

Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and often fatal form of interstitial lung disease. We hypothesized that the local pulmonary delivery of prostaglandin E2 (PGE2) by liposomes can be used for the effective treatment of IPF. To test this hypothesis, we used a murine model of bleomycin-induced IPF to evaluate liposomes which carries PGE2 topically to the lungs. Animal survival, body weight, hydroxyproline content in the lungs, lung histology, mRNA and protein expression were studied. After inhalation delivery, liposomes accumulated predominately in the lungs. In contrast, intravenous administration led to the accumulation of liposomes mainly in kidney, liver, and spleen. Liposomal PGE2 prevented the disturbances in the expression of many genes associated with the development of IPF, substantially restricted inflammation and fibrotic injury in the lung tissues, prevented decrease in body weight, limited hydroxyproline accumulation in the lungs and virtually eliminated mortality of animals after intratracheal instillation of bleomycin. In summary, our data provide evidence that pulmonary fibrosis can be effectively treated by the inhalation administration of liposomal form of PGE2 into the lungs. The results of the present investigations make the liposomal form of PGE2 an attractive drug for the effective inhalation treatment of idiopathic pulmonary fibrosis.
Keywords: Pulmonary delivery, liposomes, prostaglandin E2, fibrosis, gene and protein expression, hydroxyproline, bleomycin, body distribution of liposomes
Introduction
Idiopathic pulmonary fibrosis (IPF), a chronic, progressive, and often fatal form of interstitial lung disease, is the most common form of idiopathic interstitial pneumonia. [1] IPF causes the loss of lung epithelial cells, accumulation of fibroblasts and myofibroblasts with replacement of normal functional tissue, extracellular matrix deposition, alteration of lung architecture. These alterations lead and pulmonary hypertension leading to substantial impairment of respiration and gas exchange often resulting in patient morbidity and mortality. [2, 3] Treatment of IPF represents a major clinical challenge since this disorder does not have reliable therapeutic options and an effective therapy has yet to be identified and developed. [2, 4, 5] Patients may ultimately require supportive oxygen therapy or pulmonary transplantation. Consequently, the development of a novel effective treatment for this devastating disease is urgently needed.
Prostaglandin E2 (PGE2), a cyclooxygenase-derived lipid mediator, has attracted considerable attention for its role in the development and progression of IPF and as a possible therapeutic agent for this disease. A role for PGE2 in the treatment of IPF is based on the very specific and unique role that PGE2 plays in the lungs making “the lung as a privileged site for the beneficial actions of PGE2.” [6] In other organs and tissues, PGE2 often acts as a potent pro-inflammatory mediator and is involved in pathogenesis of many inflammatory diseases. In contrast, in the lungs, PGE2 limits the immune-inflammatory response, inhibits specific lung fibroblast functions, their proliferation and synthesis of matrix proteins such as collagen. Consequently, PGE2 potentially can be used for the treatment of IPF. [6–10] Moreover, a synthetic analog of PGE2 (16,16-dimethyl-PGE2) recently was tested using in a model of pulmonary fibrosis (intratracheal administration of bleomycin) with promising results for treatment of IPF. [11]
Systemic delivery of PGE2 has several limitations including its short half-life in the blood stream, low accumulation in the lungs and possible adverse side effects on other organs and tissues. In contrast, local inhalation delivery of PGE2 directly to the lungs has the potential to enhance the treatment of IPF by increasing its local pulmonary concentration and preventing (or at least limiting) its penetration into the bloodstream and distribution to other healthy organs. However, free native PGE2 cannot be delivered into the lungs by inhalation necessitating a special dosage form or delivery system that can be inhaled. We have shown that liposomes and some other nanoscale-based particles can be used for local inhalation delivery of drugs, antisense oligonucleotides and siRNA, vitamins and imaging agents. [12–15] It was observed that liposomes remain in the lungs after the inhalation delivery, limiting penetration of the payload into the blood stream and accumulation in other organs. Therefore, we hypothesize that the local pulmonary delivery of liposomes containing PGE2 can be used for the effective treatment of IPF and will limit its adverse side effects on other organs. To test this hypothesis, we used a standard bleomycin-induced murine model of IPF [16–20] to evaluate a liposomal drug delivery system which delivers PGE2 topically to the lungs.
Materials and Methods
Materials
Egg phosphatidylcholine and cholesterol were purchased from Avanti Polar Lipids (Alabaster, AL, USA). PGE2 was obtained from Apichem Chemical Technology Co., Ltd. (Shanghai, China), bleomycin was purchased from Sigma Aldrich (Ronkonkoma, NY, USA). Hairless SKH1 mice, 6–8 weeks-old, were purchased from Charles River Laboratories (Wilmington, MA, USA).
Liposomal Composition of PGE2
Liposomes were prepared as previously described. [13, 14, 21–23] Briefly, egg phosphatidylcholine and cholesterol were dissolved in 4.0 ml of chloroform at 55:45 ratio. The clear lipid solution was evaporated at 25 °C under reduced pressure. A thin layer was formed and rehydrated using 2.0 ml of 0.3 M sodium citrate buffer (pH=4.0). The lipid mixture was sonicated continuously for 3.0 hours. PGE2-loaded liposomes were prepared from egg phosphatidylcholine and cholesterol (55:45 ratio) using the ethanol instillation method. [22, 24] Dry lipids and PGE2 were dissolved in 98% ethanol at room temperature; 0.9% NaCl was added to the mixture to reach final lipid and PGE2 concentrations of 20 mM each. The ethanol volume was 10% of final volume. Liposomes were extruded gradually through 200 nm and 100 nm polycarbonate membranes at room temperature using an extruder device from Northern Lipids, Inc. (Vancouver, BC, Canada). Liposomes were separated from non-encapsulated PGE2 by dialysis against 100 volumes of 0.9% NaCl overnight at 4 °C. The concentration of PEG2 in the liposomes was measured using PGE2 ELISA kit (ENZO Life Sciences, Farmingdale, NY, USA). The final concentration of PGE2 in the liposomes was 0.90 – 0.95 mole of PGE2 per mole of lipids demonstrating the encapsulation efficacy of PGE2 in liposomes around 90–95%. Aliquots of each liposomal formulation were labeled with the near infrared fluorescent dye Cy5.5 Mono NHS Ester (GE Healthcare, Amersham, UK). The fluorescent dye was dissolved together with lipids in chloroform. Approximate excitation/emission maxima of Cy5.5 were 675 nm/694 nm. Portions of liposomes were labeled with osmium tetroxide (0.5%) that was added to the rehydration buffer. The size of liposomes was measured by dynamic light scattering using a 90 Plus Particle Sizer Analyzer (Brookhaven Instruments Corp., New York, NY, USA). An aliquot of 40 μL of each sample was diluted in 2 mL of saline. Zeta potential was measured on PALS Zeta Potential Analyzer (Brookhaven Instruments Corp, New York, NY, USA). Liposomes were used at a volume of 1.5 mL. All measurements were performed at room temperature. Each parameter was measured in triplicate and average values were calculated. Mean diameter of liposomes was approximately 500 ± 25 nm; average zeta potential was 5 ± 0.7mV.
Animal Model of IPF and Treatment
Experiments were performed on healthy 6–8 weeks old SKH1-hr hairless mice (20–25 g) obtained from Charles River Laboratories (Wilmington, MA, USA). Veterinary care followed the guidelines described in the guide for the care and use of laboratory animals (AAALAC) as well as the requirements established by the animal protocol approved by the Rutgers Institutional Animal Care and Use Committee (IACUC). All mice were housed in micro-isolated cages under pathogen-free conditions at room temperature with humidity of 40 ± 15% and a 12 h light/dark cycle. Mice were anesthetized via intraperitoneal injection with 80 mg/kg ketamine and 10–12 mg/kg xylazine (Butler-Schine Animal Health Inc, Dublin, Ohio, USA). Once anesthetized, the mouse was placed on the tilting rodent work stand (Hallowell EMC, Pittsfield, MA, USA) in supine position and restrained in position by an incisor loop. The tongue was then extruded via rotation with a cotton tip applicator. The larynx was visualized using a modified 4 mm ear speculum attached to the operating head of an ophthalmoscope (Wellch Allyn, Skaneateles Falls, NY, USA). The modified speculum, acting in an inverted fashion as a laryngoscope blade, provided dorsal displacement of the tongue and magnification of the laryngeal opening as described. [13, 14] Bleomycin was administered intratracheally in doses of 0.5, 1.0, 1.5, and 2.0 U/kg. The development of fibrosis was confirmed by histopathological analysis, measurement of hydroxyproline concentration in lung tissues and overexpression of genes and proteins involved in the development of IPF. Mice were treated with liposomal PGE2 by inhalation twice a week for three weeks starting one day later after the bleomycin administration. A previously developed instillation unit consisting of a Collison nebulizer connected to four-port, nose-only exposure chambers was used for inhalation delivery of liposomal PEG2. [14] Liposomes were aerosolized at the flow rate of 2 L/min for ten min. Liposomal concentration was 20 mM in the inhaled formulations. Animal weight was measured daily throughout the study. After the three weeks treatment period, all mice were anesthetized with isoflurane and euthanized. The organs (lungs, heart, liver, kidney, spleen, and brain) were excised and used for further analysis.
Gene Expression
Mouse lungs were extracted, trachea and mainstream bronchi were separated, lungs were frozen and homogenized. RNA was isolated using an RNeasy kit (Qiagen, Valencia, CA, USA) according to manufacturer’s protocol. First-strand cDNA was synthesized with Ready-To-Go You- Prime First-Strand Beads (Amersham Biosciences, Piscataway, NJ, USA) with 1 μg of total cellular RNA (from 107 cells) and 100 ng of random hexadeoxynucleotide primer (Amersham Biosciences, Piscataway, NJ, USA). After synthesis, the reaction mixture was immediately subjected to quantitative polymerase chain reaction (QPCR). A standard Mouse Fibrosis RT Profiler™ PCR Array panel from SABiosciences (Quiagen, Valencia, CA, USA) was used. The assay was performed on lung samples from healthy mice (control), mice with lung fibrosis and mice with lung fibrosis treated with liposomal PGE2. QPCR was performed using SYBER Green Master Mix as detection agent. Fold change of the gene expression was measured using SABioscience internet software which compares the expression of tested genes with that of housekeeping genes and expresses fold change in gene expression as ΔΔCt values (ΔΔCt= ΔCttreated− ΔCtcontrol). PCR specificity was verified by melting curve and gel electrophoresis. In addition to the panel of genes provided in mouse fibrosis array, the expression of genes encoding Hypoxia Inducible Factor 1α (HIF1A), von Hippel-Lindau (VHL) and β-actin (B-ACTIN, internal standard) was measured as previously described. [12]
Histopathologic Analysis
At the end of the experiments, the animals were euthanized, the lungs were extracted and immediately fixed in 10% phosphate-buffered formalin. Samples were subsequently dehydrated and embedded in Paraplast®. Five-micrometer sections were cut and stained with hematoxylin-eosin as previously described [23, 25] and analyzed.
Immunohistochemistry
To visualize the expression of proteins, immunohistochemical staining was conducted on paraffin-embedded slides of pulmonary tissue. Slides (5 μm) were deparaffiized in xylene for 5 min followed by progressive rehydration in 100%, 95%, 70%, and 50% ethanol for 3 min during each step. Endogenous peroxidase activity was blocked by incubating slides in 3% H2O2 solution in methanol at room temperature for 10 min and washing in 300 mL PBS two times for 5 min. The slides were then stained with anti-mouse monoclonal antibodies for VEGF, CCL12, MMP3 and HIF1A proteins. Antibodies against VEGF (labeled with Alexa Fluor 488 fluorescence dye) and MMP3 (labeled with FITC) were obtained from Biolegend, San Diego, CA, USA. Antibodies against CCL12 and HIF1A (both labeled with FITC) were purchased from Biorbyt, Cambridge, UK and from Novus Biologicals, Littleton, CO, USA, respectively. All antibodies were used in in the dilution of 1:100. The slides were stained using Vector M.O.M. Immunodetection Kit (Vector Lab., Inc., Burlingame, CA, USA), visualized and photographed using a fluorescence microscope (Olympus IX71, Center Valley, PA, USA).
Hydroxyproline Assay
Fourteen days after bleomycin instillation, lungs were harvested, homogenized in distilled water and examined using a Biovision hydroxyproline assay kit (Biovision, Mount View, CA, USA). Homogenized lung tissues were hydrolyzed in 12 N HCl at 120 °C for 3 h in pressure-tight vials. After this, 10 μl of samples were allocated to a 96 well plate and dried under vacuum. Oxidation buffer with chloramine T was added to each sample at room temperature for 5 min, and the samples were then incubated in dimethylaminobenzaldehyde (DMAB) reagent for 90 min at 60 °C. Samples were cooled and absorbance at 560 nm was measured using an automated microplate plate reader. Six concentrations of hydroxyproline standard dilutions (from 0 to 1 μg/well) were used to plot a hydroxyproline standard curve.
Content of Liposomes in Different Organs
The distribution of fluorescent-labeled liposomes was examined in mouse lungs, heart, liver, spleen, kidneys, and brain. The organs were excised, rinsed in saline, and fluorescence was registered using IVIS imaging system (Xenogen Corporation, Alameda, CA, USA). Visible light and fluorescence images were taken and overlaid. The intensity of fluorescence was represented on composite light/fluorescent images by different colors, with blue reflecting the lowest fluorescence intensity and red – the highest intensity. Images of each organ were then scanned and total fluorescence intensity was calculated as previously described. [13, 14, 21] Preliminary experiments showed a strong linear correlation between the total amount of labeled substance accumulated in the organ and calculated total fluorescence intensity. The fluorescence was expressed in arbitrary units with 1 units represented approximately 2 × 1010 photons/s/sr/cm2. The method allows a quantitative comparison of the concentration of the same fluorescent dye between different series of the experiments. The mass of all organs was measured. The fluorescence intensity was normalized for organ weight.
Internalization of Liposomes by Lung Cells
Internalization of osmium-labeled liposomes by lung cells was studied by electron transmission microscopy in lung tissue sections fixed prior to microscopy using standard techniques as previously described [21, 23, 26, 27]. Briefly, lung tissue slices were fixed for 2 hours in Trump’s EM Fixative (combination of formaldehyde and glutaraldehyde in 0.1 M Milloning’s Phosphate buffer, pH 7.3). Postfixation was carried out in 1% osmium tetroxide in buffer for 1 hour followed by dehydration in graded ethanol series and embedment in Spurr’s Low Viscosity Resin. Sections were prepared using a diamond knife on a LKB-2088 Ultramicrotome (LKB-Produkter/Bromma, Sweden). Observation and micrographs were made with a JEM-100CXII Electron Microscope (JEOL Ltd., Tokyo, Japan).
Statistical Analysis
Data were analyzed using descriptive statistics and single-factor ANOVA, and are presented as a mean ± SD from five independent measurements. Five to ten animals were used in each experimental group. We analyzed data sets for significance with Student’s t test and considered P values of less than 0.05 as statistically significant.
Results
Selection of Bleomycin Dose
In order to select an appropriate dose of bleomycin, four doses (0.5; 1.0; 1.5; 2.0 U/kg) were tested. Bleomycin was instilled intratracheally and mice were observed for 21 days after the instillation. The dose of 2.0 U/kg led to the death of 100% of animals within 21 days (Figure 1A). The doses of 1.5 and 1.0 U/kg induced death of 50 and 25% of animals, respectively. The lowest tested dose (0.5 U/kg) did not induce animal death. Based on these results, 1.5 U/kg dose of bleomycin was selected for the main experiments (Figure 1A and B).
Figure 1.

Survival of mice (Kaplan-Meier survival plot) after inhalation exposure to bleomycin. A – Selection of bleomycin dose. Mice were instilled intratracheally once with different concentrations of bleomycin. The dose of 1.5 U/kg that led to death of 50% of animals was selected. B – Inhalation treatment of mice with experimental lung fibrosis by liposomal PGE2 (Lip PGE2) prevents animal mortality. Inhalation of healthy mice with empty liposomes did not influence on animal survival. Lung fibrosis was induced by intratracheal instillation of 1.5 U/kg of bleomycin. Mice were treated with liposomal PGE2 twice a week for three weeks starting one day after the bleomycin administration.
Validation of IPF Model
Changes in animal body weight, hydroxyproline content in the lung tissue and lung histology were used as hallmarks of the development of IPF in experimental animals after the instillation of 1.5 U/kg bleomycin. It was found that three weeks after the instillation, the body weight of animals decreased to 75% (P < 0.05) of its initial value (Figure 2A). At the same experimental point, the concentration of hydroxyproline in the lungs increased in 2.6 times (Figure 2B). Histological analysis of control lung tissue demonstrated widely patent alveoli without inflammation or edema (Figure 3A). The bronchi were also patent. Lungs of animals instilled with bleomycin (Figure 3B) showed consolidation of the pulmonary architecture, with early fibrotic thickening of the alveolar walls, ablation of the alveolar space, and edema. Chronic inflammatory cells and fibroblasts were readily apparent within the affected areas. Taken together, these data clearly confirm the development of marked lung fibrosis in experimental animals subjected to bleomycin.
Figure 2.

Influence of inhalation treatment with liposomal PGE2 on body weight (A) and hydroxyproline content in the lungs (B). Lung fibrosis was induced by intratracheal instillation of 1.5 U/kg of bleomycin. Mice were treated with liposomal PGE2 twice a week for three weeks starting one day later after the bleomycin administration. At the end of treatment, lungs were harvested, homogenized and hydroxyproline content in the lungs was measured. 1 – Healthy mice (control); 2 – Healthy mice treated by inhalation with empty liposomes; 3 – Mice instilled with bleomycin (1.5 U/kg); 4 – Mice instilled with bleomycin (1.5 U/kg) and treated by inhalation with liposomal PGE2. Means ± S.D. are shown. *P < 0.05 when compared with control (1). †P < 0.05 when compared with mice with fibrosis (2).
Figure 3.
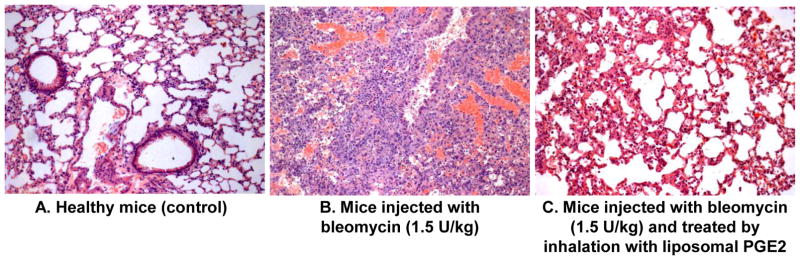
Lung histology. A- Healthy mice (control). B-Lung fibrosis was induced by intratracheal instillation of 1.5 U/kg of bleomycin. C-Mice were treated with inhalation of liposomal PGE2 within 3 weeks twice a week starting one day later after the bleomycin administration. At the end of experiment, lungs were harvested and fixed in 10% phosphate-buffered formalin. Samples were subsequently dehydrated and embedded in Paraplast®. Sections (5 μm) were cut, stained with hematoxylin and eosin. Representative images are shown (10X magnification).
Body Distribution and Accumulation of Liposomes in the Lungs
The average size of liposomes used in the present study for inhalation was 500–600 nm. Previously we demonstrated that liposomes remain predominately in the lungs for a long period of time (approximately one week) after inhalation. [13, 14] In order to confirm the preferential pulmonary accumulation of the liposomes used in the present study, we determined organ content of fluorescently-labeled liposomes using the IVIS imaging system (Figure 4). Inhalation delivery was compared with intravenous instillation of similar liposomes. It was found that 24 hours after intravenous instillation, liposomes accumulated predominately in the kidneys and liver, while substantially lesser accumulation was found in the spleen, heart and lungs. Only trace amount of liposomes registered in the brain. In contrast, after inhalation delivery, liposomes were retained in the lungs with minimal amounts found in other organs including the liver, kidneys, spleen, heart and brain. These data confirmed the favorable distribution of inhaled liposomes and formed the basis for the use of such liposomes as carriers to deliver PGE2 locally to the lungs and limitation of possible adverse systemic effects of PGE2. In order to study the penetration of liposomes into lung cells after inhalation, lipid membrane of liposomes was labeled by osmium tetroxide and visualized in lung tissues by transmission electron microscopy (Fig. 4C). These data clearly showed that liposomes did penetrated lung cells after inhalation and accumulated in the cytoplasm.
Figure 4.

Inhalation delivery of liposomes to mouse lungs. A, B - Relative tissue content of liposomes delivered to mice by intravenous instillation (A) or inhalation (B). Liposomal content was registered in organs 24 h after inhalation. C - Localization of liposomes in the mouse lung tissues after the inhalation delivery. Liposomes were labeled by osmium tetroxide and visualized by electron transmission microscopy.
Treatment of IPF with Liposomal Form of PGE2
In order to estimate anti-fibrotic effect of liposomal PGE2, we investigated the influence of this preparation on body weight, hydroxyproline content in the lungs and mortality of animals with IPF, induced by a single intratracheal instillation of bleomycin. Inhalation of healthy mice with empty liposomes did not influence on animal survival. Treatment with liposomal PGE2 prevented the decrease in the body weight of experimental animals induced by bleomycin (Figure 2A). The difference between body weight in bleomycin-treated animals with IPF (Figure 2A, bar 2) and animals instilled with bleomycin and treated with liposomal PGE2 (Figure 2A, bar 3) was statistically significant (P < 0.05). A therapeutic action of liposomal PGE2 was also confirmed by the measurement of hydroxyproline content in the lungs. Treatment of animals with IPF by liposomal PGE2 significantly decreased (P < 0.05) the hydroxyproline content in lung tissues by 1.3 fold (Figure 2B). However, the content of hydroxyproline in the lung tissues still was significantly (P < 0.05) higher when compared with healthy control mice (compare bars 3 and 1 in Figure 2B). Inhalation of healthy mice with empty liposomes did not change body weight and the hydroxyproline content in lung tissues. Inhalation treatment of animals with liposomal PGE2 substantially limited lung tissue damage induced by bleomycin (Figure 3C). Some mild thickening of the alveolar septa was noted focally, and in some areas mild fibrosis could still be observed. However, both the extent and severity of the fibrotic process were reduced in this group. Edema was minimal and only focal inflammation was present. Finally, inhalation treatment of animals with pulmonary fibrosis by liposomal PGE2 completely prevented the mortality of experimental animals (Figure 1B).
Gene and Protein Expression
In order to examine mechanisms of the development of fibrosis induced by intratracheal instillation of bleomycin and protective effect of liposomal PGE2 delivered to the lungs by inhalation, we studied the profiles of the expression of 84 key genes involved in tissue remodeling during wound repair and development of fibrosis. The data obtained using the standard Mouse Fibrosis RT Profiler™ PCR Array panel showed that after instillation of bleomycin, 24 studied genes were upregulated by more than 5 times while 7 out of 84 genes were downregulated more than 5-fold (Figure 5A). Data showed that transforming growth factor (TGF)-mediated cell signaling was impaired in mice after the instillation of bleomycin. While the expression of genes encoding different types of TGF proteins was practically unaffected, the expression of proteins associated with TGF receptors and their second messengers (ENG, TGFBR2 and SMAD6 genes, Figure 5A, #5, 65, and 73, respectively) was significantly downregulated. In addition to TGF signaling, the expression of genes encoding vascular endothelial growth factor (VEGF), integrin alpha-1 (ITGA1), calveolae protein (CAV1) signal transducer and activator of transcription 6 (STAT6) were also decreased (Figure 5A, #45, 54, 68, and 83, respectively). In contrast, the expression of genes encoding the following functional groups of proteins was substantially increased after bleomycin instillation: plasminogen and plasminogen activator (PLG and PLAU, Figure 5A, #10 and 11), several matrix metalloproteinases (MMP13, MMP1A, MMP3, MMP8, and MMP9, Figure 5A, #15, 17, 19, 20, and 21) as well as tissue inhibitor of metalloproteinases (TIMP1 - Figure 5A, #22), angiotensinogen and a member of the TGF-beta family (AGT and BMP7, Figure 5A, #26 and 27), chemokines (CCL11 and CCL12, Figure 5A. #33 and 34), gamma interferon (IFNG, Figure 4A, #36), several interleukins (IL10, IL13, IL1B, IL4, and IL5, Figure 5A, #37, 38, 40, 41, and 24, respectively) and interleukin 13 receptor (IL13RA2, Figure 5A, #53), inhibin (INHBE, Figure 5A, #43), tumor necrosis factor and its ligand (TNF and FASL, Figure 5A, #44 and 69), integrin (ITGB8, Figure 5A, #62) and transforming growth factor-beta-induced factor (TGIF1, Figure 5A, #84). The overexpression of CCL12 and MMP3 genes measured by QPCR was really significant. Consequently, in order to confirm these findings, we measured the expression of corresponding proteins. In addition, three other proteins (VEGF, hypoxia inducible factor 1 alpha, HIF1A, and von Hippel-Lindau, VHL) that are extremely important for the development of lung damage and its compensation were also analyzed. Immunohistochemical measurement of the expression of chemokine CCL12 and matrix metalloproteinase MMP3 confirmed that bleomycin instillation induced overexpression of both proteins (Figure 6). The data obtained showed that VEGF protein expression was substantially decreased after instillation of bleomycin (Figure 6). It was found that instillation of bleomycin led to the overexpression of HIF1A and suppression of VHL genes (Figure 7). Analysis of the expression of HIF1A protein (immunohistochemistry) supports RT-PCR finding and show that the expression of this protein was also upregulated after bleomycin instillation (Figure 6). Notably, that treatment of mice with liposomal PGE2 delivered by inhalation after instillation of bleomycin almost completely eliminated the aforementioned disturbances in gene and protein expression (Figures 5B, 6 and 7).
Figure 5.

Gene expression measured by the Quantitative Polymerase Chain Reaction (QPCR). The QPCR was performed using a standard Mouse Fibrosis RT Profiler™ PCR Array panel. Lung fibrosis was induced by intratracheal instillation of 1.5 U/kg of bleomycin. Mice were treated by inhalation delivery of liposomal PGE2 twice a week for three weeks starting one day later after the bleomycin administration. A – Mice instilled with bleomycin (1.5 U/kg); B – Mice instilled with bleomycin (1.5 U/kg) and treated by inhalation with liposomal PGE2.). Means ± SD are shown.
Figure 6.

Expression of proteins (immunohistochemistry) in lung tissues. Representative images of tissue sections stained with antibodies against VEGF, CCL12, MMP3 and HIF1A proteins (10X magnification) and average expression of corresponding proteins. High intensity of the color indicates high protein concentration. Lung fibrosis was induced by intratracheal instillation of 1.5 U/kg of bleomycin. Mice were treated with liposomal PGE2 twice a week for three weeks starting one day later after the bleomycin administration. 1 - Healthy mice (control); 2-Mice instilled with bleomycin (1.5 U/kg); 3 - Mice instilled with bleomycin (1.5 U/kg) and treated by inhalation with liposomal PGE2. Means ± SD from are shown. *P < 0.05 when compared with healthy mice (control). †P < 0.05 when compared with mice instilled with bleomycin.
Figure 7.

Gene expression analyzed by RT-PCR. Representative images of gel electrophoresis of RT-PCR product and average expression of genes encoding hypoxia inducible factor 1α (HIF1A), von Hippel-Lindau (VHL) and β-actin (B-ACTIN, internal standard) proteins. 1 - Healthy mice (control); 2-Mice instilled with bleomycin (1.5 U/kg); 3 - Mice instilled with bleomycin (1.5 U/kg) and treated by inhalation with liposomal PGE2. Means ± SD from are shown. *P < 0.05 when compared with healthy mice (control). †P < 0.05 when compared with mice instilled with bleomycin.
Discussion
The present study shows that intratracheal instillation of bleomycin at dose of 1.5 U/kg induces extensive lung fibrosis. As expected, the development of fibrosis was initiated by a marked pulmonary inflammation with subsequent transition into fibrosis. The sequence of the process was confirmed by morphological features of inflammation and overexpression of several genes involved in the development of inflammation. In fact, several chemokines, inflammatory cytokines and interleukins were overexpressed following bleomycin treatment. It was shown previously that chemokines are important in the pulmonary recruitment of granulocytes and are essential in the pathogenesis of bleomycin-induced lung fibrosis. [28] Moreover, interferon-gamma, an inflammatory cytokine, was implicated in the development of fibrosis in inflamed tissues. [29] The gene encoding this protein was also overexpressed after instillation of bleomycin. In addition, all analyzed genes encoding interleukins were overexpressed in the lungs after bleomycin treatment. It is well known that interleukins are important mediators of inflammation and remodeling in the lungs. In particular, it was found that the overexpression of interleukin IL10 in the lung causes mucosal metaplasia, tissue inflammation, subepithelial fibrosis and airway remodeling via IL13-dependent and -independent pathways. [30] IL13-depended pathway was definitely involved in the development of fibrosis after inflammation in the present study. The registered overexpression of the IL13 gene as well as gene encoding IL3 receptors (IL13RA2) supports this suggestion. Interleukin 13 is considered to be a major inducer of fibrosis in several different disease conditions. [31–34] The activation of inflammation and its transition to fibrosis was associated with the overexpression of several matrix metalloproteinases (collagenases/gelatinases). It is possible that such activation was compensatory and directed to the degrading of fibrillar collagens in order to limit their accumulation during pulmonary fibrosis. However, the activation of these enzymes might also enhance tissue damage during IPF. [35–37] Therefore, the present experimental data support the observation that pulmonary fibrosis is preceded by a chronic inflammatory process which induces lung injury, modulates fibrogenesis, provokes fibrosis and leads to the formation of the fibrotic scar. [36]
It is generally assumed that the transforming growth factor-beta (TGFB) family of receptors may play an important role in the initiation of the signal transduction events leading to mitogenic responses and initiation of fibrosis by induced myofibroblast differentiation. [7, 32, 38–41] However, experimental data suggest that this signal transduction pathway probably did not have a significant impact on the development of pulmonary fibrosis in the present study because genes encoding proteins and receptors involved in these pathways were either practically unchanged or downregulated after the instillation of bleomycin. Only TGIF1 and bone morphogenetic protein-7 (a member of the TGFB superfamily) were substantially upregulated, suggesting that other than TGFB receptor-initiated signaling pathways might be involved in the development of fibrosis. A second important mediator of inflammation and fibrosis, tumor necrosis factor (TNF), a multifunctional proinflammatory cytokine secreted predominantly by monocytes/macrophages, was upregulated along with tumor necrosis factor ligand superfamily member 6 (encoding by the FASL or TNFSF6 gene) after bleomycin treatment. These proteins are known to mediate the transition from pulmonary inflammation to fibrosis as well as to induce apoptosis. [42–44]
Several other signal transduction pathways activated in the lung after instillation of bleomycin, including inhibins, angiotensinogens and integrins, might also be involved in the development of pulmonary fibrosis and tissue damage in the present experimental model. [30, 39, 45–47] It is generally believed that fibrosis is accompanied by hypoxia and major hypoxic signaling pathways initiated by HIF1A and VHL proteins contribute in the development and compensation of fibrotic damage. [48–56] In many cases, it was found that tissue hypoxia promotes fibrosis and HIFA-associated signaling pathways of hypoxia are involved on the development of fibrosis in the liver and lungs. We found that bleomycin induces overexpression of HIF1A gene and protein and inhibits the expression of its counterpart – the VHL gene, supporting the role of HIF1A signaling pathways in the development of lung fibrosis. It was found that, independently of HIF1A, pVHL protein encoded by the VHL gene might be directly involved in the development of IPF. [56] However, it is unlikely that this mechanism was involved in the present study because the VHL gene was suppressed in lung tissues following instillation of bleomycin. It was also found that the overexpression of HIF1A protein can promote the development of IPF via TGF-beta-signaling pathways. [55] However, it is unlikely that this pathway was involved in the present study because, as noted above, genes encoding proteins and receptors involved in this pathway were either unchanged or downregulated after the instillation of bleomycin.
The present experimental studies demonstrate, for the first time, that treatment of pulmonary fibrosis by liposomal PGE2 delivered by inhalation results in increased survival, limitation of all studied symptoms of IPF developed after intratracheal instillation of bleomycin. The data show that liposomal PGE2 delivered locally to the lungs eliminated the decrease in the body weight, substantially limited hydroxyproline content in the lungs, disturbances in the mRNA and protein expression, restricted lung tissue damage and completely prevented animal mortality. These preventive effects of PGE2 can be a result of its ability to limit fibroblast proliferation, activation, migration, collagen secretion, and/or myofibroblast differentiation. [6–10] Similar results were recently obtained after treatment of bleomycin-induced IPF in mice by continuous subcutaneous infusion of free non-bound synthetic analog of PGE2. [11] In an independent study, orally administered losartan, an angiotensin II type 1 receptor antagonist, led to similar effect by increasing the level of PGE2 in the lungs. [57] These experimental data support our findings and underscore the therapeutic promise of treatments for IPF based on increasing the level of PGE2 in the lung tissues. It is highly likely that the normalization of the expression of many genes encoding proteins responsible for inflammation and fibrosis after bleomycin instillation observed in the present study plays a central role in the prevention of lung injury and animal mortality. Suppression of HIF1A protein by liposomal PGE2 might also play a role in limiting fibrotic damage of lung tissue. The exact mechanisms of the protective action of liposomal PGE2 require more detail study.
In summary, our data provides evidence that pulmonary fibrosis can be effectively prevented by the inhalation delivery of liposomal form of PGE2 locally into the lungs. The results of present investigations make liposomal form of PGE2 an attractive drug for inhalation treatment of idiopathic pulmonary fibrosis.
Acknowledgments
The research was supported in part by NIH R01 CA111766 and P-30 ES-005022 grants. We thank Valentin Starovoytov for his help with obtaining and providing analysis of transmission electron microscopy images.
Abbreviations
- IPF
Idiopathic pulmonary fibrosis
- PGE2
Prostaglandin E2
- QPCR
Quantitative polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 2.Gharaee-Kermani M, Gyetko MR, Hu B, Phan SH. New insights into the pathogenesis and treatment of idiopathic pulmonary fibrosis: a potential role for stem cells in the lung parenchyma and implications for therapy. Pharm Res. 2007;24:819–841. doi: 10.1007/s11095-006-9216-x. [DOI] [PubMed] [Google Scholar]
- 3.Sime PJ, O’Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol. 2001;99:308–319. doi: 10.1006/clim.2001.5008. [DOI] [PubMed] [Google Scholar]
- 4.Frankel SK, Schwarz MI. Update in idiopathic pulmonary fibrosis. Curr Opin Pulm Med. 2009;15:463–469. doi: 10.1097/MCP.0b013e32832ea4b3. [DOI] [PubMed] [Google Scholar]
- 5.Williams TJ, Wilson JW. Challenges in pulmonary fibrosis: 7--Novel therapies and lung transplantation. Thorax. 2008;63:277–284. doi: 10.1136/thx.2004.031054. [DOI] [PubMed] [Google Scholar]
- 6.Vancheri C, Mastruzzo C, Sortino MA, Crimi N. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol. 2004;25:40–46. doi: 10.1016/j.it.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Bozyk PD, Moore BB. Prostaglandin E2 and the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;45:445–452. doi: 10.1165/rcmb.2011-0025RT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang SK, Wettlaufer SH, Chung J, Peters-Golden M. Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac-1. Am J Respir Cell Mol Biol. 2008;39:482–489. doi: 10.1165/rcmb.2008-0080OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang SK, Wettlaufer SH, Hogaboam CM, Flaherty KR, Martinez FJ, Myers JL, Colby TV, Travis WD, Toews GB, Peters-Golden M. Variable prostaglandin E2 resistance in fibroblasts from patients with usual interstitial pneumonia. Am J Respir Crit Care Med. 2008;177:66–74. doi: 10.1164/rccm.200706-963OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh M, Stewart A, Tucker DE, Bonventre JV, Murphy RC, Leslie CC. Role of cytosolic phospholipase A(2) in prostaglandin E(2) production by lung fibroblasts. Am J Respir Cell Mol Biol. 2004;30:91–100. doi: 10.1165/rcmb.2003-0005OC. [DOI] [PubMed] [Google Scholar]
- 11.Failla M, Genovese T, Mazzon E, Fruciano M, Fagone E, Gili E, Barera A, La Rosa C, Conte E, Crimi N, Cuzzocrea S, Vancheri C. 16,16-Dimethyl prostaglandin E2 efficacy on prevention and protection from bleomycin-induced lung injury and fibrosis. Am J Respir Cell Mol Biol. 2009;41:50–58. doi: 10.1165/rcmb.2007-0438OC. [DOI] [PubMed] [Google Scholar]
- 12.Minko T, Stefanov A, Pozharov V. Selected contribution: Lung hypoxia: antioxidant and antiapoptotic effects of liposomal alpha-tocopherol. Journal of applied physiology. 2002;93:1550–1560. doi: 10.1152/japplphysiol.00007.2002. discussion 1549. [DOI] [PubMed] [Google Scholar]
- 13.Garbuzenko OB, Saad M, Betigeri S, Zhang M, Vetcher AA, Soldatenkov VA, Reimer DC, Pozharov VP, Minko T. Intratracheal versus intravenous liposomal delivery of siRNA, antisense oligonucleotides and anticancer drug. Pharm Res. 2009;26:382–394. doi: 10.1007/s11095-008-9755-4. [DOI] [PubMed] [Google Scholar]
- 14.Garbuzenko OB, Saad M, Pozharov VP, Reuhl KR, Mainelis G, Minko T. Inhibition of lung tumor growth by complex pulmonary delivery of drugs with oligonucleotides as suppressors of cellular resistance. Proc Natl Acad Sci U S A. 2010;107:10737–10742. doi: 10.1073/pnas.1004604107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taratula O, Garbuzenko OB, Chen AM, Minko T. Innovative strategy for treatment of lung cancer: targeted nanotechnology-based inhalation co-delivery of anticancer drugs and siRNA. J Drug Target. 2011;19:900–914. doi: 10.3109/1061186X.2011.622404. [DOI] [PubMed] [Google Scholar]
- 16.Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L152–160. doi: 10.1152/ajplung.00313.2007. [DOI] [PubMed] [Google Scholar]
- 17.Babin AL, Cannet C, Gerard C, Saint-Mezard P, Page CP, Sparrer H, Matsuguchi T, Beckmann N. Bleomycin-induced lung injury in mice investigated by MRI: model assessment for target analysis. Magn Reson Med. 2012;67:499–509. doi: 10.1002/mrm.23009. [DOI] [PubMed] [Google Scholar]
- 18.Banerjee ER, Henderson WR., Jr Characterization of lung stem cell niches in a mouse model of bleomycin-induced fibrosis. Stem Cell Res Ther. 2012;3:21. doi: 10.1186/scrt112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prata LO, Oliveira FM, Ribeiro TM, Almeida PW, Cardoso JA, da Rodrigues-Machado MG, Caliari MV. Exercise attenuates pulmonary injury in mice with bleomycin-induced pulmonary fibrosis. Exp Biol Med (Maywood) 2012;237:873–883. doi: 10.1258/ebm.2012.011334. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka K, Shirai A, Ito Y, Namba T, Tahara K, Yamakawa N, Mizushima T. Expression of 150-kDa oxygen-regulated protein (ORP150) stimulates bleomycin- induced pulmonary fibrosis and dysfunction in mice. Biochem Biophys Res Commun. 2012;425:818–824. doi: 10.1016/j.bbrc.2012.07.158. [DOI] [PubMed] [Google Scholar]
- 21.Betigeri S, Zhang M, Garbuzenko O, Minko T. Non-viral systemic delivery of siRNA or antisense oligonucleotides targeted to Jun N-terminal kinase 1 prevents cellular hypoxic damage. Drug Deliv Transl Res. 2011;1:13–24. doi: 10.1007/s13346-010-0003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saad M, Garbuzenko OB, Minko T. Co-delivery of siRNA and an anticancer drug for treatment of multidrug-resistant cancer. Nanomedicine (Lond) 2008;3:761–776. doi: 10.2217/17435889.3.6.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Saad M, Pakunlu RI, Khandare JJ, Garbuzenko OB, Vetcher AA, Soldatenkov VA, Pozharov VP, Minko T. Nonviral nanoscale-based delivery of antisense oligonucleotides targeted to hypoxia-inducible factor 1 alpha enhances the efficacy of chemotherapy in drug-resistant tumor. Clin Cancer Res. 2008;14:3607–3616. doi: 10.1158/1078-0432.CCR-07-2020. [DOI] [PubMed] [Google Scholar]
- 24.Schroeder A, Avnir Y, Weisman S, Najajreh Y, Gabizon A, Talmon Y, Kost J, Barenholz Y. Controlling liposomal drug release with low frequency ultrasound: mechanism and feasibility. Langmuir. 2007;23:4019–4025. doi: 10.1021/la0631668. [DOI] [PubMed] [Google Scholar]
- 25.Lu G, Liao J, Yang G, Reuhl KR, Hao X, Yang CS. Inhibition of adenoma progression to adenocarcinoma in a 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis model in A/J mice by tea polyphenols and caffeine. Cancer Res. 2006;66:11494–11501. doi: 10.1158/0008-5472.CAN-06-1497. [DOI] [PubMed] [Google Scholar]
- 26.Minko T, Pakunlu RI, Wang Y, Khandare JJ, Saad M. New generation of liposomal drugs for cancer. Anticancer Agents Med Chem. 2006;6:537–552. doi: 10.2174/187152006778699095. [DOI] [PubMed] [Google Scholar]
- 27.Pakunlu RI, Wang Y, Saad M, Khandare JJ, Starovoytov V, Minko T. In vitro and in vivo intracellular liposomal delivery of antisense oligonucleotides and anticancer drug. J Control Release. 2006;114:153–162. doi: 10.1016/j.jconrel.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 28.Huaux F, Gharaee-Kermani M, Liu T, Morel V, McGarry B, Ullenbruch M, Kunkel SL, Wang J, Xing Z, Phan SH. Role of Eotaxin-1 (CCL11) and CC chemokine receptor 3 (CCR3) in bleomycin-induced lung injury and fibrosis. Am J Pathol. 2005;167:1485–1496. doi: 10.1016/S0002-9440(10)61235-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Awad M, Pravica V, Perrey C, El Gamel A, Yonan N, Sinnott PJ, Hutchinson IV. CA repeat allele polymorphism in the first intron of the human interferon-gamma gene is associated with lung allograft fibrosis. Hum Immunol. 1999;60:343–346. doi: 10.1016/s0198-8859(98)00133-5. [DOI] [PubMed] [Google Scholar]
- 30.Lee CG, Homer RJ, Cohn L, Link H, Jung S, Craft JE, Graham BS, Johnson TR, Elias JA. Transgenic overexpression of interleukin (IL)-10 in the lung causes mucus metaplasia, tissue inflammation, and airway remodeling via IL-13-dependent and -independent pathways. J Biol Chem. 2002;277:35466–35474. doi: 10.1074/jbc.M206395200. [DOI] [PubMed] [Google Scholar]
- 31.Blackburn MR, Lee CG, Young HW, Zhu Z, Chunn JL, Kang MJ, Banerjee SK, Elias JA. Adenosine mediates IL-13-induced inflammation and remodeling in the lung and interacts in an IL-13-adenosine amplification pathway. J Clin Invest. 2003;112:332–344. doi: 10.1172/JCI16815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 33.Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, Elias JA, Sheppard D, Erle DJ. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. 2002;8:885–889. doi: 10.1038/nm734. [DOI] [PubMed] [Google Scholar]
- 34.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fukuda Y, Ishizaki M, Kudoh S, Kitaichi M, Yamanaka N. Localization of matrix metalloproteinases-1, -2, and -9 and tissue inhibitor of metalloproteinase-2 in interstitial lung diseases. Lab Invest. 1998;78:687–698. [PubMed] [Google Scholar]
- 36.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 37.Yaguchi T, Fukuda Y, Ishizaki M, Yamanaka N. Immunohistochemical and gelatin zymography studies for matrix metalloproteinases in bleomycin-induced pulmonary fibrosis. Pathol Int. 1998;48:954–963. doi: 10.1111/j.1440-1827.1998.tb03866.x. [DOI] [PubMed] [Google Scholar]
- 38.Ask K, Martin GE, Kolb M, Gauldie J. Targeting genes for treatment in idiopathic pulmonary fibrosis: challenges and opportunities, promises and pitfalls. Proc Am Thorac Soc. 2006;3:389–393. doi: 10.1513/pats.200602-021TK. [DOI] [PubMed] [Google Scholar]
- 39.Borok Z. Role for alpha3 integrin in EMT and pulmonary fibrosis. J Clin Invest. 2009;119:7–10. doi: 10.1172/JCI38084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu H, Peng Y, Liu F, Li J, Chen X, Liu Y, Zhang H. A selective cyclooxygenase-2 inhibitor decreases transforming growth factor-beta1 synthesis and matrix production in human peritoneal mesothelial cells. Cell Biol Int. 2007;31:508–515. doi: 10.1016/j.cellbi.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 41.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 42.Oikonomou N, Harokopos V, Zalevsky J, Valavanis C, Kotanidou A, Szymkowski DE, Kollias G, Aidinis V. Soluble TNF mediates the transition from pulmonary inflammation to fibrosis. PLoS One. 2006;1:e108. doi: 10.1371/journal.pone.0000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- 44.Zhang K, Gharaee-Kermani M, McGarry B, Remick D, Phan SH. TNF-alpha-mediated lung cytokine networking and eosinophil recruitment in pulmonary fibrosis. J Immunol. 1997;158:954–959. [PubMed] [Google Scholar]
- 45.Antoniu SA. Targeting the angiotensin pathway in idiopathic pulmonary fibrosis. Expert Opin Ther Targets. 2008;12:1587–1590. doi: 10.1517/14728220802515459. [DOI] [PubMed] [Google Scholar]
- 46.Matsuse T, Fukuchi Y, Eto Y, Matsui H, Hosoi T, Oka T, Ohga E, Nagase T, Orimo H. Expression of immunoreactive and bioactive activin A protein in adult murine lung after bleomycin treatment. Am J Respir Cell Mol Biol. 1995;13:17–24. doi: 10.1165/ajrcmb.13.1.7541220. [DOI] [PubMed] [Google Scholar]
- 47.Sheppard D. The role of integrins in pulmonary fibrosis. Eur Respir Rev. 2008;17:157–162. [Google Scholar]
- 48.Copple BL, Kaska S, Wentling C. Hypoxia-inducible factor activation in myeloid cells contributes to the development of liver fibrosis in cholestatic mice. J Pharmacol Exp Ther. 2012 doi: 10.1124/jpet.111.189340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haase VH. Pathophysiological Consequences of HIF Activation: HIF as a modulator of fibrosis. Ann N Y Acad Sci. 2009;1177:57–65. doi: 10.1111/j.1749-6632.2009.05030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, Brekken RA, Scherer PE. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. 2009;29:4467–4483. doi: 10.1128/MCB.00192-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kimura K, Iwano M, Higgins DF, Yamaguchi Y, Nakatani K, Harada K, Kubo A, Akai Y, Rankin EB, Neilson EG, Haase VH, Saito Y. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol. 2008;295:F1023–1029. doi: 10.1152/ajprenal.90209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moon JO, Welch TP, Gonzalez FJ, Copple BL. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G582–592. doi: 10.1152/ajpgi.90368.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Theilig F, Enke AK, Scolari B, Polzin D, Bachmann S, Koesters R. Tubular deficiency of von Hippel-Lindau attenuates renal disease progression in anti-GBM glomerulonephritis. Am J Pathol. 2011;179:2177–2188. doi: 10.1016/j.ajpath.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tiriveedhi V, Gelman AE, Mohanakumar T. HIF-1alpha signaling by airway epithelial cell K-alpha1-tubulin: Role in fibrosis and chronic rejection of human lung allografts. Cell Immunol. 2012;273:59–66. doi: 10.1016/j.cellimm.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K, Matsui H, Hara K, Tanaka T, Iso T, Suga T, Kurabayashi M. Hypoxia-inducible factor-1alpha mediates TGF-beta-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L740–752. doi: 10.1152/ajplung.00146.2010. [DOI] [PubMed] [Google Scholar]
- 56.Zhou Q, Pardo A, Konigshoff M, Eickelberg O, Budinger GR, Thavarajah K, Gottardi CJ, Jones J, Varga J, Selman M, Sznajder JI, Raj JU, Zhou G. Role of von Hippel-Lindau protein in fibroblast proliferation and fibrosis. Faseb J. 2011;25:3032–3044. doi: 10.1096/fj.10-177824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Molina-Molina M, Serrano-Mollar A, Bulbena O, Fernandez-Zabalegui L, Closa D, Marin-Arguedas A, Torrego A, Mullol J, Picado C, Xaubet A. Losartan attenuates bleomycin induced lung fibrosis by increasing prostaglandin E2 synthesis. Thorax. 2006;61:604–610. doi: 10.1136/thx.2005.051946. [DOI] [PMC free article] [PubMed] [Google Scholar]