

Abstract
Burkholderia pseudomallei is a Gram-negative environmental bacterium and the causative agent of melioidosis, a potentially fatal, acute or chronic disease endemic in the tropics. Acyl homoserine lactone (AHL)-mediated quorum sensing and signalling have been associated with virulence and biofilm formation in numerous bacterial pathogens. In the canonical acyl-homoserine lactone signalling paradigm, AHLs are detected by a response regulator. B. pseudomallei encodes three AHL synthases, encoded by bpsI1, bpsI2 and bpsI3, and five regulator genes. In this study, we mutated the B. pseudomallei AHL synthases individually and in double and triple combination. Five AHLs were detected and quantified by tandem liquid chromatography-mass spectroscopy. The major AHLs produced were N-octanoylhomoserine lactone and N-(3-hydroxy-decanoyl)homoserine lactone, the expression of which depended on bpsI1 and bpsI2, respectively. B. pseudomallei infection of macrophage cells causes cell fusion, leading to multinucleated cells (3 or more nuclei per cell). A triple mutant defective in production of all three AHL synthases was associated with a striking phenotype of massively enhanced host cellular fusion in macrophages. However, neither abrogation of host cell fusion, achieved by mutation of bimA or hcp1, nor enhancement of fusion altered intracellular replication of B. pseudomallei. Furthermore, when tested in murine models of acute melioidosis the AHL synthase mutants were not attenuated for virulence. Collectively, this study identifies important new aspects of the genetic basis of AHL synthesis in B. pseudomallei and the roles of these AHLs in systemic infection and in cell fusion in macrophages for this important human pathogen.
Introduction
Melioidosis is a potentially fatal disease endemic to the tropics, particularly North Eastern Thailand and Northern Australia, where the mortality rates approach 14% in Australia and 50% in Thailand [1]–[3]. The incidence of disease in the Northern Territory of Australia was 15.5 per 100,000 between 2001 and 2002 and 2.7 times that rate amongst indigenous Australians [2], [3]. Melioidosis is also considered to be under-reported and an emerging disease worldwide, with infections reported in Brazil, China, India and Malaysia [4]–[9]. Clinical presentations of melioidosis vary, ranging from chronic to acute disease and potentially rapid death due to systemic infection and septic shock [1]. Virtually any organ of the body may be infected; while skin and soft tissues including lungs, liver and spleen are often involved, the parotid gland, the central nervous system and bone may also be infected [1], [3].
Burkholderia pseudomallei, the causative agent of melioidosis, is easily isolated from soil and standing water in regions of endemicity. Due to potential infection via aerosolisation [1], [10], the current lack of a vaccine, a capacity for relapse and latent infection [11], intrinsic antibiotic resistance, and its ease of isolation and culture from the environment, B. pseudomallei is listed as a category B potential biowarfare agent [12].
Infection of humans stems from environmental exposure and is closely associated with high rainfall [10]. Two common routes of inoculation are thought to be percutaneous and inhalation. Aside from the hazards of working in standing water, such as rice paddies, inhalation of contaminated water droplets or dust (soil) is probably the most important natural route of infection. Animal studies of melioidosis using inhalational exposure and intranasal inoculation demonstrate infection of the lung and systemic infection of internal organs. Such studies have revealed previously unrecognized sites of colonization in the nasal associated lymphoid tissue (NALT) and in the nasal mucosa, especially the olfactory epithelium (OE). This suggests that respiratory infection and nasal colonization may be a portal of entry to the brain and blood without necessarily involvement of lower respiratory tract [13].
Several virulence factors of B. pseudomallei have been well defined in mice and hamsters, and in in vitro models of infection. These include the capsule, lipopolysaccharide, and Type III and VI secretion systems [14]–[16]. In vitro models of infection include murine and human phagocytic cell lines in which B. pseudomallei efficiently replicates [14]. B. pseudomallei is phagocytosed and replicates in RAW264.7 cells, a murine macrophage-like cell line in which B. pseudomallei is also noted for its capacity to induce cell fusion, leading to multinucleate cell (MNC) formation [17]–[19]. MNC formation is also observed in lung tissue in a murine model of chronic human melioidosis [20]. Both MNC formation and intercellular spread involve actin-mediated motility in epithelial cell lines [14], [15], [21]. Such motility requires the bimA gene, which encodes a polar membrane protein required for actin polymerisation [18], and one of six Type VI secretion systems (encoded by hcp1 within T6SS-1) is required for cell fusion and MNC formation [15], [22]. However, the exact role of MNC formation in pathogenesis is not established [20].
One of the most well studied intercellular bacterial communication systems is based on acyl-homoserine lactone (AHL)-mediated quorum sensing (QS). This is widespread in Gram-negative bacteria and is associated with pathogenicity in organisms such as Pseudomonas aeruginosa [23], [24]. In this system, AHLs diffuse from the cells and accumulate in a cell-density-dependent manner in the growth medium (or environment); diffusion back into the bacteria from the external milieu results in binding to a cognate regulator protein and coordinated regulation of genes in the bacterial community. In B. pseudomallei, there are three AHL synthases, encoded by the bpsI genes, and a number of regulatory genes such as bpsR [25]. Analysis of mutants lacking these genes has implicated AHL-mediated signaling in the regulation of extracellular enzyme and siderophore formation, the oxidative stress response, and biofilm formation [26]–[29]. In addition, a bpsI mutant was attenuated in a Caenorhabditis elegans model [27], and in hamsters and mice [25], [26].
In this study, we consider the hypothesis that QS plays a role in B. pseudomallei virulence in vitro, in relation to MNC formation, and in vivo, situations in which high bacterial cell densities are achieved. We focused on bpsI mutants to determine the potential roles of QS in pathogenicity, and a triple bpsI mutant in which no AHL-mediated QS is possible. For our in vivo studies we used upper respiratory tract (URT), lung, and brain infection models. The results show that whilst there is no significant role of QS for in vivo virulence in these models, there is a major effect on MNC formation in macrophage-like cells.
Materials and Methods
Bacterial Strains, Culture Conditions and Antibiotics
The bacterial strains used in this study are described in Table 1. Bacteria were grown at 37°C on lysogeny broth (LB) agar or in LB broth in shaken culture. Antibiotics, when appropriate, were added at the following concentrations: ampicillin, 100 µg ml−1, chloramphenicol, 50 µg ml−1 (E. coli) or 100 µg ml−1 (B. pseudomallei), gentamicin, 15 µg ml−1, streptomycin, 100 µg ml−1, tetracycline, 10 µg ml−1, and trimethoprim 100 µg ml−1.
Table 1. Bacterial strains used in the study.
| Species/strain | Genotype/phenotype | Reference |
| Burkholderia pseudomallei | ||
| MSHR520 | Wild-type; clinical isolate | Owen et al., 2009 |
| MSHR520 Δcap | Δcap; Tcr | Owen et al., 2009 |
| MSHR520 ΔbpsI1 | ΔbpsI1 | This study |
| MSHR520 ΔbpsI2 | ΔbpsI2 | This study |
| MSHR520 ΔbpsI3 | ΔbpsI3 | This study |
| MSHR520 ΔbpsI12 | ΔbpsI12 | This study |
| MSHR520 ΔbpsI13 | ΔbpsI13 | This study |
| MSHR520 ΔbpsI23 | ΔbpsI23 | This study |
| MSHR520 ΔbpsI123 | ΔbpsI123 | This study |
| MSHR520 Δcap ΔbpsI1 | ΔbpsI1 Δcap; Tcr | This study |
| MSHR520 Δcap ΔbpsI2 | ΔbpsI2 Δcap; Tcr | This study |
| MSHR520 Δcap ΔbpsI3 | ΔbpsI3 Δcap; Tcr | This study |
| MSHR520 Δcap ΔbpsI123 | ΔbpsI123 Δcap; Tcr | This study |
| MSHR520 Δcap bimA | ΔbimA Δcap; Tcr | This study |
| MSHR520 Δcap hcp1 | Δhcp1 Δcap; Tcr | This study |
| Escherichia coli | ||
| DH5α | SupE44 ΔlacU169 (Ф80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | |
| S17.1(λpir) | RP4-2-Tc::Mu Km::Tn7 Tp Sm (λpir) phoA20 |
Mutagenesis
Unmarked in-frame deletion mutagenesis was used to create mutants by allelic exchange [30]. Mutant alleles with a deletion within the relevant gene were synthesised and cloned in pUC57 (Genscript, USA). Deletions encompassed the following nucleotide positions, with reference to the genome sequence of K96243 [31]: bimA (BPSS1492, 2032913–2036300; deletion between 2033879 and 2035245), hcp (BPSS1498, 2042122–2044528; deletion between 2043093 and 2043496), bpsI1 (BPSS0855, 1176590–1179016; deletion between 1177565 and 1178064), bpsI2 (BPSS1180, 1587232–1589606; deletion between 1588246 and 1588739), and bpsI3 (BPSS1570, 2130500–2132616; deletion between 2131330 and 2131811). Mutant alleles were subcloned into pDM4-Tp [30] and then conjugatively mobilised from Escherichia coli S17.1 (λpir) into B. pseudomallei MSHR520, obtained from Bart Currie, Menzies School of Health Research, Darwin, Australia (previously referred to as strain 08 [19], [32]). Single cross-over integrants were selected on chloramphenicol and screened for trimethoprim resistance. Clones in which the integrated deletion construct was excised by a second recombination event was then selected as sucrose-resistant. This was confirmed by chloramphenicol and trimethoprim sensitivity and clones with the deleted allele determined by PCR analysis with appropriate primers. Double and triple mutants are denoted, for example, as BpsI12 (mutated in bpsI1 and bpsI2), bpsI123 (mutated in all three bpsI loci) etc.
Analysis of AHL Production
Bacteria were grown to late log-early stationary phase (A600 1.2) at 37°C at 180 rpm. Cultures were then centrifuged and the supernatant filtered through a 0.22 µm PVDF filter (Millipore). N-septanoyl-L-homoserine lactone (C7-HL; 100 ng ml−1) was added, to allow correction for extraction efficiency, prior to extraction twice with equal volumes of ethyl acetate acidified with 0.1% glacial acetic acid. Aspirated ethyl acetate was evaporated under nitrogen at 35°C and reconstituted in LCMS grade methanol containing N-nonanoyl-L-homoserine lactone (C9-HL; 1 µg ml−1) as internal standard. Chromatographic separation was performed using 2 µl injections onto an Agilent HPLC fitted with a Phenomenex Kinetics XB-C18 column (2.1 mm×100 mm with a 1.7 µm particle size) prior to analysis on an Agilent 6530 Q-TOF using a dual injection Jet Stream ESI source.
The chromatographic conditions consisted of the following mobile phases at a flow rate of 0.5 ml/min A: ultrapure water, and B: LCMS grade acetonitrile (Fisher), both buffered with 0.1% glacial acetic acid. The chromatographic cycle consisted of a linear gradient from 20% B to 90% B over 10 min followed by isocratic column flush at 90% B for 2 min and column re-equilibration at 20% B for a further 1 min. Mass spectroscopy (MS) analysis was performed in positive ion mode. Nitrogen was used as a drying and sheath gas, which was heated to 250°C and 325°C with flow rates of 12 l/min and 10 l/min, respectively. The capillary and fragmentor were set to 2500 V and 135 V. MS data was acquired in wide dynamic range mode between 100 and 1700 m/z with an acquisition rate of 1.4 spectrums sec−1 examining 9665 transients per spectrum. Quantitation of AHLs was performed in MS only mode against extracted ion chromatographs at 5 ppm mass accuracy with a minimum of 7 acquisitions per peak using the Agilent Mass Hunter Quantification Analysis software package. Quantification of all AHLs was performed using the ratio of peak area of analyte to that of the C9 internal standard and compared to 10 point standard curves.
Validation of analytes was done using a modified method of that reported by Gould et al. [33]. In brief, MS/MS fragmentation data was acquired at 1.4 spectrums sec−1 with 9665 transients per spectrum. The collision induced disassociation energy for fragmentation was determined according to m/z of the ion being analysed using the slope determination function of the Mass Hunter acquisition software using a slope of 3.7 and an offset of 9. MS/MS data analysis was performed using Agilent Qualitative analysis software. Positive confirmation occurred if the precursor ions examined displayed mass accuracies of 5 ppm or less to the analyte of interest and the 102.0550 m/z fragment ion corresponding to the lactone ring structure common to the AHLs was present.
In vitro Infection of Macrophage-like Cells
RAW264.7 cells (ATCC TIB-71) were grown in DMEM (Gibco), supplemented with 10% heat-inactivated FBS (Gibco) at 37°C with 5% CO2. Cells were seeded at 5×104 cells per 2.0 cm2 growth area into 96 or 24 well cell culture plates 24 h prior to infection. Cells were infected with bacteria grown for 16 h at a multiplicity of infection of 10 bacteria per cell and incubated at 37°C with 5% CO2 for 1 h. Culture medium was removed and replaced with medium containing 10 µg ml−1 imipenem (Sigma) to suppress extracellular bacterial replication [19], [34]. Incubation was continued for up to 24 h. To quantify intracellular survival and replication infected RAW264.7 cells in 96 well tissue culture plates were washed with phosphate buffered saline (PBS) and permeabilised using 0.1% triton X-100. Released cell–associated bacteria were enumerated by plating and colony counts after 18–20 h growth.
Assessment of Multinucleated Cell Formation
Infected RAW264.7 cells in 24 well tissue culture plates were washed with PBS and immediately fixed with 3.7% (v/v) formalin and stored at 4°C with added PBS until use. For enumeration of nuclei and multinucleation, cells were stained in the wells using 10% Giemsa for 10 min, washed with PBS and air-dried before viewing under 200× magnification. Ten fields of view or at least 1000 nuclei were counted for each well. Both numbers and sizes of MNCs, defined as containing 3 or more nuclei per cell, were recorded. For immunofluorescence, the monolayers were grown and infected on glass coverslips within 24 well plates, washed 3 times with PBS and permeabilised for 10 min with 0.1% triton X-100. All subsequent stages took place at room temperature and were each followed by 3 washes with PBS. Blocking was 30 min with 5% (w/v) BSA in PBS, followed by incubation with a polyclonal rabbit B. pseudomallei antisera [19] for 30 min, followed by incubation with phalloidin-alexa 568 conjugate (Invitrogen) and an anti-rabbit IgG Alexa 488 conjugate (Invitrogen) for 30 min and then 10 min incubation with DAPI (Invitrogen). Coverslips were dried and mounted onto slides using n-propyl gallate (Sigma). Slides were stored at 4°C until viewing.
Animal Studies
BALB/c 5 week old mice (n = 9) were infected intranasally with 3×105 cfu for each strain of B. pseudomallei, with inoculating doses confirmed by serial dilutions and enumeration on agar. For investigation of URT, mice were euthanised at the stated time intervals post-infection by lethabarb (pentobarbital sodium) injection, and blood, NALT, olfactory bulbs (OB) and OE were collected for enumeration of B. pseudomallei; for investigation of systemic infection, blood, lungs, liver and spleen were harvested [13]. Ethical approval for animal experimentation was granted by the Griffith University Animal Ethics Committee, in accordance with National Health and Medical Research Council (NHMRC) of Australia guidelines.
Statistics
Data were compared between groups using a non-parametric ANOVA (Kruskal-Wallis test) using a Dunns post test with significance set at p<0.05. Analyses were carried out using Prism version 5.0 (GraphPad Software).
Results
B. pseudomallei-triggered MNC Formation is Abrogated by Deletion of bimA or hcp1 and is Independent of Intracellular Replication
In the course of utilising RAW264.7 cells to investigate the role of QS in intracellular replication, we examined whether cell fusion is required for optimal intracellular replication. We first confirmed that a mutant lacking Hcp1 [22], encoded within the T6SS-1 locus, failed to induce significant MNC formation in infected RAW264.7 cells up to 12 h post-infection (Figure 1a; Figures 2, panel D, & Figure 3 panel D). Similarly, almost complete abrogation of MNC formation was seen when actin-based motility was prevented by deletion of bimA (Figure 1a; Figures 2, panel C, & Figure 3 panel C). However, no significant differences in numbers of intracellular bacteria were seen between the wild-type (parental; wt) and mutant strains (Figure 1b), indicating that intracellular replication is independent of MNC formation.
Figure 1. Infection of RAW264.7 cells by B. pseudomallei MSHR520, MSHR520ΔbimA, and MSHR520Δhcp1.

Infections were carried out over a period of 12 h and fixed for Giemsa staining (a) or lysed to release internalised bacteria, which were then plated for enumeration of cfu per well (b). (a) Formation of MNCs in cells infected with ΔbimA and Δhcp1 mutants as compared with wt (MSHR520). Comparisons between mutants and wt were statistically significant (P<0.001). >1000 nuclei per well were counted and are expressed as number of nuclei in each category. Columns show the mean values with SEM. (b) Intracellular bacteria in cells infected with wt, ΔbimA and Δhcp1 mutants as compared with wt (MSHR520). No significant difference (P = 0.46) is observed between wt and ΔbimA or wt and Δhcp1. Scatter plots show each well as an individual point with the mean value represented by the horizontal bar. Data are derived from 4 separate experiments, each including duplicate or triplicate wells.
Figure 2. Representative views of B. pseudomallei infected RAW264.7 cells at 12 h post infection.
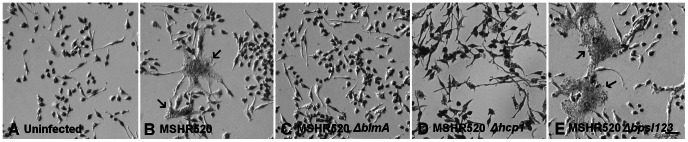
A) Uninfected; B) MSHR520 (wt) infected; C) MSHR520 ΔbimA; D) MSHR520 Δhcp1; E) MSHR520 ΔbpsI123. Cells were fixed with 3.7% (w/v) PFA and stained with Giemsa. Arrows in B and E indicate MNCs. Scale bar in E represents 50 µm.
Figure 3. Representative views of B. pseudomallei infected RAW264.7 cells at 12 h post infection.

A) & B) MSHR520 (wt) infected; C) MSHR520 ΔbimA; D) MSHR520 Δhcp1; E) & F) MSHR520 ΔbpsI123. Nuclei are stained blue with DAPI; bacteria are labelled green with rabbit anti-B. pseudomallei IgG and anti-rabbit IgG-alexa 568; and actin is labelled red with phalloidin-alexa 488. Red actin tails, at the poles of bacteria, are visible in all panels with the exception of C. Scale bar represents 25 µm (A, C, D, E), 12.5 µm (B), 250 µm (F).
Effect of Deletion of bpsI1, 2 & 3 on AHL Production
Homologues of LuxI proteins are responsible for the biosynthesis of AHLs that mediate bacterial QS. In-frame deletion mutants of all three B. pseudomallei bpsI homologues (BPSS0855, BPSS1180, BPSS1570) were constructed singly and in combination, and the AHLs produced by these strains assessed by LCMS/MS analysis of culture supernatant (Table 2). N-octanoylhomoserine lactone (C8HL) and N-(3-hydroxy-decanoyl)homoserine lactone (OHC10HL) were the predominant AHLs found, constituting nearly 90% of the total AHLs in the wt strain. The AHLs, N-(3-hydroxy-octanoyl)homoserine lactone (OHC8HL), N-decanoylhomoserinelactone (C10HL) and N-(3-hydroxy-dodecanoyl)homoserine lactone (OHC12HL) were also present in significant amounts. C8HL was absent (<2%) in the bpsI1 mutant, and all other bpsI1-deficient strains (bpsI12, bpsI13, bpsI123). Likewise, C10HL, OHC10HL and OHC12HL were absent in the single bpsI2 mutant and all other bpsI2 mutant strains. The production of OHC8HL by the mutant strains was more complex: All three single mutants and bpsI31 expressed less OHC8HL but this AHL was not detected in cultures of bpsI12, bpsI32 and bpsI123; together, these data show a role for bpsI2 in OHC8HL synthesis, but in conjunction with bpsI1 and/or bpsI3.
Table 2. AHLs produced by B. pseudomallei MSHR520 and its derived mutants.
| AHL | Extracellular AHLs detected (nM) | |||||||
| MSHR520 | bpsI1 | bpsI2 | bpsI3 | bpsI12 | bpsI13 | bpsI23 | bpsI123 | |
| C8HSL | 8647±1222 | 129±191 | 5178±2496 | 7002±4841 | ND | ND | 12110±4218 | ND |
| OHC8HSL | 1350±587 | 778±224 | 486±227 | 455±307 | ND | 650±45 | ND | ND |
| C10HSL | 623±142 | 335±178 | ND | 469±260 | ND | 390±70 | ND | ND |
| OHC10HSL | 10966±4233 | 8583±3697 | ND | 6421±5164 | ND | 9561±1520 | ND | ND |
| C12HSL | ND | ND | ND | ND | ND | ND | ND | ND |
| OHC12HSL | 284±229 | 399±175 | ND | ND | ND | 449±177 | ND | ND |
Mutant strains are deleted in each AHL synthase gene indicated, either singly or in double or triple combination. Each value is presented as the mean ± SD. ND, not detected.
AHL Synthase-deficient B. pseudomallei Induces Mass MNC Formation in Macrophages
Since cultured cells may involve cell density-mediated QS, we next examined the effect of abrogation of AHL synthesis on the capacity of RAW264.7 cells to support replication of B. pseudomallei, and undergo cell fusion leading to MNC formation. We quantified MNC formation by enumerating the numbers of cells with multiple nuclei, in cultures infected with the wt strain or mutants with bpsI deleted individually, in pairs, or mutation in all three bpsI loci. We observed a remarkable increase in cellular fusion induced by the triple bpsI mutant, (Figure 4 a, c, d; and see Figures 2, panel E, and 3 panels E & F). This was manifested as a marked and significant increase in the number of nuclei associated with cells containing >10 nuclei, as compared to the wt. Experiments using the mutants lacking only one or two AHL synthases failed to show any significant differences compared to the wt strain in terms of MNC formation (Figure 4 c, d). Interestingly, enumeration of intracellular bacteria showed no significant differences in intracellular survival/replication between the wt strain and mutants lacking AHL synthases, including the triple bpsI mutant (Figure 4b).
Figure 4. Infection of RAW264.7 cells by B. pseudomallei MSHR520 and related mutants.

Infections were carried out over a period of 12 h and fixed for Giemsa staining (a), (c), (d), or lysed to release internalised bacteria which were then plated for enumeration of cfu per well (b). (a) Formation of MNCs is increased in the triple ΔbpsI123 mutant as compared with wt (MSHR520); comparison of mutant to wt is statistically significant for the 51+ group. >1000 nuclei were counted for each well and are expressed as the number of nuclei per group. Columns show the mean values with SEM. (b) No significant variation in intracellular bacteria is seen between wt and ΔbpsI1, ΔbpsI2, ΔbpsI3, ΔbpsI123. (c) MNC formation in single AHL mutants: In contrast to the triple mutant strain (comparison of mutant to wt is statistically significant for the 51+ group), formation of MNCs does not significantly differ between the wt and any of the single AHL mutants. (d) MNC formation in double AHL mutants: In contrast to the triple mutant strain (comparison of mutant to wt is statistically significant for the 11–25 and 26–50 groups), formation of MNCs does not differ between the wt and any of the double AHL mutants. Data in each figure are derived from 3 separate experiments, each including duplicate or triplicate wells.
Representative images of RAW264.7 cells infected with MSHR520 and isogenic mutants are shown in Figures 2 and 3, either Geimsa stained or subject to IHC, respectively. Typical MSHR520-induced MNCs are shown with actin tail formation (Figure 2 panel B; Figure 3 panels A & B); MNCs were not evident following infection with bimA or hcp1 mutants (nor were actin tails observed in the former case), although infected cells were observed (Figures 2 & 3 panels C & D). Very large MNCs (>10–50 nuclei), induced following MSHR520 infection were substantially more numerous in bpsI123-infected cells (Figure 4), and are shown in Figures 2 (panel E) and 3 (panels E & F).
AHL Synthase Mutants Retain Virulence Following Intranasal Infection in Mice
To assess whether abolishing AHL synthesis might affect pathogenicity in an acute infection model, BALB/c mice were challenged intranasally and organ loads were enumerated post-infection. No significant differences were observed in bacterial numbers between mice infected with the wt strain, and the bpsI123 mutant in lung, liver, spleen, or blood (Figure 5a).
Figure 5. Infection of BALB/c mice with B. pseudomallei MSHR520 and related mutants.

(a) Infection with MSHR520 and MSHR520ΔbpsI123 followed by determination of bacterial loads in systemic sites. (b) Infection with MSHR520Δcap and MSHR520Δcap ΔbpsI123 followed by determination bacterial loads in the upper respiratory tract (URT), olfactory epithelium (OE), olfactory bulb (OB), nasal-associated lymphoid tissue (NALT), and blood. No significant differences were observed between wt and mutant strains for equivalent tissues and time points.
Finally, to determine if infection of the URT, and subsequent infection of the OB, might be affected by disrupting QS signalling in B. pseudomallei, infections with acapsulate AHL synthase mutants were assessed. In these assays, the confounding factor of haematogenous spread to the brain was reduced, since acapsulate strains have a lower capacity to survive in the blood, as we previously demonstrated [13]. However, as observed with capsulate strains, the capacity of AHL synthase mutants to colonise and survive in the URT, and the OB, was not significantly different compared to the wt strain (Figure 5b).
Discussion
This study establishes that C8HL and OHC10HL are the predominant AHLs produced by B. pseudomallei MSHR520, constituting 90% of the total detected AHLs, and are produced in approximately equal concentrations in aerated broth cultures. The AHLs OHC8HL, C10HL and OHC12HL are also present in significant amounts. A difference in our findings compared to a prior study [29] is that we observed distinct relative amounts of C8HL and OHC10HL. Since C8HL is virtually absent in the bpsI1 mutant, and not detected in all other bpsI1-deficient strains (bpsI12, bpsI13, and bpsI123; Table 2), we conclude that BpsI1 is responsible for synthesis of C8HL. Likewise, we conclude that BpsI2 is responsible for the synthesis of C10HL, OHC10HL and OHC12HL since they are absent in the single bpsI2 mutant and all other bpsI2 mutant strains. The pattern for OHC8HL is more complex, with reduced production but not absence in each of the single bpsI mutants and in bpsI31. OHC8HL was not detected in cultures of bpsI12, bpsI32 and bpsI123, indicating that bpsI2 is required but that bpsI1 and/or bpsI3 are also required for full production. It was reported that transcription of bpsI3 depends on bpsI1 in stationary phase [29] and thus it may be that OHC8HL depends on both bpsI2 and bpsI3. Our results are consistent with prior data derived using single mutants, although the AHL levels of two of the single mutants were undetected rather than reduced [29]. Dependence on two synthases might be via an unknown mechanism, or via both acting independently in the synthesis of OHC8HL.
B. pseudomallei, B. thailandensis and B. mallei can invade and replicate inside mammalian cells, and induce cellular fusion. A striking and novel finding in the current work was that complete abolition of AHL synthesis in the triple mutant (bpsI123) resulted in a remarkable enhancement of cellular fusion in RAW264.7 cells. By comparing the AHL mutants with the wt strain, and two mutants (bimA and hcp1) previously reported to be unable to induce cellular fusion in epithelial cell lines [15], [18], [21], [22] we showed this phenotype to be independent of intracellular bacterial survival and replication. Despite inducing increased MNC formation, the AHL synthase mutants were not more proficient in intracellular replication. The hcp1 and bimA mutants, which did not induce MNCs, were also unaffected in intracellular replication. These results establish for the first time that MNC formation induced by B. pseudomallei is not the result of enhanced bacterial replication or survival in host cells, nor is it required to support bacterial growth. The mechanism by which cell fusion is induced by B. pseudomallei and enhanced by the absence of AHL synthesis remains for investigation.
Another interesting finding in our study is that neither the bpsI12 nor the bpsI123 mutants produced detectable AHLs. The lack of AHLs in the bpsI12 strain may be due to loss of, or reduced, transcription in stationary phase of bpsI3 as result of loss of bpsI1, as noted above. Thus the bpsI12 mutant may represent a phenotypically triple mutant under these conditions. However, despite this similarity in AHL profile the bpsI12 strain did not show the increased MNC phenotype of the triple mutant. We hypothesise that, in cells infected with the bpsI12 mutant, BpsI3 may contribute to production of an AHL that was not detected in our analyses.
Typically, loss of QS results in attenuation of indicators of virulence [23], [24], though an increase in morbidity in vivo has also been reported [35]. Previous studies on B. pseudomallei have implicated AHL signalling in virulence in the murine model: Mutants in bpsI1 and bpsI3 have an increased LD50 in hamsters, although the bpsI1 mutant showed a survival curve similar to wt [25]. Valade et al. [26] showed that a bpsI1 (“pmlI”) mutant had increased LD50 in Swiss mice. Conversely, our study found no effect on virulence in mice when all AHL signal production is abolished. Using our in-frame deletion mutants we showed that a variety of AHLs are normally produced but none are detectable in the triple mutant lacking all three synthases. Hence, since the triple mutant showed a major phenotype in cultured cells, but colonization was not significantly reduced as a result of abrogation of AHL production, we conclude that AHL-mediated QS is not necessary for successful acute infection. However, melioidotic lesions, particularly from skin or chronic infections, are noted to contain fused host cells, or granulomas [36]. It thus seems plausible that there remains a role for AHL-mediated QS in chronic infection. Indeed, a recently developed model of murine chronic infection was characterised by MNC-containing lesions [20]. Increased cell fusion in the absence of QS might enhance the formation of such lesions and/or granulomas. The role of AHL synthesis in chronic infection remains to be assessed and the studies reported herein will be useful to test this hypothesis in the setting of chronic disease.
AHL-mediated QS is probably important in survival of B. pseudomallei in the environment [26], [29] which might also include resistance to predation by, and/or growth in, amoebae [37]–[40]. In this case, a putative role in chronic infection might be viewed as ‘accidental’ or ‘environmental opportunism’ [16], [37], [41], [42]. It seems likely that QS regulates a gene(s) which is involved in the suppression of MNC formation, identification of which could provide new insight into bacterial mechanisms underlying macrophage cell fusion.
Funding Statement
This work has been supported by the National Health and Medical Research Council and by Griffith University. GCU is supported by a Future Fellowship from the Australian Research Council. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Currie BJ, Ward L, Cheng AC (2010) The epidemiology and clinical spectrum of melioidosis: 540 cases from the 20 year Darwin prospective study. PLoS Negl Trop Dis 4: e900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cheng AC, Hanna JN, Norton R, Hills SL, Davis J, et al. (2003) Melioidosis in northern Australia, 2001–02. Commun Dis Intell 27: 272–277. [DOI] [PubMed] [Google Scholar]
- 3. White NJ (2003) Melioidosis. Lancet 361: 1715–1722. [DOI] [PubMed] [Google Scholar]
- 4. Dance DA (2000) Melioidosis as an emerging global problem. Acta Trop 74: 115–119. [DOI] [PubMed] [Google Scholar]
- 5. Currie BJ, Dance DA, Cheng AC (2008) The global distribution of Burkholderia pseudomallei and melioidosis: an update. Trans R Soc Trop Med Hyg 102 Suppl 1S1–4. [DOI] [PubMed] [Google Scholar]
- 6. Chugh TD (2008) Emerging and re-emerging bacterial diseases in India. J Biosci 33: 549–555. [DOI] [PubMed] [Google Scholar]
- 7. Rolim DB, Vilar DC, Sousa AQ, Miralles IS, de Oliveira DC, et al. (2005) Melioidosis, northeastern Brazil. Emerg Infect Dis 11: 1458–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saravu K, Mukhopadhyay C, Vishwanath S, Valsalan R, Docherla M, et al. (2010) Melioidosis in southern India: epidemiological and clinical profile. Southeast Asian J Trop Med Public Health 41: 401–409. [PubMed] [Google Scholar]
- 9. Hassan MR, Pani SP, Peng NP, Voralu K, Vijayalakshmi N, et al. (2010) Incidence, risk factors and clinical epidemiology of melioidosis: a complex socio-ecological emerging infectious disease in the Alor Setar region of Kedah, Malaysia. BMC Infect Dis 10: 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Currie BJ, Jacups SP (2003) Intensity of rainfall and severity of melioidosis, Australia. Emerg Infect Dis 9: 1538–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ngauy V, Lemeshev Y, Sadkowski L, Crawford G (2005) Cutaneous melioidosis in a man who was taken as a prisoner of war by the Japanese during World War II. J Clin Microbiol 43: 970–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jeddeloh JA, Fritz DL, Waag DM, Hartings JM, Andrews GP (2003) Biodefense-driven murine model of pneumonic melioidosis. Infect Immun 71: 584–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Owen SJ, Batzloff M, Chehrehasa F, Meedeniya A, Casart Y, et al. (2009) Nasal-associated lymphoid tissue and olfactory epithelium as portals of entry for Burkholderia pseudomallei in murine melioidosis. J Infect Dis 199: 1761–1770. [DOI] [PubMed] [Google Scholar]
- 14. Galyov EE, Brett PJ, DeShazer D (2010) Molecular insights into Burkholderia pseudomallei and Burkholderia mallei pathogenesis. Annu Rev Microbiol 64: 495–517. [DOI] [PubMed] [Google Scholar]
- 15. French CT, Toesca IJ, Wu TH, Teslaa T, Beaty SM, et al. (2011) Dissection of the Burkholderia intracellular life cycle using a photothermal nanoblade. Proc Natl Acad Sci U S A 108: 12095–12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beacham IR, Nathan S, Hara Y, Peak IR (2012) Virulence determinants in Burkholderia pseudomallei: Opportunist or accidental pathogen? In: Ketheesan N, editor. Melioidosis: A century of observation and research. Amsterdam: Elsevier B. V: 87–99. [Google Scholar]
- 17. Kespichayawattana W, Rattanachetkul S, Wanun T, Utaisincharoen P, Sirisinha S (2000) Burkholderia pseudomallei induces cell fusion and actin-associated membrane protrusion: a possible mechanism for cell-to-cell spreading. Infect Immun 68: 5377–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stevens MP, Stevens JM, Jeng RL, Taylor LA, Wood MW, et al. (2005) Identification of a bacterial factor required for actin-based motility of Burkholderia pseudomallei. Mol Microbiol 56: 40–53. [DOI] [PubMed] [Google Scholar]
- 19. Boddey JA, Day CJ, Flegg CP, Ulrich RL, Stephens SR, et al. (2007) The bacterial gene lfpA influences the potent induction of calcitonin receptor and osteoclast-related genes in Burkholderia pseudomallei-induced TRAP-positive multinucleated giant cells. Cell Microbiol 9: 514–531. [DOI] [PubMed] [Google Scholar]
- 20. Conejero L, Patel N, de Reynal M, Oberdorf S, Prior J, et al. (2011) Low-dose exposure of C57BL/6 mice to Burkholderia pseudomallei mimics chronic human melioidosis. Am J Pathol 179: 270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sitthidet C, Korbsrisate S, Layton AN, Field TR, Stevens MP, et al. (2011) Identification of motifs of Burkholderia pseudomallei BimA required for intracellular motility, actin binding, and actin polymerization. J Bacteriol 193: 1901–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burtnick MN, Brett PJ, Harding SV, Ngugi SA, Ribot WJ, et al. (2011) The cluster 1 type VI secretion system is a major virulence determinant in Burkholderia pseudomallei . Infect Immun 79: 1512–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de Kievit TR, Iglewski BH (2000) Bacterial quorum sensing in pathogenic relationships. Infect Immun 68: 4839–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fuqua C, Greenberg EP (2002) Listening in on bacteria: acyl-homoserine lactone signalling. Nat Rev Mol Cell Biol 3: 685–695. [DOI] [PubMed] [Google Scholar]
- 25. Ulrich RL, Deshazer D, Brueggemann EE, Hines HB, Oyston PC, et al. (2004) Role of quorum sensing in the pathogenicity of Burkholderia pseudomallei . J Med Microbiol 53: 1053–1064. [DOI] [PubMed] [Google Scholar]
- 26. Valade E, Thibault FM, Gauthier YP, Palencia M, Popoff MY, et al. (2004) The PmlI-PmlR quorum-sensing system in Burkholderia pseudomallei plays a key role in virulence and modulates production of the MprA protease. J Bacteriol 186: 2288–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Song Y, Xie C, Ong YM, Gan YH, Chua KL (2005) The BpsIR quorum-sensing system of Burkholderia pseudomallei . J Bacteriol 187: 785–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lumjiaktase P, Diggle SP, Loprasert S, Tungpradabkul S, Daykin M, et al. (2006) Quorum sensing regulates dpsA and the oxidative stress response in Burkholderia pseudomallei . Microbiology 152: 3651–3659. [DOI] [PubMed] [Google Scholar]
- 29. Gamage AM, Shui G, Wenk MR, Chua KL (2011) N-Octanoylhomoserine lactone signalling mediated by the BpsI-BpsR quorum sensing system plays a major role in biofilm formation of Burkholderia pseudomallei . Microbiology 157: 1176–1186. [DOI] [PubMed] [Google Scholar]
- 30. Logue CA, Peak IR, Beacham IR (2009) Facile construction of unmarked deletion mutants in Burkholderia pseudomallei using sacB counter-selection in sucrose-resistant and sucrose-sensitive isolates. J Microbiol Methods 76: 320–323. [DOI] [PubMed] [Google Scholar]
- 31. Holden MT, Titball RW, Peacock SJ, Cerdeno-Tarraga AM, Atkins T, et al. (2004) Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei . Proc Natl Acad Sci U S A 101: 14240–14245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brown NF, Beacham IR (2000) Cloning and analysis of genomic differences unique to Burkholderia pseudomallei by comparison with B. thailandensis. J Med Microbiol 49: 993–1001. [DOI] [PubMed] [Google Scholar]
- 33. Gould TA, Herman J, Krank J, Murphy RC, Churchill ME (2006) Specificity of acyl-homoserine lactone synthases examined by mass spectrometry. J Bacteriol 188: 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miyagi K, Kawakami K, Saito A (1997) Role of reactive nitrogen and oxygen intermediates in gamma interferon-stimulated murine macrophage bactericidal activity against Burkholderia pseudomallei . Infect Immun 65: 4108–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Coulthurst SJ, Clare S, Evans TJ, Foulds IJ, Roberts KJ, et al. (2007) Quorum sensing has an unexpected role in virulence in the model pathogen Citrobacter rodentium . EMBO Rep 8: 698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wong KT, Puthucheary SD, Vadivelu J (1995) The histopathology of human melioidosis. Histopathology 26: 51–55. [DOI] [PubMed] [Google Scholar]
- 37. Casadevall A, Pirofski LA (2007) Accidental virulence, cryptic pathogenesis, martians, lost hosts, and the pathogenicity of environmental microbes. Eukaryot Cell 6: 2169–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brussow H (2007) Bacteria between protists and phages: from antipredation strategies to the evolution of pathogenicity. Mol Microbiol 65: 583–589. [DOI] [PubMed] [Google Scholar]
- 39. Hilbi H, Weber SS, Ragaz C, Nyfeler Y, Urwyler S (2007) Environmental predators as models for bacterial pathogenesis. Environ Microbiol 9: 563–575. [DOI] [PubMed] [Google Scholar]
- 40. Inglis TJ, Sagripanti JL (2006) Environmental factors that affect the survival and persistence of Burkholderia pseudomallei . Appl Environ Microbiol 72: 6865–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moxon ER, Jansen VA (2005) Phage variation: understanding the behaviour of an accidental pathogen. Trends Microbiol 13: 563–565. [DOI] [PubMed] [Google Scholar]
- 42. Brown SP, Cornforth DM, Mideo N (2012) Evolution of virulence in opportunistic pathogens: generalism, plasticity, and control. Trends Microbiol 20: 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]