

Polycystic kidney disease (PKD) represents a family of genetic disorders characterized by renal cystic growth and progression to kidney failure1. No treatment is currently available for people with PKD, although possible therapeutic interventions are emerging2, 3, 4, 5, 6, 7, 8. Despite genetic and clinical heterogeneity, PKDs have in common defects of cystic epithelia, including increased proliferation, apoptosis and activation of growth regulatory pathways1. Sphingolipids and glycosphingolipids are emerging as major regulators of these cellular processes9. We sought to evaluate the therapeutic potential for glycosphingolipid modulation as a new approach to treat PKD. Here we demonstrate that kidney glucosylceramide (GlcCer) and ganglioside GM3 levels are higher in human and mouse PKD tissue as compared to normal tissue, regardless of the causative mutation. Blockade of GlcCer accumulation with the GlcCer synthase inhibitor Genz-123346 effectively inhibits cystogenesis in mouse models orthologous to human autosomal dominant PKD (Pkd1 conditional knockout mice) and nephronophthisis (jck and pcy mice). Molecular analysis in vitro and in vivo indicates that Genz-123346 acts through inhibition of the two key pathways dysregulated in PKD: Akt protein kinase–mammalian target of rapamycin signaling and cell cycle machinery. Taken together, our data suggest that inhibition of GlcCer synthesis represents a new and effective treatment option for PKD.
PKD is transmitted as an autosomal dominant (AD) or autosomal recessive (AR) trait. Mutations in polycystin-1 and polycystin-2 are responsible for ADPKD, the most common form of PKD1, 10, 11. Recessive forms of PKD include ARPKD, a childhood disease, and nephronophthisis, the most frequent genetic cause of end-stage renal disease in the first three decades of life11, 12. Although multiple forms of PKD have differing clinical manifestations, common mechanisms promote cystogenesis at the cellular and molecular levels1. The products of the various genes mutated in PKD are expressed in primary cilia or centrosomes12. Multiple molecular mechanisms contribute to PKD, including aberrant cilia–cell cycle signaling, intracellular calcium dysregulation, Wnt pathways, cAMP-activated proliferation and the Akt–mammalian target of rapamycin (mTOR) pathway13, 14, 15, 16. Enhanced understanding of these mechanisms and development of animal models orthologous to human ADPKD, ARPKD and nephronophthisis has led to the discovery of new potential therapies1, 17. At present, there is no mechanism-based treatment available for PKD.
Sphingolipids and glycosphingolipids regulate many cellular processes, including proliferation, apoptosis and modulation of cell signaling pathways18,19, 20, 21. It is becoming increasingly recognized that glycosphingolipids have key roles in the progression of a number of diseases, including diabetes and cancer18. Glycosphingolipids are key components of membrane rafts, modulating cell surface receptors, including the epidermal growth factor, insulin and insulin-like growth factor-1 receptors22, 23, 24, 25. Alterations of glycosphingolipid metabolism, with elevated GlcCer and lactosylceramide (LacCer) abundance, have been documented in human ADPKD and the cpk mouse model, suggesting a role in cystogenesis19, 26. LacCer can act as a growth factor on kidney epithelial cells, thereby directly contributing to cystogenesis19. Recently, the gangliosides GM3 and GM1 were detected in primary cilium of epithelial cells27. Therefore, changes in glycosphingolipid metabolism in cystic epithelial cells may have a major role in driving cyst growth.
We hypothesized that inhibiting glycosphingolipid synthesis and lowering the abundance of GlcCer and its derivatives with specific GlcCer inhibitors might effectively treat PKD. We used Genz-123346, an orally available inhibitor of GlcCer synthase that shares some structural features with previously developed compounds and blocks the conversion of ceramide to GlcCer28, 29, 30. A consequence of inhibiting GlcCer is decreased expression of downstream lipids, including GM3, which has been linked to a positive effect on glycemic control in rodent models of diabetes30. A similar compound very effectively reduced GlcCer accumulation in preclinical models of Gaucher’s disease and was well tolerated in phase 1 and 2 clinical trials31, 32.
To determine whether abnormal glycosphingolipid metabolism is a common feature of PKD, we compared GlcCer and GM3 levels in normal and cystic kidneys of human ADPKD, its orthologous Pkd1 conditional knockout mouse model and jck and pcy mice orthologous to human nephronophthisis33, 34, 35 (Fig. 1). We observed significantly increased GlcCer and GM3, but not ceramide, abundance in all cystic samples analyzed compared to normal controls (Fig. 1b). These data reveal that altered glycosphingolipid metabolism is a hallmark of human and mouse PKD and may be mechanistically involved in dysregulation of cell cycle progression and proliferation. If so, lowering GlcCer abundance by specifically targeting GlcCer synthase should directly affect cell cycle progression in vitro. Indeed, depletion of GlcCer and GM3 from cultured rat kidney epithelial cells with either Genz-123346 or GlcCer synthase–specific siRNA delayed cell cycle progression (Supplementary Fig. 1).
Figure 1. Altered glycosphingolipid metabolism in human and mouse PKD kidneys.
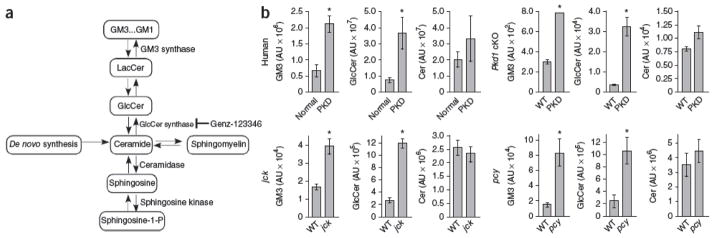
(a) Schematic representation of glycosphingolipid metabolism. The de novo synthesis of sphingolipids is a multistep process that results in the production of ceramide from serine and palmitoyl-CoA. Ceramide can be deacylated by ceramidase to generate sphingosine, which can be phosphorylated by sphingosine kinase to generate sphingosine-1-phosphate. Alternatively, ceramide can be converted to or generated from sphingomyelin, a membrane lipid, or glycosylated by UDP-glucose:ceramide glucosyltransferase (GlcCer synthase) to generate GlcCer. GlcCer can be further modified by the addition of galactose to LacCer, which can be modified by GM3 synthase to GM3. GM3 can subsequently be modified to generate other gangliosides. Genz-123346 is a direct inhibitor of GlcCer synthase. (b) Kidney GM3, GlcCer and ceramide levels measured by liquid chromatography–mass spectrometry analysis in human ADPKD, 24-day-old Pkd1 conditional knockout (cKO) mice (deleted on P1 and P2), 9-week-old jck mice and 30-week-old pcy mice relative to normal controls. AU = arbitrary units. Data are expressed as means ± s.e.m. n = 10 humans with PKD and 3 normal humans; n = 3 for all mouse samples. *P < 0.05 compared to control.
We reasoned that reducing GlcCer levels by treatment with the GlcCer synthase inhibitor Genz-123346 may block cell cycle and proliferation and attenuate cystogenesis in vivo. We gave jck mice 0.1% or 0.2% Genz-123346 in their feed from 4 to 9 weeks of age. Genz-123346 treatment resulted in a dose-dependent reduction of renal GlcCer and GM3 levels (Fig. 2a) that translated into effective inhibition of cystic disease (Fig. 2b,c and Supplementary Table 1). Because PKD develops over the life of an individual, safety is a major consideration. Genz-123346 was generally well tolerated in mice, although we noted a slight reduction in body weight gain at the highest dose (Supplementary Table 1). Notably, efficacy was achieved at lower doses as well, with no associated body weight loss (Supplementary Table 1). A similar compound has recently proven to be well tolerated in people with Gaucher’s disease in phase 1 and 2 clinical trials32.
Figure 2. Blockade of GlcCer synthase activity with Genz-123346 lowers renal GlcCer abundance and effectively inhibits PKD in jck mice.

(a) Kidney GlcCer and GM3 amounts in male mice treated with 0.1% or 0.2% Genz-123346. *P < 0.05 compared to vehicle controls. n = 3 per group. (b) Dose-dependent effect of Genz-123346 on PKD progression in jck males. Shown are quantitative analyses of kidney to body weight ratio (Kidney / BW), cystic volume and BUN in jck males. *P < 0.05 compared to vehicle controls. (c) Representative kidney sections from jck mice treated with 0.2% Genz-123346 and vehicle controls. Data are expressed as means ± s.e.m. The number of mice in each group is shown in Supplementary Table 1.
We have previously shown that mechanisms of cystogenesis in jck mice have multiple similarities to human ADPKD, including activation of mitogenic signaling pathways, dysregulated cell cycle and increased apoptosis33, 36. To determine primary molecular targets affected by glycosphingolipid modulation in response to Genz-123346 in vivo, we acutely treated jck mice with established disease (7 weeks of age) with Genz-123346 for only 5 d (Fig. 3a–d). Such short-term treatment proved sufficient to reduce kidney GlcCer levels without considerably affecting cystic growth (data not shown). Therefore, we could assess primary targets responsible for the treatment effects. Western blot analysis of treated kidneys showed a direct effect of Genz-123346 on the cell cycle machinery, as evidenced by reduced cyclin D expression and reduced Rb phosphorylation, suggesting G1/S cell cycle arrest (Fig. 3b). Reduced proliferating cell nuclear antigen (PCNA) levels confirmed the inhibitory effect of Genz-123346 on proliferation (Fig. 3b). This mechanistic effect of GlcCer synthase inhibition might be responsible for its therapeutic efficacy in PKD, as we have previously demonstrated that direct blockade of the cell cycle with the CDK inhibitor roscovitine results in a robust arrest of PKD in preclinical models2. A large body of evidence suggests that glycosphingolipids play a key part in mediating cell proliferation9. Exogenous LacCer can increase kidney proximal tubular cell proliferation19. Also, increasing GlcCer levels in Madin-Darby canine kidney cells with a β-glucosidase inhibitor promotes proliferation, whereas inhibition of GlcCer synthase activity with 1-phenyl-2-decanoylamino-3-morpholino-1-propanol hydrochloride (PDMP) decreases proliferation37. PDMP treatment of NIH3T3 cells also results in effective inhibition of proliferation through blockade of the cell cycle20.
Figure 3. Molecular pathways of cystogenesis affected by inhibition of GlcCer synthase in vivo.

(a–d) The effect of acute inhibition of GlcCer synthase. Representative immunoblots of protein extracts from 55-day-old male jck kidneys after 5 d of treatment with vehicle or 0.2% Genz-123346. (a) Schematic of the treatment regimen. (b) Immunoblots of cell cycle markers in kidneys of vehicle-treated (V) and Genz-123346–treated (T) mice. (c) The effect of Genz-123346 on apoptosis. Immunoblots of apoptotic markers expressed in kidneys of vehicle-treated and Genz-123346–treated mice. (d) Western blot analysis of Akt-mTOR and MAP kinase pathways. (e–h) The effect of long-term inhibition of GlcCer synthase activity on cystogenesis. Western blot analyses of whole kidney lysates from wild type (WT) and jck males treated with either vehicle or 0.2% Genz-123346 from 26 to 64 d of age. (e) Schematic of the treatment regimen. (f) Cell cycle marker analysis. (g) Apoptotic marker analysis. (h) Analysis of cell signaling pathway activity. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; S6, ribosomal protein S6; P-S6, phosphorylated ribosomal protein S6; Cyc, cyclin; Bcl-xL, Bcl-2–like protein-1; ApaF-1, apoptotic peptidase activating factor-1; IGF-1R, insulin-like growth factor-1 receptor.
We also observed a direct effect of Genz-123346 on the Akt-mTOR signaling pathway, with reduced phosphorylation of Akt and ribosomal protein S6 (Fig. 3d and Supplementary Fig. 2). Furthermore, mTOR has also been validated as a target for PKD through multiple preclinical trials3, 7, 8. In contrast, apoptosis or mitogen-activated protein kinase kinase (MEK)-extracellular signal–regulated kinase (ERK) signaling were not directly affected by Genz-123346 (Fig. 3c,d).
Because primary targeting of the Akt-mTOR and cell cycle pathways by Genz-123346 may indirectly affect other pathways of cystogenesis upon chronic treatment, we analyzed the long-term effects of GlcCer synthase inhibition in jck mice treated with Genz-123346 from 4 to 9 weeks (Fig. 3e–h). In addition to the inhibition of the cell cycle and Akt-mTOR pathways, chronic administration of Genz-123346 indirectly inhibited apoptosis and MEK-ERK signaling in jck kidneys (Fig. 3g,h). Of note, Genz-123346 had no effect on any molecular pathway analyzed in wild-type treated kidneys, suggesting its effect is limited to diseased kidneys (Fig. 3f–h).
To determine whether cyst-lining cells are responsible for elevated glycosphingolipid levels in diseased kidneys, we derived immortalized epithelial cells from wild-type and jck kidneys. Cultured cystic cells showed high expression of GlcCer synthase mRNA (Supplementary Fig. 3a) and elevated GlcCer and GM3 levels (Supplementary Fig. 3b) compared to wild-type cells. Notably, Akt-mTOR signaling was activated in cultured cystic cells and was attenuated in response to Genz-123346 treatment (Supplementary Fig. 3c). The in vitro data support the idea that altered glycosphingolipid metabolism modulates cell signaling and proliferation cell autonomously in kidney epithelial cells. Although our data support a link between aberrant glycosphingolipid synthesis and cystogenesis, the exact mechanisms are largely unknown and need to be investigated.
To further evaluate whether GlcCer synthase inhibition can effectively treat a slowly progressive, adult form of PKD characterized by cyst formation and fibrosis, we tested efficacy in pcy mice with 0.2% Genz-123346 in feed between 4 and 15 weeks of age. Genz-123346 lowered kidney GlcCer and GM3 abundance (Fig. 4a) and effectively inhibited cystogenesis and fibrogenesis in pcy mice (Fig. 4b–d and Supplementary Table 2). Therefore, inhibition of GlcCer synthase activity retards PKD progression in two different models of nephronophthisis.
Figure 4. Genz-123346 effectively inhibits PKD in pcy mice and in Pkd1 conditional knockout mice.
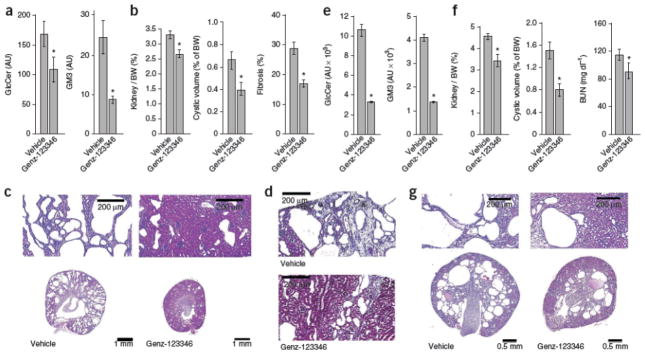
(a–d) The effect of Genz-123346 on cystogenesis in pcy mice. (a) Kidney GlcCer and GM3 levels after Genz-123346 treatment from 4 to 15 weeks of age. n = 3 per group. (b) Quantitative analysis of kidney/body weight (BW) ratio, cystic volume and fibrosis in treated pcy mice. (c) Representative kidney sections from treated pcy mice and vehicle controls at 15 weeks. (d) Mallory’s trichrome–stained kidney sections showing reduced fibrosis in Genz-123346 treated pcy mice. (e–g) The effect of Genz-123346 on cystogenesis in Pkd1 conditional knockout mice. (e) Kidney GlcCer and GM3 abundance in conditional knockout mice treated from 7 to 33 d of age. n = 3 per group. (f) Kidney/body weight ratio, cyst volume and BUN in treated Pkd1 conditional knockout mice. (g) Representative kidney sections from vehicle-treated and Genz-123346–treated Pkd1 conditional knockout mice. *P < 0.05 compared to vehicle controls. Data are expressed as means ± s.e.m. The number of mice per group for each experiment is indicated in Supplementary Tables 2 and 3.
To strengthen the argument for glycosphingolipid modulation as a new therapeutic strategy for ADPKD, we tested Genz-123346 efficacy in an orthologous mouse model with a conditionally inactivated Pkd1 gene. Such models have only recently been produced, and confirmatory trials of drugs shown to be effective in nonorthologous models are emerging4, 8. We generated mice with a germline null allele for Pkd1 (Pkd1tm1Gzbd), a conditional knockout allele with lox sites flanking exons 21–23 (Pkd1tm1Gztn) and a tamoxifen-regulatable Cre gene36, 38. Cystogenesis was induced by injecting tamoxifen into nursing females at postnatal day 5 (P5) and progressed with renal functional decline over 4–5 weeks. Genz-123346 treatment between day 7 and day 33 significantly lowered kidney GlcCer and GM3 amounts (Fig. 4e) and inhibited cystogenesis, as evidenced by reduced kidney to body weight ratio, cystic volume and blood urea nitrogen (BUN) (Fig. 4f,g and Supplementary Table 3).
The data presented here show that glycosphingolipid metabolism is altered in jck and pcy mouse models of nephronophthisis and the Pkd1 conditional knockout mouse model of ADPKD. Inhibition of GlcCer synthase alters glycosphingolipid metabolism and effectively blocks disease progression in mouse PKD. Mechanism-of-action studies suggest that GlcCer synthase inhibition results in effective cell cycle arrest and inhibition of the Akt-mTOR pathway, ultimately leading to decreased apoptosis and mitogenic signaling. Together, these results demonstrate that modulation of glycosphingolipid metabolism is a new and effective approach for the treatment of PKD.
Methods
Mouse handling and treatment
Mice were handled in accordance with Genzyme Institutional Animal Care and Use Committee guidelines. Genz-123346 is a specific GlcCer synthase inhibitor that does not inhibit other enzymes such as nonlysosomal glucocerebrosidase, acid β-glucosidase, digestive saccharases and debranching enzyme at concentrations that effectively inhibit GlcCer synthase activity31. We administered Genz-123346 ad libitum to jck mice by mixing in powdered 5053 diet (Pharmaserv) at 0.225% or 0.1125% (wt/wt) from 26 to 64 d of age or 50 to 55 d of age, as indicated in the Results and Figures 2 and 3 legends. Because jck mice show sex dimorphism in disease progression, we analyzed males and females separately33. Fibrosis is an insignificant component of PKD in jck mice and was therefore not tested in this model. We performed jck genotyping as previously described33. We maintained pcy mice on a CD1 genetic background by intercrossing homozygous mice34. We administered Genz-123346 ad libitum to pcy mice from 4 to 15 weeks of age by mixing in powdered 5053 diet at 0.225% (wt/wt). Pkd1 conditional knockout mouse generation is described in the Supplementary Methods (Pkd1tm1Gztn allele). We bred females homozygous for the Pkd1 conditional knockout allele to males homozygous for a tamoxifen-inducible Cre allele38 and heterozygous for a Pkd1 germline mutation36 (Pkd1tm1Gzbd allele) to generate mice heterozygous for the Cre allele, heterozygous for the Pkd1tm1Gztn conditional allele and either heterozygous for the Pkd1tm1Gzbd germline allele (mutant) or carrying a wild-type Pkd1 allele (wild-type controls). We induced Cre recombinase activity by injecting the nursing females with tamoxifen (250 mg per kg body weight in sunflower oil) on P5 to deliver it to the pups in the milk. We treated pups with Genz-123346 at 25 mg per kg body weight per dose twice daily from P7 to P20 and then administered 0.15% Genz-123346 in feed from P21 to P33. We killed the mice by CO2 asphyxiation before organ harvest. BUN was determined with a VetAce analyzer (Alfa Wasserman).
Histological analysis
We quantified cystic volume as described previously2. To quantify fibrosis, we digitized Mallory’s trichrome–stained kidney sections with an ACIS II system (Chromavision) and used Metamorph software (Molecular Devices) to quantify the percentage of fibrotic area to total tissue area.
Glycosphingolipid analysis
We obtained kidney samples from people with ADPKD undergoing nephrectomy for end-stage renal disease from Bioserve Biotechnologies and Cooperative Histology Tissue Network (CHTN). We obtained normal human kidney samples from Bioserve Biotechnologies. Informed consent was obtained by Bioserve Biotechnologies and the Cooperative Histology Tissue Network prior to sample collection, following approval by the relevant Institutional Review Boards. Kidney samples were homogenized at 100 mg ml 1 in distilled water with a Mini Beadbeater (Biospec Products) following the manufacturer’s protocol. We extracted sphingolipids with a modified Folch method39. We dried the supernatant under a stream of nitrogen, reconstituted in a methanol-chloroform-water mixture, which we then diluted fivefold with 0.2% formic acid (vol/vol) and 5 mM ammonium formate in 1:1 methanol-acetonitrile. Sphingolipids were seperated with an Agilent 1100 HPLC system (Agilent) equipped with a Waters Xbridge Phenyl 3.0 × 100 mm 3.5-μm column (Waters) and analyzed the eluent by electrospray ionization mass spectrometry with an API-4000 mass spectrometer (Applied Biosystems). Measurements took place in positive ion mode. All multiple reaction monitoring transitions included m/z 264.2 as the product ions. We normalized the results to total phosphate level, determined with a previously developed method40. The sphingolipid extract was digested in 15% nitric acid in a microwave oven and then analyzed by inductively coupled plasma atomic emission spectroscopy (Varian Instruments). We verified the results of relative quantification by performing absolute quantification of GlcCer levels in kidney tissues using HPLC as described previously31 (data not shown).
Western blot analysis
We homogenized samples on ice in RIPA buffer (Boston BioProducts) containing 1 mM dithiothreitol, 5 mM EDTA, 2 mM NaF, 1 mM Na3VO4 (all supplied by Sigma-Aldrich), Pefabloc SC and Complete protease inhibitor cocktail (both from Roche Applied Science). We determined protein concentrations by BCA protein assay (Pierce). We loaded equal amounts of protein on 4–12% NuPage Bis-Tris gels following the manufacturer’s protocols (Invitrogen). We performed electrophoretic transfer onto polyvinylidene difluoride membranes (Millipore) in a semi-dry apparatus according to the manufacturer’s instructions (Genomic Solutions). We blocked membranes with 5% nonfat milk in Tris-buffered saline (TBS) containing 0.1% Tween-20 and incubated with primary antibodies overnight at 4 °C. Primary antibodies were detected with horseradish peroxidase–labeled secondary antibodies at 1 in10,000 dilution (antibody to rabbit or mouse IgG: Promega; antibody to rat IgG: eBioscience). Immunoreactive proteins were elucidated by enhanced chemiluminescence (GE Healthcare). Primary antibodies to the following proteins were used: Bcl-xL, Akt, ApaF-1, cleaved poly-(ADP-ribose) polymerase, cyclin D3 (all from BD Biosciences), phospho-Akt (Ser473), IGF-IRβ, S6 ribosomal protein, phospho-S6 ribosomal protein (Ser235/236), cyclin D1, phospho-Cyclin D1 (Thr286), phospho-Rb (Ser780), total ERK, phospho-ERK (Thr202/Tyr204) (all from Cell Signaling Technologies), cyclin D2 (Biosource International), caspase-2, caspase-3 proform, MEK1/2, epidermal growth factor receptor (all from Millipore), PCNA and GAPDH (US Biological).
Statistical analysis
Data are expressed as means ± s.e.m. Comparisons were made by two-tailed t tests with GraphPad Prism software (GraphPad Software, Inc.); significance was accepted at the 0.05 level of probability (P < 0.05).
Additional methods
Detailed methodology is described in the Supplementary Methods.
Supplementary Material
Acknowledgments
We thank V. Gattone (Indiana University School of Medicine) for the kind gift of pcy breeding pairs and advice on colony maintenance. We thank S. Jones and the staff of Rodent Experimental Models (Worcester, Massachusetts) for production of the Pkd1 conditional knockout mice. We thank the staff of the Genzyme Departments of Comparative Medicine and Histology for help with in vivo studies and sample preparations. We thank S. Moreno for expert technical assistance. We are grateful to K. McEachern, R. Sacchiero, D. Copeland, S. Cheng, N. Yew, A. Smith, R. Gregory, T. Sybertz, K. Klinger and J. Burns for helpful discussions and comments on this manuscript.
References
- 1.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–168. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006;444:949–952. doi: 10.1038/nature05348. [DOI] [PubMed] [Google Scholar]
- 3.Tao Y, Kim J, Schrier RW, Edelstein CL. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol. 2005;16:46–51. doi: 10.1681/ASN.2004080660. [DOI] [PubMed] [Google Scholar]
- 4.Leuenroth SJ, Bencivenga N, Chahboune H, Hyder F, Crews CM. Triptolide reduces cyst formation in a neonatal to adult transition Pkd1 model of ADPKD. Nephrol Dial Transplant. 2010 Feb 4; doi: 10.1093/ndt/gfp777. published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gattone VH, II, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. 2003;9:1323–1326. doi: 10.1038/nm935. [DOI] [PubMed] [Google Scholar]
- 6.Ruggenenti P, et al. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int. 2005;68:206–216. doi: 10.1111/j.1523-1755.2005.00395.x. [DOI] [PubMed] [Google Scholar]
- 7.Wahl PR, et al. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD) Nephrol Dial Transplant. 2006;21:598–604. doi: 10.1093/ndt/gfi181. [DOI] [PubMed] [Google Scholar]
- 8.Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. 2010;21:489–497. doi: 10.1681/ASN.2009040421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bieberich E. Integration of glycosphingolipid metabolism and cell-fate decisions in cancer and stem cells: review and hypothesis. Glycoconj J. 2004;21:315–327. doi: 10.1023/B:GLYC.0000046274.35732.47. [DOI] [PubMed] [Google Scholar]
- 10.Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329:332–342. doi: 10.1056/NEJM199307293290508. [DOI] [PubMed] [Google Scholar]
- 11.Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13:2384–2398. doi: 10.1097/01.asn.0000028643.17901.42. [DOI] [PubMed] [Google Scholar]
- 12.Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20:23–35. doi: 10.1681/ASN.2008050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quarmby LM, Parker JD. Cilia and the cell cycle? J Cell Biol. 2005;169:707–710. doi: 10.1083/jcb.200503053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simons M, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamaguchi T, et al. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004;279:40419–40430. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 16.Wahl PR, et al. Mitotic activation of Akt signalling pathway in Han:SPRD rats with polycystic kidney disease. Nephrology (Carlton) 2007;12:357–363. doi: 10.1111/j.1440-1797.2007.00811.x. [DOI] [PubMed] [Google Scholar]
- 17.Masoumi A, Reed-Gitomer B, Kelleher C, Schrier RW. Potential pharmacological interventions in polycystic kidney disease. Drugs. 2007;67:2495–2510. doi: 10.2165/00003495-200767170-00004. [DOI] [PubMed] [Google Scholar]
- 18.Lahiri S, Futerman AH. The metabolism and function of sphingolipids and glycosphingolipids. Cell Mol Life Sci. 2007;64:2270–2284. doi: 10.1007/s00018-007-7076-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chatterjee S, Shi WY, Wilson P, Mazumdar A. Role of lactosylceramide and MAP kinase in the proliferation of proximal tubular cells in human polycystic kidney disease. J Lipid Res. 1996;37:1334–1344. [PubMed] [Google Scholar]
- 20.Rani CS, et al. Cell cycle arrest induced by an inhibitor of glucosylceramide synthase. Correlation with cyclin-dependent kinases. J Biol Chem. 1995;270:2859–2867. doi: 10.1074/jbc.270.6.2859. [DOI] [PubMed] [Google Scholar]
- 21.Cuvillier O, et al. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- 22.Hong S, Huo H, Xu J, Liao K. Insulin-like growth factor-1 receptor signaling in 3T3–L1 adipocyte differentiation requires lipid rafts but not caveolae. Cell Death Differ. 2004;11:714–723. doi: 10.1038/sj.cdd.4401405. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita T, et al. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc Natl Acad Sci USA. 2003;100:3445–3449. doi: 10.1073/pnas.0635898100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rebbaa A, Hurh J, Yamamoto H, Kersey DS, Bremer EG. Ganglioside GM3 inhibition of EGF receptor mediated signal transduction. Glycobiology. 1996;6:399–406. doi: 10.1093/glycob/6.4.399. [DOI] [PubMed] [Google Scholar]
- 25.Tagami S, et al. Ganglioside GM3 participates in the pathological conditions of insulin resistance. J Biol Chem. 2002;277:3085–3092. doi: 10.1074/jbc.M103705200. [DOI] [PubMed] [Google Scholar]
- 26.Deshmukh GD, Radin NS, Gattone VH, II, Shayman JA. Abnormalities of glycosphingolipid, sulfatide and ceramide in the polycystic (cpk/cpk) mouse. J Lipid Res. 1994;35:1611–1618. [PubMed] [Google Scholar]
- 27.Janich P, Corbeil D. GM1 and GM3 gangliosides highlight distinct lipid microdomains within the apical domain of epithelial cells. FEBS Lett. 2007;581:1783–1787. doi: 10.1016/j.febslet.2007.03.065. [DOI] [PubMed] [Google Scholar]
- 28.Lee L, Abe A, Shayman JA. Improved inhibitors of glucosylceramide synthase. J Biol Chem. 1999;274:14662–14669. doi: 10.1074/jbc.274.21.14662. [DOI] [PubMed] [Google Scholar]
- 29.Abe A, et al. Improved inhibitors of glucosylceramide synthase. J Biochem. 1992;111:191–196. doi: 10.1093/oxfordjournals.jbchem.a123736. [DOI] [PubMed] [Google Scholar]
- 30.Zhao H, et al. Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes. 2007;56:1210–1218. doi: 10.2337/db06-0719. [DOI] [PubMed] [Google Scholar]
- 31.McEachern KA, et al. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Mol Genet Metab. 2007;91:259–267. doi: 10.1016/j.ymgme.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Lukina E, et al. A phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood. 2010 May 3; doi: 10.1182/blood-2010-03-273151. published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith LA, et al. Development of polycystic kidney disease in juvenile cystic kidney mice: insights into pathogenesis, ciliary abnormalities and common features with human disease. J Am Soc Nephrol. 2006;17:2821–2831. doi: 10.1681/ASN.2006020136. [DOI] [PubMed] [Google Scholar]
- 34.Takahashi H, et al. A hereditary model of slowly progressive polycystic kidney disease in the mouse. J Am Soc Nephrol. 1991;1:980–989. doi: 10.1681/ASN.V17980. [DOI] [PubMed] [Google Scholar]
- 35.Otto EA, et al. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol. 2008;19:587–592. doi: 10.1681/ASN.2007040490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Natoli TA, et al. Pkd1 and Nek8 mutations affect cell-cell adhesion and cilia in cysts formed in kidney organ cultures. Am J Physiol Renal Physiol. 2008;294:F73–F83. doi: 10.1152/ajprenal.00362.2007. [DOI] [PubMed] [Google Scholar]
- 37.Shayman JA, et al. Modulation of renal epithelial cell growth by glucosylceramide. Association with protein kinase C, sphingosine, and diacylglycerol. J Biol Chem. 1991;266:22968–22974. [PubMed] [Google Scholar]
- 38.Seibler J, et al. Rapid generation of inducible mouse mutants. Nucleic Acids Res. 2003;31:e12. doi: 10.1093/nar/gng012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 40.Jankowski K. Microdetermination of phosphorus in organic materials from polymer industry by microwave-induced plasma atomic emission spectrometry after microwave digestion. Microchem J. 2001;70:41–49. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.