophenols, and dioxins: two nested case-control studies. Epidemiology. 1995;6(4):396–402. [PubMed] [Google Scholar]
- 26.Hardell L, Eriksson M. The association between soft tissue sarcomas and exposure to phenoxyacetic acids. A new case-referent study. Cancer. 1988;62(3):652–656. doi: 10.1002/1097-0142(19880801)62:3<652::aid-cncr2820620334>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 27.Smith JG, Christophers AJ. Phenoxy herbicides and chlorophenols: a case control study on soft tissue sarcoma and malignant lymphoma. Br J Cancer. 1992;65(3):442–448. doi: 10.1038/bjc.1992.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woods JS, Polissar L, Severson RK, Heuser LS, Kulander BG. Soft tissue sarcoma and non-Hodgkin’s lymphoma in relation to phenoxyherbicide and chlorinated phenol exposure in western Washington. J Natl Cancer Inst. 1987;78(5):899–910. [PubMed] [Google Scholar]
- 29.Ognjanovic S, Martel G, Manivel C, Olivier M, Langer E, Hainaut P. Low Prevalence of TP53 Mutations and MDM2 Amplifications in Pediatric Rhabdomyosarcoma. Sarcoma. 2012;2012:492086. doi: 10.1155/2012/492086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen MF, Koufos A, Gallie BL, et al. Osteosarcoma and retinoblastoma: a shared chromosomal mechanism revealing recessive predisposition. Proc Natl Acad Sci USA. 1985;82(18):6216–6220. doi: 10.1073/pnas.82.18.6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu JJ, Kamath-Loeb AS, Glick E, et al. Werner syndrome gene variants in human sarcomas. Mol Carcinog. 2010;49(2):166–174. doi: 10.1002/mc.20586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res. 2006;21(Suppl 2):P58–P63. doi: 10.1359/jbmr.06s211. [DOI] [PubMed] [Google Scholar]
- 33.Ottaviani G, Jaffe N. The etiology of osteosarcoma. Cancer Treat Res. 2009;152:15–32. doi: 10.1007/978-1-4419-0284-9_2. [DOI] [PubMed] [Google Scholar]
- 34.Savage SA, Mirabello L. Using epidemiology and genomics to understand osteosarcoma etiology. Sarcoma. 2011;2011:548151. doi: 10.1155/2011/548151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clinton C, Gazda HT. Diamond-blackfan anemia. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews. Seattle WA: University of Washington, Seattle; 1993. [Google Scholar]
- 36.Fioretti F, Tavani A, Gallus S, Negri E, Franceschi S, La Vecchia C. Menstrual and reproductive factors and risk of soft tissue sarcomas. Cancer. 2000;88(4):786–789. doi: 10.1002/(sici)1097-0142(20000215)88:4<786::aid-cncr8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 37.Anfinsen KP, Devesa SS, Bray F, et al. Age-period-cohort analysis of primary bone cancer incidence rates in the United States (1976–2005) Cancer Epidemiol Biomarkers Prev. 2011;20(8):1770–1777. doi: 10.1158/1055-9965.EPI-11-0136. [DOI] [PubMed] [Google Scholar]
- 38.Winn DM, Li FP, Robison LL, Mulvihill JJ, Daigle AE, Fraumeni JF., Jr A case-control study of the etiology of Ewing’s sarcoma. Cancer Epidemiol Biomarkers Prev. 1992;1(7):525–532. [PubMed] [Google Scholar]
- 39.Grufferman S, Wang HH, DeLong ER, Kimm SY, Delzell ES, Falletta JM. Environmental factors in the etiology of rhabdomyosarcoma in childhood. J Natl Cancer Inst. 1982;68(1):107–113. [PubMed] [Google Scholar]
- 40.Mirabello L, Pfeiffer R, Murphy G, et al. Height at diagnosis and birth-weight as risk factors for osteosarcoma. Cancer Causes Control. 2011;22(6):899–908. doi: 10.1007/s10552-011-9763-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spector LG, Puumala SE, Carozza SE, et al. Cancer risk among children with very low birth weights. Pediatrics. 2009;124(1):96–104. doi: 10.1542/peds.2008-3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ognjanovic S, Carozza SE, Chow EJ, et al. Birth characteristics and the risk of childhood rhabdomyosarcoma based on histological subtype. Bri J Cancer. 2010;102(1):227–231. doi: 10.1038/sj.bjc.6605484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Von Behren J, Spector LG, Mueller BA, et al. Birth order and risk of childhood cancer: a pooled analysis from five US States. Int J Cancer. 2011;128(11):2709–2716. doi: 10.1002/ijc.25593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14. doi: 10.1186/2045-3329-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arndt CA, Crist WM. Common musculoskeletal tumors of childhood and adolescence. N Engl J Med. 1999;341(5):342–352. doi: 10.1056/NEJM199907293410507. [DOI] [PubMed] [Google Scholar]
- 46.Malempati S, Hawkins DS. Rhabdomyosarcoma: review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer. 2012;59(1):5–10. doi: 10.1002/pbc.24118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crist W, Gehan EA, Ragab AH, et al. The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol. 1995;13(3):610–630. doi: 10.1200/JCO.1995.13.3.610. [DOI] [PubMed] [Google Scholar]
- 48.Maurer HM, Beltangady M, Gehan EA, et al. The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer. 1988;61(2):209–220. doi: 10.1002/1097-0142(19880115)61:2<209::aid-cncr2820610202>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 49.Maurer HM, Gehan EA, Beltangady M, et al. The Intergroup Rhabdomyosarcoma Study-II. Cancer. 1993;71(5):1904–1922. doi: 10.1002/1097-0142(19930301)71:5<1904::aid-cncr2820710530>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 50.Dantonello TM, Int-Veen C, Winkler P, et al. Initial patient characteristics can predict pattern and risk of relapse in localized rhabdomyosarcoma. J Clin Oncol. 2008;26(3):406–413. doi: 10.1200/JCO.2007.12.2382. [DOI] [PubMed] [Google Scholar]
- 51.Breneman JC, Lyden E, Pappo AS, et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma – a report from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol. 2003;21(1):78–84. doi: 10.1200/JCO.2003.06.129. [DOI] [PubMed] [Google Scholar]
- 52.Oberlin O, Rey A, Lyden E, et al. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J Clin Oncol. 2008;26(14):2384–2389. doi: 10.1200/JCO.2007.14.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodeberg DA, Garcia-Henriquez N, Lyden ER, et al. Prognostic significance and tumor biology of regional lymph node disease in patients with rhabdomyosarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2011;29(10):1304–1311. doi: 10.1200/JCO.2010.29.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weigel B, Lyden E, Anderson JR, et al. Early results from Children’s Oncology Group (COG) ARST0431: intensive multidrug therapy for patients with metastatic rhabdomyosarcoma (RMS). 2010 ASCO Annual Meeting. J Clin Oncol. 2010;28(Suppl 15):A-9503. [Google Scholar]
- 55.Peinemann F, Kroger N, Bartel C, et al. High-dose chemotherapy followed by autologous stem cell transplantation for metastatic rhabdomyosarcoma – a systematic review. PloS One. 2011;6(2):e17127. doi: 10.1371/journal.pone.0017127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weigel BJ, Breitfeld PP, Hawkins D, Crist WM, Baker KS. Role of high-dose chemotherapy with hematopoietic stem cell rescue in the treatment of metastatic or recurrent rhabdomyosarcoma. J Pediatr Hematol Oncol. 2001;23(5):272–276. doi: 10.1097/00043426-200106000-00007. [DOI] [PubMed] [Google Scholar]
- 57.Benjamin RS, Patel SR. Pediatric and adult osteosarcoma: comparisons and contrasts in presentation and therapy. Cancer Treat Res. 2009;152:355–363. doi: 10.1007/978-1-4419-0284-9_19. [DOI] [PubMed] [Google Scholar]
- 58.Link MP, Goorin AM, Miser AW, et al. The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med. 1986;314(25):1600–1606. doi: 10.1056/NEJM198606193142502. [DOI] [PubMed] [Google Scholar]
- 59.Eilber F, Giuliano A, Eckardt J, Patterson K, Moseley S, Goodnight J. Adjuvant chemotherapy for osteosarcoma: a randomized prospective trial. J Clin Oncol. 1987;5(1):21–26. doi: 10.1200/JCO.1987.5.1.21. [DOI] [PubMed] [Google Scholar]
- 60.Kager L, Zoubek A, Potschger U, et al. Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol. 2003;21(10):2011–2018. doi: 10.1200/JCO.2003.08.132. [DOI] [PubMed] [Google Scholar]
- 61.Meyers PA, Heller G, Healey JH, et al. Osteogenic sarcoma with clinically detectable metastasis at initial presentation. J Clin Oncol. 1993;11(3):449–453. doi: 10.1200/JCO.1993.11.3.449. [DOI] [PubMed] [Google Scholar]
- 62.Brandal P, Bjerkehagen B, Bruland OS, Skjeldal S, Bogsrud TV, Hall KS. Synchronous and metachronous skeletal osteosarcomas: the Norwegian Radium Hospital experience. Acta Oncol. 2009;48(8):1165–1172. doi: 10.3109/02841860903032809. [DOI] [PubMed] [Google Scholar]
- 63.Chou AJ, Kleinerman ES, Krailo MD, et al. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: a report from the Children’s Oncology Group. Cancer. 2009;115(22):5339–5348. doi: 10.1002/cncr.24566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meyers PA, Schwartz CL, Krailo MD, et al. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival – a report from the Children’s Oncology Group. J Clin Oncol. 2008;26(4):633–638. doi: 10.1200/JCO.2008.14.0095. [DOI] [PubMed] [Google Scholar]
- 65.Janeway KA, Barkauskas DA, Krailo MD, et al. Outcome for adolescent and young adult patients with osteosarcoma: a report from the Children’s Oncology Group. Cancer. 2012;118(18):4597–4605. doi: 10.1002/cncr.27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aljubran AH, Griffin A, Pintilie M, Blackstein M. Osteosarcoma in adolescents and adults: survival analysis with and without lung metastases. Ann Oncol. 2009;20(6):1136–1141. doi: 10.1093/annonc/mdn731. [DOI] [PubMed] [Google Scholar]
- 67.Harting MT, Lally KP, Andrassy RJ, et al. Age as a prognostic factor for patients with osteosarcoma: an analysis of 438 patients. J Cancer Res Clin Oncol. 2010;136(4):561–570. doi: 10.1007/s00432-009-0690-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ebb D, Meyers P, Grier H, et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: a report from the children’s oncology group. J Clin Oncol. 2012;30(20):2545–2551. doi: 10.1200/JCO.2011.37.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O’Day K, Gorlick R. Novel therapeutic agents for osteosarcoma. Expert Rev Anticancer Ther. 2009;9(4):511–523. doi: 10.1586/era.09.7. [DOI] [PubMed] [Google Scholar]
- 70.Kempf-Bielack B, Bielack SS, Jurgens H, et al. Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS) J Clin Oncol. 2005;23(3):559–568. doi: 10.1200/JCO.2005.04.063. [DOI] [PubMed] [Google Scholar]
- 71.Gelderblom H, Jinks RC, Sydes M, et al. Survival after recurrent osteosarcoma: data from 3 European Osteosarcoma Intergroup (EOI) randomized controlled trials. Eur J Cancer. 2011;47(6):895–902. doi: 10.1016/j.ejca.2010.11.036. [DOI] [PubMed] [Google Scholar]
- 72.Letourneau PA, Xiao L, Harting MT, et al. Location of pulmonary metastasis in pediatric osteosarcoma is predictive of outcome. J Pediatr Surg. 2011;46(7):1333–1337. doi: 10.1016/j.jpedsurg.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bielack SS, Kempf-Bielack B, Branscheid D, et al. Second and subsequent recurrences of osteosarcoma: presentation, treatment, and outcomes of 249 consecutive cooperative osteosarcoma study group patients. J Clin Oncol. 2009;27(4):557–565. doi: 10.1200/JCO.2008.16.2305. [DOI] [PubMed] [Google Scholar]
- 74.Nathan SS, Gorlick R, Bukata S, et al. Treatment algorithm for locally recurrent osteosarcoma based on local disease-free interval and the presence of lung metastasis. Cancer. 2006;107(7):1607–1616. doi: 10.1002/cncr.22197. [DOI] [PubMed] [Google Scholar]
- 75.Scholz RB, Kabisch H, Weber B, Roser K, Delling G, Winkler K. Studies of the RB1 gene and the p53 gene in human osteosarcomas. Pediatr Hematol Oncol. 1992;9(2):125–137. doi: 10.3109/08880019209018328. [DOI] [PubMed] [Google Scholar]
- 76.Choy E, Hornicek F, MacConaill L, et al. High-throughput genotyping in osteosarcoma identifies multiple mutations in phosphoinositide-3-kinase and other oncogenes. Cancer. 2012;118(11):2905–2914. doi: 10.1002/cncr.26617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144(1):27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Okouneva T, Azarenko O, Wilson L, Littlefield BA, Jordan MA. Inhibition of centromere dynamics by eribulin (E7389) during mitotic metaphase. Mol Cancer Ther. 2008;7(7):2003–2011. doi: 10.1158/1535-7163.MCT-08-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rodriguez-Galindo C, Navid F, Liu T, Billups CA, Rao BN, Krasin MJ. Prognostic factors for local and distant control in Ewing sarcoma family of tumors. Ann Oncol. 2008;19(4):814–820. doi: 10.1093/annonc/mdm521. [DOI] [PubMed] [Google Scholar]
- 80.Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol. 2000;18(17):3108–3114. doi: 10.1200/JCO.2000.18.17.3108. [DOI] [PubMed] [Google Scholar]
- 81.Esiashvili N, Goodman M, Marcus RB., Jr Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol. 2008;30(6):425–430. doi: 10.1097/MPH.0b013e31816e22f3. [DOI] [PubMed] [Google Scholar]
- 82.Ladenstein R, Potschger U, Le Deley MC, et al. Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol. 2010;28(20):3284–3291. doi: 10.1200/JCO.2009.22.9864. [DOI] [PubMed] [Google Scholar]
- 83.Tap WD, Demetri G, Barnette P, et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol. 2012;30(15):1849–1856. doi: 10.1200/JCO.2011.37.2359. [DOI] [PubMed] [Google Scholar]
- 84.Olmos D, Postel-Vinay S, Molife LR, et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol. 2010;11(2):129–135. doi: 10.1016/S1470-2045(09)70354-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Malempati S, Weigel B, Ingle AM, et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(3):256–262. doi: 10.1200/JCO.2011.37.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Juergens H, Daw NC, Geoerger B, et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol. 2011;29(34):4534–4540. doi: 10.1200/JCO.2010.33.0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pappo AS, Patel SR, Crowley J, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol. 2011;29(34):4541–4547. doi: 10.1200/JCO.2010.34.0000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Atzori F, Tabernero J, Cervantes A, et al. A phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clinical Cancer Res. 2011;17(19):6304–6312. doi: 10.1158/1078-0432.CCR-10-3336. [DOI] [PubMed] [Google Scholar]
- 89.Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Soldatenkov VA, Albor A, Patel BK, Dreszer R, Dritschilo A, Notario V. Regulation of the human poly(ADP-ribose) polymerase promoter by the ETS transcription factor. Oncogene. 1999;18(27):3954–3962. doi: 10.1038/sj.onc.1202778. [DOI] [PubMed] [Google Scholar]
- 91.Grohar PJ, Woldemichael GM, Griffin LB, et al. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. J Nat Cancer Inst. 2011;103(12):962–978. doi: 10.1093/jnci/djr156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barber-Rotenberg JS, Selvanathan SP, Kong Y, et al. Single enantiomer of YK-4-279 demonstrates specificity in targeting the oncogene EWS-FLI1. Oncotarget. 2012;3(2):172–182. doi: 10.18632/oncotarget.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Erkizan HV, Kong Y, Merchant M, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat Med. 2009;15(7):750–756. doi: 10.1038/nm.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Erkizan HV, Scher LJ, Gamble SE, et al. Novel peptide binds EWS-FLI1 and reduces the oncogenic potential in Ewing tumors. Cell Cycle. 2011;10(19):3397–3408. doi: 10.4161/cc.10.19.17734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garrido-Laguna I, Janku F. Ridaforolimus in advanced sarcomas: a leap forward or missed opportunity? J Clin Oncol. 2012;30(8):892–893. doi: 10.1200/JCO.2011.40.6538. [DOI] [PubMed] [Google Scholar]
- 96.Kofman S, Perlia CP, Economou SG. Mithramycin in the treatment of metastatic Ewing’s sarcoma. Cancer. 1973;31(4):889–893. doi: 10.1002/1097-0142(197304)31:4<889::aid-cncr2820310419>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 97.Al-Refaie WB, Andtbacka RH, Ensor J, et al. Lymphadenectomy for isolated lymph node metastasis from extremity soft-tissue sarcomas. Cancer. 2008;112(8):1821–1826. doi: 10.1002/cncr.23363. [DOI] [PubMed] [Google Scholar]
- 98.Mazeron JJ, Suit HD. Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer. 1987;60(8):1800–1808. doi: 10.1002/1097-0142(19871015)60:8<1800::aid-cncr2820600822>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 99.Coindre JM, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer. 2001;91(10):1914–1926. doi: 10.1002/1097-0142(20010515)91:10<1914::aid-cncr1214>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 100.Bleyer A, Choi M, Fuller CD, Thomas CR, Jr, Wang SJ. Relative lack of conditional survival improvement in young adults with cancer. Semin Oncol. 2009;36(5):460–467. doi: 10.1053/j.seminoncol.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 101.Bleyer A, Montello M, Budd T, Saxman S. National survival trends of young adults with sarcoma: lack of progress is associated with lack of clinical trial participation. Cancer. 2005;103(9):1891–1897. doi: 10.1002/cncr.20995. [DOI] [PubMed] [Google Scholar]
- 102.Zbuk K, Sidebotham EL, Bleyer A, La Quaglia MP. Colorectal cancer in young adults. Semin Oncol. 2009;36(5):439–450. doi: 10.1053/j.seminoncol.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 103.Albritton KH, Eden T. Access to care. Pediatr Blood Cancer. 2008;50(Suppl 5):1094–1098. doi: 10.1002/pbc.21461. [DOI] [PubMed] [Google Scholar]
- 104.Butow P, Palmer S, Pai A, Goodenough B, Luckett T, King M. Review of adherence-related issues in adolescents and young adults with cancer. J Clin Oncol. 2010;28(32):4800–4809. doi: 10.1200/JCO.2009.22.2802. [DOI] [PubMed] [Google Scholar]
- 105.Dang-Tan T, Trottier H, Mery LS, et al. Delays in diagnosis and treatment among children and adolescents with cancer in Canada. Pediatr Blood Cancer. 2008;51(4):468–474. doi: 10.1002/pbc.21600. [DOI] [PubMed] [Google Scholar]
- 106.Downs-Canner S, Shaw PH. A comparison of clinical trial enrollment between adolescent and young adult (AYA) oncology patients treated at affiliated adult and pediatric oncology centers. J Pediatr Hematol Oncol. 2009;31(12):927–929. doi: 10.1097/MPH.0b013e3181b91180. [DOI] [PubMed] [Google Scholar]
- 107.Ferrari A, Montello M, Budd T, Bleyer A. The challenges of clinical trials for adolescents and young adults with cancer. Pediatr Blood Cancer. 2008;50(Suppl 5):1101–1104. doi: 10.1002/pbc.21459. [DOI] [PubMed] [Google Scholar]
- 108.Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049–1054. doi: 10.1002/cncr.25538. [DOI] [PubMed] [Google Scholar]
- 109.Penel N, Glabbeke MV, Mathoulin-Pelissier S, et al. Performance status is the most powerful risk factor for early death among patients with advanced soft tissue sarcoma: the European Organisation for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group (STBSG) and French Sarcoma Group (FSG) study. Br J Cancer. 2011;104(10):1544–1550. doi: 10.1038/bjc.2011.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meric F, Hess KR, Varma DG, et al. Radiographic response to neoadjuvant chemotherapy is a predictor of local control and survival in soft tissue sarcomas. Cancer. 2002;95(5):1120–1126. doi: 10.1002/cncr.10794. [DOI] [PubMed] [Google Scholar]
- 111.Kim S, Ott HC, Wright CD, et al. Pulmonary resection of metastatic sarcoma: prognostic factors associated with improved outcomes. Ann Thorac Surg. 2011;92(5:):1780–1786. 1786–1787. doi: 10.1016/j.athoracsur.2011.05.081. discussion. [DOI] [PubMed] [Google Scholar]
- 112.Kon Z, Martin L. Resection for thoracic metastases from sarcoma. Oncology (Williston Park) 2011;25(12):1198–1204. [PubMed] [Google Scholar]
- 113.Predina JD, Puc MM, Bergey MR, et al. Improved survival after pulmonary metastasectomy for soft tissue sarcoma. J Thorac Oncol. 2011;6(5):913–919. doi: 10.1097/JTO.0b013e3182106f5c. [DOI] [PubMed] [Google Scholar]
- 114.Benjamin RS, Wiernik PH, Bachur NR. Adriamycin: a new effective agent in the therapy of disseminated sarcomas. Med Pediatr Oncol. 1975;1(1):63–76. doi: 10.1002/mpo.2950010109. [DOI] [PubMed] [Google Scholar]
- 115.Demetri GD, Elias AD. Results of single-agent and combination chemotherapy for advanced soft tissue sarcomas. Implications for decision making in the clinic. Hematol Oncol Clin North Am. 1995;9(4):765–785. [PubMed] [Google Scholar]
- 116.Edmonson JH, Ryan LM, Blum RH, et al. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol. 1993;11(7):1269–1275. doi: 10.1200/JCO.1993.11.7.1269. [DOI] [PubMed] [Google Scholar]
- 117.Verma S, Younus J, Stys-Norman D, Haynes AE, Blackstein M. Ifosfamide-based combination chemotherapy in advanced soft-tissue sarcoma: a practice guideline. Curr Oncol. 2007;14(4):144–148. doi: 10.3747/co.2007.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Verma S, Younus J, Stys-Norman D, Haynes AE, Blackstein M. Meta-analysis of ifosfamide-based combination chemotherapy in advanced soft tissue sarcoma. Cancer Treat Rev. 2008;34(4):339–347. doi: 10.1016/j.ctrv.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 119.Anderson SE, Keohan ML, D’Adamo DR, Maki RG. A retrospective analysis of vinorelbine chemotherapy for patients with previously treated soft-tissue sarcomas. Sarcoma. 2006;2006:15947. doi: 10.1155/SRCM/2006/15947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Antman KH, Ryan L, Elias A, Sherman D, Grier HE. Response to ifosfamide and mesna: 124 previously treated patients with metastatic or unresectable sarcoma. J Clin Oncol. 1989;7(1):126–131. doi: 10.1200/JCO.1989.7.1.126. [DOI] [PubMed] [Google Scholar]
- 121.Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol. 2002;20(12):2824–2831. doi: 10.1200/JCO.2002.11.050. [DOI] [PubMed] [Google Scholar]
- 122.Kuttesch JF, Jr, Krailo MD, Madden T, Johansen M, Bleyer A. Phase II evaluation of intravenous vinorelbine (Navelbine) in recurrent or refractory pediatric malignancies: a Children’s Oncology Group study. Pediatr Blood Cancer. 2009;53(4):590–593. doi: 10.1002/pbc.22133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Leu KM, Ostruszka LJ, Shewach D, et al. Laboratory and clinical evidence of synergistic cytotoxicity of sequential treatment with gemcitabine followed by docetaxel in the treatment of sarcoma. J Clin Oncol. 2004;22(9):1706–1712. doi: 10.1200/JCO.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 124.Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected] J Clin Oncol. 2007;25(19):2755–2763. doi: 10.1200/JCO.2006.10.4117. [DOI] [PubMed] [Google Scholar]
- 125.Nielsen OS, Judson I, van Hoesel Q, et al. Effect of high-dose ifosfamide in advanced soft tissue sarcomas. A multicentre phase II study of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2000;36(1):61–67. doi: 10.1016/s0959-8049(99)00240-3. [DOI] [PubMed] [Google Scholar]
- 126.van Oosterom AT, Mouridsen HT, Nielsen OS, et al. Results of randomised studies of the EORTC Soft Tissue and Bone Sarcoma Group (STBSG) with two different ifosfamide regimens in first- and second-line chemotherapy in advanced soft tissue sarcoma patients. Eur J Cancer. 2002;38(18):2397–2406. doi: 10.1016/s0959-8049(02)00491-4. [DOI] [PubMed] [Google Scholar]
- 127.Van Glabbeke M, van Oosterom AT, Oosterhuis JW, et al. Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens – a European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol. 1999;17(1):150–157. doi: 10.1200/JCO.1999.17.1.150. [DOI] [PubMed] [Google Scholar]
- 128.Skubitz KM, D’Adamo DR. Sarcoma. Mayo Clin Proc. 2007;82(11):1409–1432. doi: 10.4065/82.11.1409. [DOI] [PubMed] [Google Scholar]
- 129.Karavasilis V, Seddon BM, Ashley S, Al-Muderis O, Fisher C, Judson I. Significant clinical benefit of first-line palliative chemotherapy in advanced soft-tissue sarcoma: retrospective analysis and identification of prognostic factors in 488 patients. Cancer. 2008;112(7):1585–1591. doi: 10.1002/cncr.23332. [DOI] [PubMed] [Google Scholar]
- 130.Spunt SL, Hill DA, Motosue AM, et al. Clinical features and outcome of initially unresected nonmetastatic pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Clin Oncol. 2002;20(15):3225–3235. doi: 10.1200/JCO.2002.06.066. [DOI] [PubMed] [Google Scholar]
- 131.Spunt SL, Pappo AS. Childhood nonrhabdomyosarcoma soft tissue sarcomas are not adult-type tumors. J Clin Oncol. 2006;24(12:):1958–1959. 1959–1960. doi: 10.1200/JCO.2005.05.4957. author reply. [DOI] [PubMed] [Google Scholar]
- 132.Spunt SL, Poquette CA, Hurt YS, et al. Prognostic factors for children and adolescents with surgically resected nonrhabdomyosarcoma soft tissue sarcoma: an analysis of 121 patients treated at St Jude Children’s Research Hospital. J Clin Oncol. 1999;17(12):3697–3705. doi: 10.1200/JCO.1999.17.12.3697. [DOI] [PubMed] [Google Scholar]
- 133.Spunt SL, Skapek SX, Coffin CM. Pediatric nonrhabdomyosarcoma soft tissue sarcomas. Oncologist. 2008;13(6):668–678. doi: 10.1634/theoncologist.2007-0182. [DOI] [PubMed] [Google Scholar]
- 134.van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–1886. doi: 10.1016/S0140-6736(12)60651-5. [DOI] [PubMed] [Google Scholar]
- 135.Herzog CE. Overview of sarcomas in the adolescent and young adult population. J Pediatr Hematol Oncol. 2005;27(4):215–218. doi: 10.1097/01.mph.0000161762.53175.e4. [DOI] [PubMed] [Google Scholar]
- 136.National Cancer Institute Surveillance Epidemiology End Results Available from: http://seer.cancer.govAccessed Dec 11, 2012
- 137.Tunn PU, Andreou D, Illing H, Fleige B, Dresel S, Schlag PM. Sentinel node biopsy in synovial sarcoma. Eur J Surg Oncol. 2008;34(6):704–707. doi: 10.1016/j.ejso.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 138.Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627–634. doi: 10.1002/cncr.20386. [DOI] [PubMed] [Google Scholar]
- 139.Lewis JJ, Antonescu CR, Leung DH, et al. Synovial sarcoma: a multivariate analysis of prognostic factors in 112 patients with primary localized tumors of the extremity. J Clin Oncol. 2000;18(10):2087–2094. doi: 10.1200/JCO.2000.18.10.2087. [DOI] [PubMed] [Google Scholar]
- 140.Spurrell EL, Fisher C, Thomas JM, Judson IR. Prognostic factors in advanced synovial sarcoma: an analysis of 104 patients treated at the Royal Marsden Hospital. Ann Oncol. 2005;16(3):437–444. doi: 10.1093/annonc/mdi082. [DOI] [PubMed] [Google Scholar]
- 141.Clark J, Rocques PJ, Crew AJ, et al. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet. 1994;7(4):502–508. doi: 10.1038/ng0894-502. [DOI] [PubMed] [Google Scholar]
- 142.Ferrari A, De Salvo GL, Oberlin O, et al. Synovial sarcoma in children and adolescents: a critical reappraisal of staging investigations in relation to the rate of metastatic involvement at diagnosis. Eur J Cancer. 2012;48(9):1370–1375. doi: 10.1016/j.ejca.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 143.Ladanyi M, Antonescu CR, Leung DH, et al. Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: a multi-institutional retrospective study of 243 patients. Cancer Res. 2002;62(1):135–140. [PubMed] [Google Scholar]
- 144.Lee SH, Chang MH, Baek KK, et al. High-dose ifosfamide as second-or third-line chemotherapy in refractory bone and soft tissue sarcoma patients. Oncology. 2011;80(3–4):257–261. doi: 10.1159/000328795. [DOI] [PubMed] [Google Scholar]
- 145.Sleijfer S, Ray-Coquard I, Papai Z, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043) J Clin Oncol. 2009;27(19):3126–3132. doi: 10.1200/JCO.2008.21.3223. [DOI] [PubMed] [Google Scholar]
- 146.Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Herbst RS, Eckhardt SG, Kurzrock R, et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol. 2010;28(17):2839–2846. doi: 10.1200/JCO.2009.25.1991. [DOI] [PubMed] [Google Scholar]
- 148.Fox E, Aplenc R, Bagatell R, et al. A phase 1 trial and pharmacokinetic study of cediranib, an orally bioavailable pan-vascular endothelial growth factor receptor inhibitor, in children and adolescents with refractory solid tumors. J Clin Oncol. 2010;28(35):5174–5181. doi: 10.1200/JCO.2010.30.9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kamran SC, Howard SA, Shinagare AB, et al. Malignant peripheral nerve sheath tumors: Prognostic impact of rhabdomyoblastic differentiation (malignant triton tumors), neurofibromatosis 1status and location. Eur J Surg Oncol. 2013;39(1):46–52. doi: 10.1016/j.ejso.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 150.Katz D, Lazar A, Lev D. Malignant peripheral nerve sheath tumour (MPNST): the clinical implications of cellular signalling pathways. Expert Rev Mol Med. 2009;11:e30. doi: 10.1017/S1462399409001227. [DOI] [PubMed] [Google Scholar]
- 151.Feldkamp MM, Angelov L, Guha A. Neurofibromatosis type 1 peripheral nerve tumors: aberrant activation of the Ras pathway. Surg Neurol. 1999;51(2):211–218. doi: 10.1016/s0090-3019(97)00356-x. [DOI] [PubMed] [Google Scholar]
- 152.Legius E, Marchuk DA, Collins FS, Glover TW. Somatic deletion of the neurofibromatosis type 1 gene in a neurofibrosarcoma supports a tumour suppressor gene hypothesis. Nat Genet. 1993;3(2):122–126. doi: 10.1038/ng0293-122. [DOI] [PubMed] [Google Scholar]
- 153.Xu GF, O’Connell P, Viskochil D, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62(3):599–608. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]
- 154.Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311–314. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ferrari A, Miceli R, Casanova M, et al. Adult-type soft tissue sarcomas in paediatric age: a nomogram-based prognostic comparison with adult sarcoma. Eur J Cancer. 2007;43(18):2691–2697. doi: 10.1016/j.ejca.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 156.Kattan MW, Leung DH, Brennan MF. Postoperative nomogram for 12-year sarcoma-specific death. J Clin Oncol. 2002;20(3):791–796. doi: 10.1200/JCO.2002.20.3.791. [DOI] [PubMed] [Google Scholar]
- 157.Treglia G, Taralli S, Bertagna F, et al. Usefulness of whole-body fluorine-18-fluorodeoxyglucose positron emission tomography in patients with neurofibromatosis type 1: a systematic review. Radiol Res Pract. 2012;2012:431029. doi: 10.1155/2012/431029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Melean G, Hernandez AM, Valero MC, Hernandez-Imaz E, Martin Y, Hernandez-Chico C. Monozygotic twins with Neurofibromatosis type 1, concordant phenotype and synchronous development of MPNST and metastasis. BMC Cancer. 2010;10:407. doi: 10.1186/1471-2407-10-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sawada S, Florell S, Purandare SM, Ota M, Stephens K, Viskochil D. Identification of NF1 mutations in both alleles of a dermal neurofibroma. Nat Genet. 1996;14(1):110–112. doi: 10.1038/ng0996-110. [DOI] [PubMed] [Google Scholar]
- 160.Serra E, Puig S, Otero D, et al. Confirmation of a double-hit model for the NF1 gene in benign neurofibromas. Am J Hum Genet. 1997;61(3):512–519. doi: 10.1086/515504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Eckert JM, Byer SJ, Clodfelder-Miller BJ, Carroll SL. Neuregulin-1 beta and neuregulin-1 alpha differentially affect the migration and invasion of malignant peripheral nerve sheath tumor cells. Glia. 2009;57(14):1501–1520. doi: 10.1002/glia.20866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Miller SJ, Jessen WJ, Mehta T, et al. Integrative genomic analyses of neurofibromatosis tumours identify SOX9 as a biomarker and survival gene. EMBO Mol Med. 2009;1(4):236–248. doi: 10.1002/emmm.200900027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Brems H, Beert E, de Ravel T, Legius E. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol. 2009;10(5):508–515. doi: 10.1016/S1470-2045(09)70033-6. [DOI] [PubMed] [Google Scholar]
- 164.Hilton DA, Ristic N, Hanemann CO. Activation of ERK, AKT and JNK signalling pathways in human schwannomas in situ. Histopathology. 2009;55(6):744–749. doi: 10.1111/j.1365-2559.2009.03440.x. [DOI] [PubMed] [Google Scholar]
- 165.Wee S, Jagani Z, Xiang KX, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69(10):4286–4293. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- 166.Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci USA. 2005;102(24):8573–8578. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ducimetiere F, Lurkin A, Ranchere-Vince D, et al. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. PloS One. 2011;6(8):e20294. doi: 10.1371/journal.pone.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hernando E, Charytonowicz E, Dudas ME, et al. The AKT-mTOR pathway plays a critical role in the development of leiomyosarcomas. Nat Med. 2007;13(6):748–753. doi: 10.1038/nm1560. [DOI] [PubMed] [Google Scholar]
- 169.Hensley ML. Update on gemcitabine and docetaxel combination therapy for primary and metastatic sarcomas. Curr Opin Oncol. 2010;22(4):356–361. doi: 10.1097/CCO.0b013e32833aafef. [DOI] [PubMed] [Google Scholar]
- 170.Yang J, Du X, Chen K, et al. Genetic aberrations in soft tissue leiomyosarcoma. Cancer Lett. 2009;275(1):1–8. doi: 10.1016/j.canlet.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Setsu N, Yamamoto H, Kohashi K, et al. The Akt/mammalian target of rapamycin pathway is activated and associated with adverse prognosis in soft tissue leiomyosarcomas. Cancer. 2012;118(6):1637–1648. doi: 10.1002/cncr.26448. [DOI] [PubMed] [Google Scholar]
- 172.Mita MM, Poplin E, Britten CD, et al. Phase I/IIa trial of the mammalian target of rapamycin inhibitor ridaforolimus (AP23573; MK-8669) administered orally in patients with refractory or advanced malignancies and sarcoma. Ann Oncol. 2012 doi: 10.1093/annonc/mds602. [DOI] [PubMed] [Google Scholar]
- 173.Chawla SP, Staddon AP, Baker LH, et al. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J Clin Oncol. 2012;30(1):78–84. doi: 10.1200/JCO.2011.35.6329. [DOI] [PubMed] [Google Scholar]
- 174.Demetri GD, Chawla SP, von Mehren M, et al. Efficacy and safety of trabectedin in patients with advanced or metastatic liposarcoma or leiomyosarcoma after failure of prior anthracyclines and ifosfamide: results of a randomized phase II study of two different schedules. J Clin Oncol. 2009;27(25):4188–4196. doi: 10.1200/JCO.2008.21.0088. [DOI] [PubMed] [Google Scholar]
- 175.Sanfilippo R, Grosso F, Jones RL, et al. Trabectedin in advanced uterine leiomyosarcomas: a retrospective case series analysis from two reference centers. Gynecol Oncol. 2011;123(3):553–556. doi: 10.1016/j.ygyno.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 176.Monk BJ, Blessing JA, Street DG, Muller CY, Burke JJ, Hensley ML. A phase II evaluation of trabectedin in the treatment of advanced, persistent, or recurrent uterine leiomyosarcoma: a gynecologic oncology group study. Gynecol Oncol. 2012;124(1):48–52. doi: 10.1016/j.ygyno.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Schoffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histological subtypes. Lancet Oncol. 2011;12(11):1045–1052. doi: 10.1016/S1470-2045(11)70230-3. [DOI] [PubMed] [Google Scholar]
- 178.Italiano A, Toulmonde M, Cioffi A, et al. Advanced well- differentiated/dedifferentiated liposarcomas: role of chemotherapy and survival. Ann Oncol. 2012;23(6):1601–1607. doi: 10.1093/annonc/mdr485. [DOI] [PubMed] [Google Scholar]
- 179.Asano N, Susa M, Hosaka S, et al. Metastatic patterns of myxoid/round cell liposarcoma: a review of a 25-year experience. Sarcoma. 2012;2012:345161. doi: 10.1155/2012/345161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Haniball J, Sumathi VP, Kindblom LG, et al. Prognostic factors and metastatic patterns in primary myxoid/round-cell liposarcoma. Sarcoma. 2011;2011:538085. doi: 10.1155/2011/538085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sleijfer S, Ouali M, van Glabbeke M, et al. Prognostic and predictive factors for outcome to first-line ifosfamide-containing chemotherapy for adult patients with advanced soft tissue sarcomas: an exploratory, retrospective analysis on large series from the European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group (EORTC-STBSG) Eur J Cancer. 2010;46(1):72–83. doi: 10.1016/j.ejca.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 182.Gronchi A, Bui BN, Bonvalot S, et al. Phase II clinical trial of neoadjuvant trabectedin in patients with advanced localized myxoid liposarcoma. Ann Oncol. 2012;23(3):771–776. doi: 10.1093/annonc/mdr265. [DOI] [PubMed] [Google Scholar]
- 183.Grosso F, Sanfilippo R, Virdis E, et al. Trabectedin in myxoid liposarcomas (MLS): a long-term analysis of a single-institution series. Ann Oncol. 2009;20(8):1439–1444. doi: 10.1093/annonc/mdp004. [DOI] [PubMed] [Google Scholar]
- 184.Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol. 2007;8(7):595–602. doi: 10.1016/S1470-2045(07)70175-4. [DOI] [PubMed] [Google Scholar]
- 185.Mahmood ST, Agresta S, Vigil C, et al. Phase II study of sunitinib malate, a multi-targeted tyrosine kinase inhibitor in patients with relapsed or refractory soft tissue sarcomas. Focus on 3 prevalent histologies: Leiomyosarcoma, liposarcoma, and malignant fibrous histiocytoma. Int J Cancer. 2011;129(8):1963–1969. doi: 10.1002/ijc.25843. [DOI] [PMC free article] [PubMed] [Google Scholar]