

Abstract
Background and objective
Majeed syndrome is an autosomal recessive disorder characterised by the triad of chronic recurrent multifocal osteomyelitis, congenital dyserythropoietic anaemia and a neutrophilic dermatosis that is caused by mutations in LPIN2. Long-term outcome is poor. This is the first report detailing the treatment of Majeed syndrome with biological agents and demonstrates clinical improvement with IL-1blockade.
Methods
We describe the clinical presentation, genetic analysis, cytokine profiles and response to biological therapy in two brothers with Majeed syndrome.
Results
Both boys were homozygous for a novel 2-base pair deletion in LPIN2 (c.1312_1313delCT; p.Leu438fs+16X), confirming the diagnosis. Their bone disease and anaemia were refractory to treatment with corticosteroids. Both siblings had elevated proinflammatory cytokines in their serum, including tumour necrosis factor α (TNF-α), however a trial of the TNF inhibitor etanercept resulted in no improvement. IL-1 inhibition with either a recombinant IL-1 receptor antagonist (anakinra) or an anti-IL-1β antibody (canakinumab) resulted in dramatic clinical and laboratory improvement.
Conclusions
The differential response to treatment with TNF-α or IL-1 blocking agents sheds light into disease pathogenesis; it supports the hypothesis that Majeed syndrome is an IL-1β dependent autoinflammatory disorder, and further underscores the importance of IL-1 in sterile bone inflammation.
Majeed syndrome (OMIM #609628) is a rare autosomal recessive disorder that presents with early onset chronic recurrent multifocal osteomyelitis (CRMO) and microcytic congenital dyserythropoietic anaemia, often accompanied by recurrent fever or neutrophilic dermatosis.1 Affected children present with bone pain, sometimes with fever. The radiographic findings resemble bacterial osteomyelitis, but the lesions are sterile and there is no improvement with antibiotic therapy. Corticosteroids provide only partial improvement in both the bone and skin disease.1–3 There is no effective treatment and affected individuals have persistent inflammation and go on to develop permanent joint contractures and growth deformities.1,2
Autoinflammatory disorders are innate immune system disorders that present with recurrent bouts of inflammation.4 Most are due to dysregulation of the interleukin-1 (IL-1) pathway,4 which can result in a feedback loop where IL-1 induces its own production.5 Most of the autoinflammatory disorders are responsive to IL-1 blockade.4,5 DIRA (deficiency of the IL-1 receptor antagonist) is an autoinflammatory disease of the skin and bone that presents in the first weeks of life with pustulosis, sterile osteitis and periosteitis.6,7 The syndrome is caused by mutations in IL1RN, the gene that encodes the IL-1 receptor antagonist (IL-1Ra); treatment with recombinant IL-1Ra (anakinra) results in dramatic and rapid clinical improvement.6,7 The pathophysiology of DIRA unequivocally implicates IL-1 in sterile bone inflammation. Majeed syndrome is also thought to be an autoinflammatory bone disorder, yet there is no definitive evidence for this assumption and the role of IL-1 in the disease remains unknown.8
In this report, we describe two siblings with Majeed syndrome who have a novel 2-base pair (bp) deletion in LPIN2. After failure of tumour necrosis factor (TNF) inhibition to improve their inflammatory bone disease, both boys had a dramatic clinical response to IL-1 inhibition, first with anakinra and later to canakinumab, providing the first evidence of the importance of IL-1β in Majeed syndrome pathogenesis and confirming the autoinflammatory nature of the disease.
METHODS
Informed consent
Informed consent was obtained from the parents and from the Danish National Board of Health for off-label use of canakinumab as well as for the immunological and genetic testing.
Gene sequencing
DNA extracted from whole blood and the coding regions and splice sites of LPIN2 were sequenced as previously described.9
Multiplex cytokine analysis
Blood was drawn in EDTA and centrifuged at 2000 g for 10 min within half an hour of sample collection. Plasma was stored in NuncCryo tubes (Nunc, Roskilde, Denmark) at −80°C. Plasma cytokines (IL-1β, IL-1Ra, IL-6, IL-8, IL-17, INF-γ and TNF-α) were measured in a magnetic Bio-Plex Pro Assay (Bio-Rad, Hercules, California, USA), according to the manufacturer’s instructions. Plasma samples were diluted 1:1 with sample buffer and incubated for 1 h in darkness, with rotation (approximately 400 rpm), and at room temperature. The responses were analysed at the Luminex100 using the BioPlex Manager V.6.0 software (BioRad). Detection limits were 2–7 pg/ml.
RESULTS
Clinical information
Two brothers (sibling A: 29 months old; and sibling B: 13 months old) born to related parents of Turkish ancestry were admitted with relapsing episodes of severe pain and ‘pseudoparalysis’ of upper and lower extremities since the age of 6 months and 3 months, respectively. Three months later, sibling B began having recurrent fevers lasting 1–3 days with a maximum of 38.8°C, but the boy was afebrile in between attacks. Sibling A never developed recurrent fevers. On presentation sibling A had warmth and swelling over the distal right tibia and sibling B had a swollen, tender third left phalanx. Radiographs of the affected areas were normal. Whole body MRI in sibling A performed at presentation revealed increased signal intensity on short tau inversion recovery (STIR) images and decreased signal intensity on T1 weighted images localised in the proximal and distal metaphyses of both tibiae (figure 1A), distally in the left fibula and left radius and ulna. Biopsy revealed sterile chronic non-granulomatous inflammation.
Figure 1.
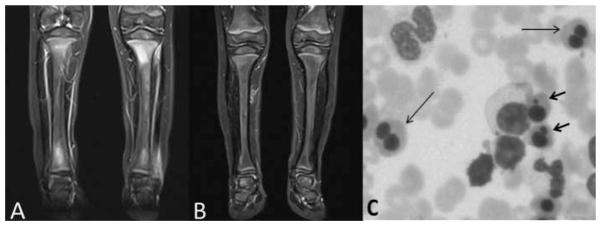
Clinical findings: MRI abnormalities improve with IL-1 inhibition. (A) MRI with coronal (short tau inversion recovery (STIR)) sequence of sibling A obtained at age 29 months before starting treatment; shows increased signal intensity on STIR images predominantly affecting both metaphyseal regions of the tibiae but with patchy involvement of the diaphyses and epiphyses as well. There is evidence of soft tissue inflammation adjacent to an area of affected bone in the metaphyseal region of the left fibula. (B) MRI STIR sequence 3 months after treatment with canakinumab. (C) Bone marrow from sibling A shows erythroblasts with binucleated (long thin arrows) and nuclear budding (short thick arrows).
Both boys had elevated erythrocyte sedimentation rates (ESR 92 mm/h and 96 mm/h, for sibling A and B, respectively), elevated C-reactive protein (19.6 and 23.7 mg/l), normal leucocyte count, slight thrombocytosis (503 and 444×109/l) and moderate anaemia (Hb 9.7 and 9.0 g/dl). Haemoglobin electrophoresis and ferritin levels were normal. Bone marrow aspirations revealed 6% and 9%, respectively, bi- or multinucleated erythroblasts. Majeed syndrome was diagnosed clinically because of the combination of CRMO and dyserythropoietic anaemia. Cumulatively, sibling A had involvement of the tibiae, left fibula, radii and left lower ribs. Sibling B had involvement of multiple phalangeal bones, right humerus, elbows, knees and ankles. Sibling B also had evidence of synovitis of the right ankle and left knee, and a temporary flexion contracture of the right knee.
The disease was refractory to treatment with corticosteroids and the TNF-α inhibitor etanercept in both boys. However, for sibling A, a rapid clinical and laboratory improvement was observed after introduction of anakinra (1.7 mg/kg/day) for 6 weeks. Symptoms flared after cessation of anakinra. To avoid daily injections, both siblings were then treated with canakinumab (anti-IL-1β antibody) 4 mg/kg/4 weeks, resulting in a dramatic effect and normalising of ESR within 1 month (figure 3) After 3 months, nearly complete resolution of bone lesions in the tibiae, left fibula (figure 1B), left radius and ulna was seen. The effect on the anaemia was less clear (figure 1C) as the anaemia is likely a mixed picture of chronic inflammation and dyserythropoiesis. Repeat bone marrow biopsies were not clinically indicated due to the mild anaemia and were not repeated after treatment.
Figure 3.

Inflammatory cytokine response over time relative to treatment of Majeed syndrome with biological agents. Cytokine profiles A and B reflect serum cytokine changes in sibling A and sibling B, respectively. Time zero reflects serum cytokines prior to the initiation of biological therapy. The right-hand panel demonstrates erythrocyte sedimentation rate (ESR) and haemoglobin over time in relation to treatment in sibling A (top) and sibling B (bottom). E, etanercept, A, anakinra, C, canakinumab. Canakinumab dosing was 4 mg/kg every 4 weeks.
LPIN2 gene resequencing of each affected child revealed a homozygous 2-base pair (bp) deletion (c.1312_1313delCT) resulting in a premature stop codon (p.Leu438fs+16X) (figure 2). The variation has not been reported in >12 000 control chromosomes (1000Genomes (http://browser.1000genomes.org/Homo_sapiens/Transcript/ProtVariations)10 and Exome Variation (Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, Washington, USA (http://evs.gs.washington.edu/EVS/); accessed 19 December 2011; n=10 753 control chromosomes)) or the In fevers database (http://fmf.igh.cnrs.fr/ISSAID/infevers/index.php). Both siblings had elevated pro-inflammatory cytokine response (IL-1β, IL-6, IL-8 and TNF-α) compared with normal. No significant change was observed in these cytokines during treatment with the TNF inhibitor etanercept (figure 3). However, treatment with IL-1 blockade was accompanied by a decline in inflammatory cytokine levels.
Figure 2.

Novel mutation in LPIN2: exon 9 forward and reverse sequences are shown for sibling A, sibling B and wild-type. There is a homozygous 2-base pair deletion in exon 9 of LPIN2 seen both in the forward and reverse directions (c.1312_1313delCT; p.Leu438fs+16X).
DISCUSSION
Majeed syndrome is a rare disorder, yet phenotypically, the bone disease resembles the more common paediatric disease CRMO.1,2,8 CRMO often occurs together with pustulosis palmoplantaris, psoriasis vulgaris or Crohn’s disease,8 suggesting a shared pathogenesis. SAPHO (synovitis, acne, pustulosis, hyperostosis, osteitis) syndrome shares many features with CRMO and is the term utilised in the adult literature; it is likely that CRMO and SAPHO syndrome share similar disease pathogenesis. Treatment for CRMO and SAPHO syndrome consists of non-steroidal anti-inflammatory drugs alone or short courses of corticosteroids, but for many this does not provide adequate disease control.11 There are reports of symptomatic improvement with methotrexate, sulfasalazine, colchicine and azathioprine, but also reports of treatment failures with each of these agents.11 For refractory cases, bisphosphonates and TNF inhibitors have been tried, with reports of efficacy in some, but treatment failures are also reported.11 There are only a few reports of the use of anakinra in CRMO and SAPHO, with mixed results.12,13
Only three mutations in LPIN2 have been identified in patients with Majeed syndrome.3,9 A (c.540–541delAT; p.Cys181*) frame shift mutation,9 a missense mutation S734L changes a highly conserved serine to a leucine,9 and a mutation in the 5′ (donor) splice site of exon 17 (c.2327+1G>C; p.Arg776Serfs*66). Here we report a novel 2 bp deletion mutation (c.1312_1313delCT) predicted to produce a truncated protein, 454 amino acids in length. LIPIN2 is an 896-amino acid protein that derives its name from its highly conserved N-terminal and C-terminal LIP domains. LIPIN-1, -2 and -3 are phosphatidate phosphatases (PAPs) important in glycerolipid biosynthesis and as transcription co-activators regulating lipid metabolism genes.14 In vitro assays demonstrate that the S734L change reported in Majeed syndrome abolishes its PAP activity but not the ability of LIPIN-2 to associate with microsomal membranes, suggesting that the absence of PAP activity is important in disease pathogenesis.15 In addition, LIPIN-2 may play an important role in the response to oxidative stress as it is highly up-regulated in several murine models of tissue injury.8 LIPIN-2 may play a role in mitosis as mutations in Ned1, a LPINortholog, cause chromosomal missegregation in yeast which may explain the dyserythropoiesis seen in Majeed syndrome.8 How an absence of PAP activity in LIPIN-2 leads to inflammation in the skin and bone remains unclear. Perhaps a defect in PAP activity results in reduced IL-1Ra production as the levels detected in the boys’ serum seem inappropriately low given the degree of inflammation.
In summary, we describe two brothers with Majeed syndrome whose systemic and bone inflammation was controlled by utilising either anakinra or canakinumab, which supports the hypothesis that IL-1 is important in disease pathogenesis. It is the response to canakinumab that specifically implicates IL-1β rather than IL-1α in disease pathogenesis. Given that both DIRA and Majeed syndrome are IL-1 pathway disorders and both share the phenotype of sterile osteitis, it is suggested that IL-1 dysregulation may play an important role in the pathogenesis of non-syndromic CRMO and SAPHO syndrome.
Acknowledgments
We would like to thank the family for participating.
Funding PJF is funded by the NIH/NIAMS: 1R01AR059703-01A1.
Footnotes
Contributors BF: interpretation of data, critical review of the manuscript with important intellectual contribution to its content, approval of the final version. MB: acquisition and interpretation of these data; critical input into the content of the paper, approval of the final version to be published. GK and HH: acquisition of data, critical input into paper content, approval of final version to be published. TH: involved in conception and design, interpretation of data, co-authored first draft and helped with revision of manuscript, approved final version. XB: acquisition of genetic sequencing data, analysis and interpretation of the data, critical review of the manuscript, final approval of the final version. PJF: involved with conception, acquisition of data, analysis and interpretation. Co-wrote, edited and revised the manuscript, data interpretation, approved final draft.
Competing interests None.
Patient consent Obtained.
Ethics approval University of Iowa Institutional Review Board for Biomedical Research.
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Majeed HA, Kalaawi M, Mohanty D, et al. Congenital dyserythropoietic anemia and chronic recurrent multifocal osteomyelitis in three related children and the association with Sweet syndrome in two siblings. J Pediatr. 1989;115:730–4. doi: 10.1016/s0022-3476(89)80650-x. [DOI] [PubMed] [Google Scholar]
- 2.Majeed HA, Al-Tarawna M, El-Shanti H, et al. The syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia. Report of a new family and a review. Eur J Pediatr. 2001;160:705–10. doi: 10.1007/s004310100799. [DOI] [PubMed] [Google Scholar]
- 3.Al-Mosawi ZS, Al-Saad KK, Ijadi-Maghsoodi R, et al. A splice site mutation confirms the role of LPIN2 in Majeed syndrome. Arthritis Rheum. 2007;56:960–4. doi: 10.1002/art.22431. [DOI] [PubMed] [Google Scholar]
- 4.Masters SL, Simon A, Aksentijevich I, et al. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annu Rev Immunol. 2009;27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dinarello CA. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol. 2011;41:1203–17. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- 6.Reddy S, Jia S, Geoffrey R, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009;360:2438–44. doi: 10.1056/NEJMoa0809568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360:2426–37. doi: 10.1056/NEJMoa0807865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferguson PJ, El-Shanti HI. Autoinflammatory bone disorders. Curr Opin Rheumatol. 2007;19:492–8. doi: 10.1097/BOR.0b013e32825f5492. [DOI] [PubMed] [Google Scholar]
- 9.Ferguson PJ, Chen S, Tayeh MK, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome) J Med Genet. 2005;42:551–7. doi: 10.1136/jmg.2005.030759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Twilt M, Laxer RM. Clinical care of children with sterile bone inflammation. Curr Opin Rheumatol. 2011;23:424–31. doi: 10.1097/BOR.0b013e328349c363. [DOI] [PubMed] [Google Scholar]
- 12.Rech J, Manger B, Lang B, et al. Adult-onset Still’s disease and chronic recurrent multifocal osteomyelitis: a hitherto undescribed manifestation of autoinflammation. Rheumatol Int. 2012;32:1827–9. doi: 10.1007/s00296-011-2020-x. [DOI] [PubMed] [Google Scholar]
- 13.Eleftheriou D, Gerschman T, Sebire N, et al. Biologic therapy in refractory chronic non-bacterial osteomyelitis of childhood. Rheumatology. 2010;49:1505–12. doi: 10.1093/rheumatology/keq122. [DOI] [PubMed] [Google Scholar]
- 14.Reue K. The lipin family: mutations and metabolism. Curr Opin Lipidol. 2009;20:165–70. doi: 10.1097/MOL.0b013e32832adee5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donkor J, Zhang P, Wong S, et al. A conserved serine residue is required for the phosphatidate phosphatase activity but not the transcriptional coactivator functions of lipin-1 and lipin-2. J Biol Chem. 2009;284:29968–78. doi: 10.1074/jbc.M109.023663. [DOI] [PMC free article] [PubMed] [Google Scholar]