

Abstract
The organelle paralogy hypothesis is one model for the acquisition of non-endosymbiotic organelles, generated from molecular evolutionary analyses of proteins encoding specificity in the membrane traffic system. GTPase Activating Proteins (GAPs) for the ADP-ribosylation factor (Arfs) GTPases are additional regulators of the kinetics and fidelity of membrane traffic. Here we describe molecular evolutionary analyses of Arf GAP protein family. Of the ten subfamilies previously defined in humans, we find that five were likely present in the Last Eukaryotic Common Ancestor (LECA). Of the three more recently derived subfamilies, one was likely present in the ancestor of opisthokonts (animals and fungi) and apusomonads (flagellates classified as the sister lineage to opisthokonts), while two arose in the holozoan lineage. We also propose to have identified a novel ancient subfamily (ArfGAPC2), present in diverse eukaryotes but which is lost frequently, including in the opisthokonts. Surprisingly few ancient domains accompanying the ArfGAP domain were identified, in marked contrast to the extensively decorated human Arf GAPs. Phylogenetic analyses of the subfamilies reveal patterns of single and multiple gene duplications specific to the Holozoa, to some degree mirroring evolution of Arf GAP targets, the Arfs. Conservation, and lack thereof, of various residues in the ArfGAP structure provide contextualization of previously identified functional amino acids and their application to Arf GAP biology in general. Overall, our results yield insights into current Arf GAP biology, reveal complexity in the ancient eukaryotic ancestor, and integrate the Arf GAP family into a proposed mechanism for the evolution of non-endosymbiotic organelles.
Keywords: membrane traffic, phylogeny, comparative genomics, vesicle transport, GTPase, ADP-ribosylation factors (Arfs), GTPase activating proteins (GAPs), evolutionary cell biology
INTRODUCTION
Eukaryotic organelles have arisen by one of two evolutionary mechanisms. Mitochondria and plastids are derived from ancient endosymbiosis of a proto-eukaryote with an α-proteobacterium or a cyanobacterium, respectively (1). Other organelles appear to have arisen autogenously, i.e. without major endosymbiotic contribution, from building blocks present within the earliest eukaryotes. While the process of endosymbiotic organelle evolution is becoming increasingly well characterized (2, 3), our mechanistic understanding of autogenous organelle evolution is only beginning to coalesce. These details are emerging from evolutionary studies of organelles of autogenous origin, the best candidates of which are those of the membrane traffic system.
Composed of the endoplasmic reticulum (ER), the Golgi body, the various endosomes (early, late, recycling, multi-vesicular body), the lysosome/vacuole, and the plasma membrane, the membrane traffic system is involved in the production of membrane and secretory proteins, in endocytosis and exocytosis, and in cell movement: they are crucial in maintaining basic cellular function (4). Traffic of material between cellular organelles involves a similar set of interacting protein machinery, with organelle- or pathway-specific homologs for each individual transport step (4). Vesicle formation involves small GTPases (Arf, Sar1), proteins for cargo selection (e.g. Adaptins) and for membrane deformation, while vesicle fusion involves proteins for tethering (e.g. Rab GTPases), membrane fusion (e.g. SNAREs) and regulation. The specificity of each transport step is encoded in the combinatorial interactions of these various, organelle-specific proteins (5).
Importantly, this model of membrane traffic has been developed through experimental studies in animal and yeast model systems. This is only a small fraction of overall eukaryotic cellular diversity, which is now classified into six supergroups; Opisthokonta, Amoebozoa, Excavata, Archaeplastida, SAR, and the contentious CCTH, plus a few lineages that do not fit clearly into any of these, e.g. the apusomonads that appear sister to the opisthokonts (6–8). Questions of eukaryotic cellular evolution are inextricably linked to questions concerning the modern cell biology of these diverse organisms and comparison to the cell biology of model organisms. Broad surveys allow us to both infer the state of the Last Eukaryotic Common Ancestor (LECA), and derive a generalized model for membrane traffic in eukaryotes, not simply one applicable to yeast and mammals.
Comparative genomic and molecular phylogenetic analyses have shown that the major protein families involved in vesicle formation and fusion are broadly conserved across the available genomic diversity of eukaryotes (9). Furthermore, as much of the machinery is composed of protein families, these analyses have revealed that the organelle-specific paralogs of these proteins are conserved as well, implying the presence of sophisticated membrane traffic machinery in the LECA. One currently proposed mechanism of autogenous organelle evolution invokes an increase in number and complexity of endomembrane organelles via gene duplication and co-evolution of the interacting proteins encoding organelle identity/traffic-specificity (9). Therefore, understanding the evolutionary history of these individual protein families will further elucidate the mechanism through which the organelles of the membrane traffic system evolved. This mechanism also predicts a mixed phylogenetic pattern with ancient membrane traffic homologs being widely distributed across eukaryotes, and with additional complexity arising independently in the various descendent lineages (10). This has been borne out in analyses of the three major membrane traffic protein families examined thus far, adaptins (10, 11), Rabs (12–14) and SNAREs (15–17), showing ancient complexity in some families along with lineage-specific expansions.
One additional important set of components to examine in the context of this theory are the small GTPases involved in vesicle formation, in particular the ADP-ribosylation factors (Arfs). Arfs are ~21 kDa GTPases within the larger Ras superfamily. The greater Arf family is highly conserved throughout eukaryotic evolution (18). The Arf family has been functionally divided into three main sub-groups; the Arf proteins share >60% primary sequence identity and a number of biochemical and cellular activities (4), the Arf-like (Arl) proteins that are more numerous and divergent in both sequence and activities, and the Sar1 proteins are the most divergent but with clearly defined functions in export of proteins from the ER to cis-Golgi, acting at ER exit sites (18). Arfs act as regulators of a number of cellular processes and can dually serve to help integrate them; including membrane traffic, phospholipid metabolism, cell adhesion, and cell motility (19). Six Arfs exist in vertebrates, but only five are encoded in the human genome (20). However, while at least six members of the greater Arf family were likely present in the LECA, it appears to have possessed few Arfs, possibly only one (18). Arf activities are initiated in cells as a result of interaction with Arf guanine nucleotide exchange factors (Arf GEFs) and are terminated as a consequence of GTP hydrolysis, resulting from interaction with an Arf GTPase activating protein (Arf GAP).
Originally thought to play primary roles as terminators of Arf signaling, Arf GAPs have more recently emerged with the potential to serve as essential Arf effectors that provide added elements of specificity and localization to those signals (21–26). These more divergent properties are, in part, provided by the presence of additional domains in some Arf GAP proteins while the catalytic (GTP hydrolysis promoting) actions have been strongly linked to a minimal zinc-binding cysteine cluster and “arginine finger” with specific spacing (CX2CX16CX2CX4R) within the larger Arf GAP domain of ~130 residues (27). The four cysteines chelate a single zinc atom and act as a classic zinc finger at the core of the Arf GAP domain to stabilize the folded structure. In contrast, the arginine finger is highly exposed on the protein surface and has been shown to act analogously to those in some other Ras superfamily GAPs by insertion into the catalytic site of the cognate GTPase, resulting in neutralization of accumulating negative charge and stabilization of the transition state in the hydrolysis of the β-γ phosphate bond (28, 29). Mutation of the catalytic arginine has been found to decrease catalytic power in every case tested and typically by several orders of magnitude (28, 30, 31). While a few exceptions are likely to emerge (e.g., see reference (32)), for the most part Arf GAPs act only on the Arf proteins and not on the closely related Arls. Arf GAPs can regulate membrane traffic through effects on activated Arfs, membrane deformation (33, 34), cargo selection (35), or act as platforms for signal integration between the membrane traffic system and the cytoskeleton (36). The Arf GAP family of proteins is composed of multiple paralogous members, thus prompting evolutionary questions regarding the extent of ancient diversity, timing of paralog emergence and degree of evolutionary novelty in eukaryotes.
The goals of an earlier analysis of Arf GAP evolution (37) were to focus on organisms used extensively in cell biological and genetic studies while generating a more consistent system for naming genes and proteins within the Arf GAP subfamilies. This resulted in the identification of ten subfamilies in humans (Figure 1A), based on domain structure and the sequence of their Arf GAP domains, as well as generation of hypotheses for the timing of origins of five of them. Herein we assess the conservation and evolution of these ten Arf GAP subfamilies across eukaryotes using comparative genomic and phylogenetic methods. The detailed analyses of the evolution, expansion and acquisition or loss of additional domains in Arf GAP subfamilies, allowed us to determine that at least five of the ten Arf GAP subfamilies found in humans are sufficiently conserved in eukaryotes to be inferred as present in the LECA. Conversely, three of the previously identified subfamilies appear to be more recent, lineage-specific innovations. We also report the presence of a proposed 11th subfamily that is ancient and present in diverse eukaryotes but not animals or fungi. With at least six Arf GAPs present in the LECA and as few as one Arf we predict that the emergence of Arf GAPs provided among the earliest sources of diversity and specificity in Arf signaling. However, our analysis also reveals patterns to the way in which Holozoa have substantially increased the size of their Arf GAP complement, suggestive of more complex roles for Arf GAPs within the lineage leading to multicellular animals.
Figure 1. Distribution of Arf GAP subfamilies across eukaryotic taxa.

A) Tabulation of the 11 Arf GAP subfamilies with acronyms, full names and the latest deduced point of origin. B) Six subfamilies are present in at least three eukaryotic supergroups, based on the five-orders criterion, and are presumed as present in the LECA. AGAP is likely present in the opisthokont and amoebozoan ancestor, and is ancient, if not necessarily in the LECA. GIT and ARAP are specific to the Filozoa (Holozoa except Ichthyosporea; (53)), while ASAP is found opisthokonts and apusomonads (in addition to the questionable T. vaginalis sequences-see figure S1). Large taxonomic groupings are color coded, with taxonomic key on the left. Numbers in brackets indicate the total number of Arf GAPs identified in the corresponding genome. Sectors with solid colors indicate those homologs identified using the five-orders criterion. The pale colored sectors indicate those identified by the two-orders criterion. Grey sectors indicate that no ortholog was found in the genome of the organism in question, but was found in the genome of closely related organism through nr-BLAST at the two-orders criterion (see methods). Open sectors indicate that no ortholog was found using BLAST or HMMer to probe the genome in question or through nr-BLASTs. For the ArfGAPC2 column (boxed), the solid colours represent the presence of at least one ortholog meeting a criterion of a bi-directional retrieval of another ArfGAPC2 ortholog at a five-orders criterion.
RESULTS
Comparative genomics reveals ancient versus more recent origins of the ten Arf GAP subfamilies found in humans
In order to assess the conservation and diversity of the eukaryotic proteins containing the ArfGAP domain, comparative genomic analyses were undertaken. Homology searching via BLASTp and HMMer was performed on the genomes of 38 organisms spanning eukaryotic diversity (listed in Table S1) using each of the known human and S. cerevisiae Arf GAP protein sequences. In total, 446 candidate sequences were identified. Of these, 334 were unambiguously assigned, based on the Reciprocal Best Hit (RBH) method of homology assessment (38, 39) and using the five-orders criterion (see Materials and Methods for details), to one of the ten previously identified Arf GAP subfamilies found in humans. However, if the two-orders criterion is applied, then an additional 76 sequences could be more tentatively classified into one of the ten human subfamilies. The remaining 36 unclassified sequences were designated as orphans and set aside for further analyses. Phylogenetic analysis of all ArfGAP domain-containing proteins was undertaken to further verify the classification, but little resolution was obtained (data not shown) so this approach was not pursued further.
Based on our analyses, five of the ten human Arf GAP subfamilies (SMAP, ArfGAP1, ArfGAP2/3, ACAP, and AGFG) were found in four or more of the eukaryotic supergroups with orthologs identified at the five-orders criterion (Figure 1). The results from the nr-BLASTs, additionally identified a number of candidate orthologs and gave us further confidence in the broad distribution of these Arf GAPs. Thus, these five subfamilies are presumed to have been present in the LECA. By contrast three Arf GAP subfamilies arose later. ARAP and GIT appear to be specific to Capsaspora owczarzaki, Monosiga brevicolis, and Metazoa. ASAP is present throughout the Holozoa and in the apusomonad representative, Thecamonas trahens. Several of the Arf GAP sequences from T. vaginalis returned ASAP as their top hit in reciprocal BLAST analyses into humans. While these could represent horizontal gene transfer events it is more likely, based on our phylogenetic analyses (Figure S1), that these are highly divergent members of the ACAP subfamily because they cluster with the sequences classified as T. vaginalis ACAPs. ACAP is clearly an ancient subfamily, and T. vaginalis is well known to have highly divergent protein sequences. We therefore treat ASAP as a more recent subfamily, present in the ancestor of apusomonads and opisthokonts.
Two of the human subfamilies (AGAP and ADAP) had distributions that were equivocal and thus the proposed origins are somewhat more speculative. Orthologs of AGAP were identified at the five-orders criterion in members of the Holozoa and the Amoebozoa. This represents an ancient eukaryotic ancestor, but may not be the LECA, depending on the placement of the root of eukaryotes, which is currently a matter of open scientific debate. We also found sequences from three other eukaryotic supergroups, but these were classified only at the two-orders criterion. Thus while AGAP clearly has ancient origins, it does not meet our stringent requirements for concluding its presence in the LECA (see materials and methods). The tenth subfamily identified in humans is ADAP. We found orthologs for the ADAP subfamily only in holozoan representatives at the five-orders criterion but note that if the two-order criterion was applied, we found putative ADAP sequences in representatives from three supergroups. Because the distribution is inconclusive, we do not feel confident in predicting an origin for this subfamily.
In all of the Arf GAP subfamilies, lineage-specific loss is apparent. This is especially prevalent in fungi, with AGFG present only in the basal fungus genome in our analysis, Batrachochytrium dendrobatidis, and AGAP present only in the zygomycete fungal genome of R. delemar. ASAP and ADAP were not identified in any of the fungal genomes sampled. We also failed to identify ADAP homologs in archaeplastids. Depending on whether this subfamily is ancient or not, this may represent a second independent incidence of loss (Figure 2).
Figure 2. Schematization of Arf GAP gains and losses in eukaryotes.

A) Tree of eukaryotes depicting domains and Arf GAP subfamilies present in the LECA, as well as gains or losses of additional domains and subfamilies throughout eukaryotes. To increase the confidence of predictions, losses are only proposed when the deduction is based on two genomes of the relevant lineage. The symbols denoting the origins of ADAP and AGAP are set to the side, illustrating the equivocal nature of the deduced origins hypotheses as either present in LECA or more recently derived in Metazoa and opisthokonts + amoebozoa, respectively. B) Gain and loss of Arf GAP subfamilies and domains in Holozoa. Symbol legend for both panels is inset in B and the subfamily in which the domain was gained or lost is indicated in brackets. PH = Pleckstrin Homology domain, ANK = Ankyrin Repeat, BAR = Bin-Amphiphysin-Rvs domain, C2 =calcium dependent membrane-targeting domain, SAM = Sterile alpha motif, SH3 = Src homology-3 domain, GIT = G protein-coupled receptor kinase-interacting protein domain, UBA = ubiquitin associated/translation elongation factor EF1B N-terminal domain (definitions are taken from InterproScan results).
Identification of a novel, ancient subfamily of Arf GAP proteins: ArfGAPC2
One possible explanation for the ArfGAP domain containing proteins that could not be classified into the ten human subfamilies (i.e. the orphans) is that some may be representatives of additional subfamilies, present in diverse eukaryotes but that had been lost in humans. In order to assess this possibility, we performed BLASTp analyses with each of the 36 ‘orphan’ sequences into the genomes containing at least one such unclassified sequence. For the majority of the sequences, no best hits meeting either RBH criteria were found. However, for six sequences we found reciprocal best hits at the five-orders criterion to other sequences also classified as orphans (Figure S2A). Primary structure investigation revealed that these six sequences share a conserved domain architecture with a Ca2+ dependent-membrane targeting (C2) domain C-terminal to the ArfGAP domain (Figure 3). Together this provides a strong indication that these sequences are orthologs. We therefore designate this new subfamily as ArfGAPC2.
Figure 3.

Evidence of the newly described ArfGAPC2 subfamily. Domain organization of each ArfGAPC2 subfamily member is illustrated. Each sequence contains an ArfGAP domain followed by a C2 domain. Sequences are drawn to scale.ArfGAP = ArfGAP domain; C2 = Calcium Dependent Membrane Targetting; PH = Pleckstrin Homology
In addition to the six sequences above, a orphan sequence in A. thaliana (designated AtAGC2_A) retrieved putative ArfGAPC2 sequences in other genomes at the five-orders criterion. Upon reciprocal BLAST, however, the ArfGAPC2 sequences from other taxa retrieved a cluster including AtAGC2_A and four sequences designated as SMAPs, each of which contain the same domain organization, the ArfGAP domain with a C2 domain C-terminal to it. When taken as a group, these meet not only the five-orders criteria, but are in fact at least 23 orders of magnitude better than sequences of any other subfamily. To assess whether other ArfGAPC2 orthologs had been mis-classified, we re-examined our set of classified sequences looking for ArfGAPs with this distinctive architecture. In addition to the four above A. thaliana sequences, a single putative SMAP sequence from Physcomitrella patens, two putative ACAP sequences from Emiliania huxleyi and an additional orphan sequence from A. thaliana were found to contain the C-terminal C2 domain. Upon BLAST analysis, these did retrieve ArfGAPC2 sequences at the five-orders criterion. In order to further classify these sequences, we performed phylogenetic analysis on all SMAP, ACAP and putative ArfGAPC2 sequences from the taxa in question (Figure S2). While the phylogeny was largely unresolved, the plant ArfGAPC2 sequences did cluster together with moderate support (0.80/61/50; Figure S2), as did the three E. huxleyi sequences (0.96/66/59). This suggests that these additional sequences are the result of expansions of the ArfGAPC2 complement in these genomes.
Thus, we propose that we have identified a novel Arf GAP subfamily present in diverse eukaryotes but lost in animals and fungi (Figure 1). All proposed orthologs meet the five-orders criterion and share a common domain organization and show homology across their entire length. Based upon the presence of ArfGAPC2 sequences in archaeplastids, apusomonads, heterokonts and haptophytes, we predict it to have been present in the LECA, making it the sixth subfamily likely present in LECA. Not only do we find members in diverse taxa, but we have observed expansions of the family in plants and in E. huxleyi.
Domain conservation
There is growing and compelling evidence (21, inter alia) that other domains can influence both Arf GAP activity and effector functions. This was an important reason for us to explore the domain organization of the Arf GAP subfamilies. In order to assess evolution of domains within the Arf GAP subfamilies, we established a diagnostic domain structure for each of the ten human subfamilies, based on the sequences classified by the five-orders criterion. After bioinformatic domain identification of each validated sequence (Table S1), the domain profiles for the Arf GAP subfamilies emerged (Figure 3, 4). In stark contrast to the complex array of diagnostic domains in each of the human Arf GAPs (Figure 4), the only domains consistently conserved across eukaryotes are: ArfGAP, pleckstrin homology (PH), and BAR domains (solid colour in Figure 4), plus the C2 domain discussed above. This suggests that these domains became associated with the ArfGAP domain, in their respective proteins, prior to the LECA (Figure 2). While this finding in no way undermines the utility of the naming conventions for the subfamilies, which is based upon domain organization in the mammalian Arf GAPs, it highlights the information that may be gleaned from examining protein families across a broad evolutionary context. Furthermore, by assessing the domain architecture of the Arf GAPs at various evolutionary nodes, we were able to reconstruct the stepwise acquisition of domain complexity (Figure 2).
Figure 4.
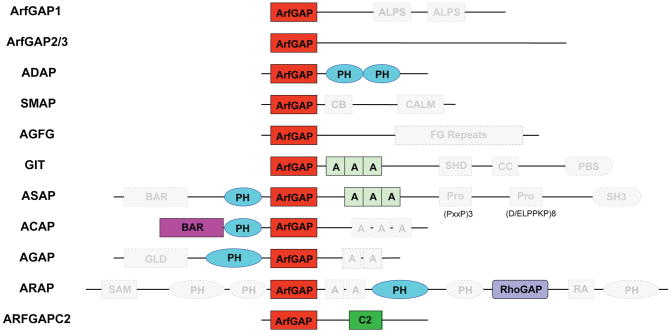
Conservation of the Arf GAP subfamilies and their domains. Conserved domains of each Arf GAP subfamily as defined by this study are shown in color; those identified in humans (as defined by Kahn et al. (2008)), but not conserved are shown in grey and bounded by a dashed outline. Very few domains are conserved across eukaryotes. ArfGAP = ArfGAP domain; ALPS = ArfGAP1 Lipid Packing Sensor; C2 = Calcium Dependent Membrane Targetting; CB = Clathrin-Box; CALM = CALM binding domain; SHD = Spa-homology domain; CC = Coiled-coil; PBS = Paxillin Binding Site; BAR = Bin/Amphiphysin/Rvs; PH = Pleckstrin Homology; Pro = Proline rich regions (motifs and number of repeats illustrated yellow domain); SH3 = Src homology-3 domain; GLD = GTPase-like domain; SAM = Sterile alpha motif; RhoGAP = RhoGAP domain; RA = Ras-association. Modified from Kahn et al. (37).
Phylogenetic analysis of individual subfamilies reveal similar evolutionary patterns between Arf GAPs and Arfs in vertebrates
In a number of instances studies of paralogs within a subfamily in human cells have been shown to bind to distinct protein partners (37). This increasing diversity raises questions of substrate specificity and of when these gene duplications occurred. In order to address the latter issue, phylogenetic analyses of the ten Arf GAP subfamilies found in humans were undertaken.
Our analyses (Figures S3A-C) revealed some expansion in non-opisthokont lineages, notably expansion of ArfGAP2/3, ACAP and AGFG in the archaeplastids. However, the most striking result was that all Arf GAP subfamilies, with the exception of ArfGAP1, have undergone at least a single duplication at the base of vertebrates, giving rise to two or more paralogs. With additional Danio rerio paralogs (ACAP_C and ARAP_C) grouping basal to the three clades containing the human sequences, the phylogenies of ACAP (Figure S3D) and ARAP (Figure 5A) are not straightforward to interpret. The simplest explanation for these phylogenetic patterns would either disregard the additional D. rerio paralog as a highly divergent sequence artifactually excluded from the three vertebrate clades or else requires invoking three duplication events prior to the divergence of the taxa sampled and subsequent loss of the basal clade paralogs from all sampled taxa other than D. rerio. The ADAP phylogeny (Figure S3E) also requires a somewhat involved ‘most-parsimonious’ explanation, with at least two duplications needing to be invoked and two major losses, D. rerio losing ADAP1 and the entire mammalian line losing the clade landmarked by D. rerio ADAP_A. The final phylogeny requiring further explanation is that of ASAP (Figure S3F), which at its most basic requires two duplications followed by loss of the clade landmarked by D. rerio ASAP_B in mammals and independent losses of ASAP3 in D. rerio, and G. gallus, at least. As there are several additional examples where individual organisms lack specific paralogs (Figure S3), there may have been further lineage-specific losses and/or inaccuracies in genome databases.
Figure 5.
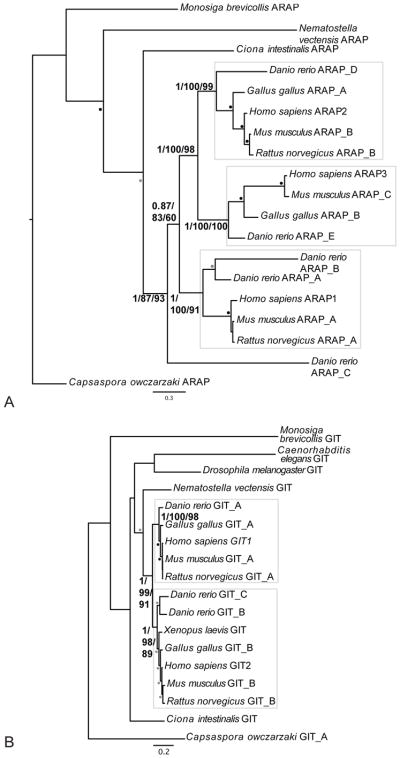
Phylogenetic reconstruction of GIT and ARAP summarizing the two patterns of gene duplication observed in vertebrates. A) ARAP has undergone at least two duplications near the base of vertebrates producing at least three paralogs, while B) GIT has undergone a single duplication near the base of vertebrates producing two. For both panels the best Bayesian topology is shown. Numerical values represent Bayesian posterior probabilities/Maximum-Likelihood bootstrap values (PhyML)/Maximum-Likelihood bootstrap values (RAxML). Nodes of interest are in bold. Values for other supported nodes have been replaced by symbols: closed, dark circles = 1.00/95/95; closed, light circles = 0.95/75/75; open circles = 0.8/50/50.
However, despite this complexity, a clear pattern does exist whereby four of the subfamilies (SMAP (Figure S3G), ArfGAP2/3 (Figure S3H), AGFG (Figure S3I) and GIT (Figure 5B)) have each undergone a single duplication resulting in two paralogs, whereas the additional five subfamilies (i.e. ADAP (Figure S3E), ACAP (Figure S3D), ASAP (Figure S3F), AGAP (Figure S3J), and ARAP (Figure 5A)) have each undergone more than one duplication, resulting in (at least) three paralogs.
Identification and comparisons of highly conserved residues in Arf GAP subfamilies
The only biochemical activity currently ascribed to the Arf GAP domain is the ability to increase the rate of GTP hydrolysis by bound GTPases. This is a consequence of the optimal positioning of the catalytic arginine from the ArfGAP into the catalytic site in the Arf GTPase. In the ASAP3-Arf6 complex this was found to help neutralize negative charges and stabilize the transition state (28). We sought to identify residues from our taxonomically diverse set of Arf GAPs that were critical to function, as seen by their conservation across deep evolutionary time. We anticipated these conserved residues to lie within the structural core of the ArfGAP domain and at the substrate/Arf interaction interface (40, 41) and hoped that they may provide some insights into residues critical to the binding specificity to Arf family GTPases or potentially even within the Arf family. In order to identify the conserved functionally critical residues, we examined only the ten Arf GAP subfamilies for which functional information is available, leaving aside the above newly described ArfGAPC2.
We found essentially identical results from analysis of the ancient five plus ADAP and AGAP, the later three, or all ten subfamilies combined. Consequently, all ten are used and a single consensus of the ten consensuses is shown in Figure S4. Conserved residues within each subfamily and between subfamilies were assessed for their conservation. We chose the 80% cutoff for stringency, admittedly arbitrarily, in efforts to focus on and highlight those most likely to be important for conserved functions. Only the four cysteine residues were found to be absolutely conserved within all Arf GAP subfamily consensus sequences. This was expected, as this zinc finger motif is known to be critical to stabilization of protein folds, particularly when present in small protein domains such as ArfGAP. The catalytic arginine, which together with the cysteines originally defined the ArfGAP motif, was also highly conserved but fell just short of our cut-off in AGFG. Overall, at the 80% level we found 18 residues that emerged as very highly conserved within and between the Arf GAP subfamilies (Figure S4).
These most conserved residues were then analyzed for function by examination of their role in the structure of an Arf·GTP-Arf GAP complex, as recently reported for the ArfGAP domain of human ASAP3 with Arf6·GDP·AlF (28). When the 18 most conserved residues were mapped onto the structure (Figure 6), we found an overwhelming majority of them to be involved in stabilization of the zinc-binding cysteine motif and overall ArfGAP domain fold. A description of our interpretations of functional roles for each of the conserved residues is presented in Figure S4. Note that in Ismail et. al (2010) they identify ten residues in the ArfGAP domain of ASAP3 that are directly involved in binding to Arf6. Seven of those residues (identified in their Fig. 1D) are not conserved in Arf GAP evolution. The three that are found in our analyses (W451, R469, and D484 in ASAP3 correspond to W14, R32, and D47 in Figure S4) are each closely involved in catalysis. R469/R32 is the arginine finger. D484/D47 contacts the main chain of Arf6-Q67 plus D68 and the side chain of Q67, stabilizing switch 2 and catalytic glutamine in Arf6. W451/W14 is centrally located in the binding interface between the Arf and Arf GAP. Mutation of any one of these three residues leads to severe loss in Arf GAP activity (28). All of the other highly conserved residues emerging from our analyses (Figure S4) are predicted to be involved in stabilization of the ArfGAP fold as a result of either direct chelation of one tightly bound zinc atom (four cysteines) or side chain interactions that are seen in the ASAP3-Arf6 crystal structure to stabilize the core of the domain. In doing so, several of them also contribute to stabilization and orientation of the catalytic arginine. The high degree of conservation of residues involved in catalysis or stabilization of the fold of the domain were expected and reassuring to emerge so clearly from our analyses. However, we were surprised at the absence of conserved residues involved directly in the binding to substrate, Arf·GTP (28, 40, 41).
Figure 6.

The previously determined ([28]); PDB 3LVQ) structure of the ArfGAP domain of human ASAP3 (cyan), complexed with Arf6 (purple) bound to GDP-AlF3 (yellow) was used to map the most highly conserved residues from the Arf GAP subfamilies. The zinc atom bound in the ArfGAP domain is shown as a grey sphere. The 15 most conserved residues implicated in stabilization of the ArfGAP domain fold are colored red with side chains visible and the three conserved residues involved in GTP hydrolysis, either directly or indirectly, are colored green. Conserved residues are labeled using the numbering in the aligned sequences shown in Fig. S4. Conserved glycines and serines are not labeled due to limitations on space. Note that the conserved (red) residues are overwhelmingly buried in the core of the folded domain and the paucity of conserved residues at the protein-protein interface. This figure was generated using The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC.
Examination of the consensus sequences also revealed some subfamily specific differences that are predicted to result in differences in function, including the complete lack of Arf GAP activity. For example, nine of the 40 AGFG sequences lack the catalytic arginine and would be expected to be incapable of supporting robust GTP hydrolysis, even if binding to Arf is conserved. Only two of the 40 AGFG sequences contain an aspartate at the position homologous to D47 in the other subfamilies (D484 in ASAP3 structure) and that Ismail, et. al (2010) show contacting the glutamine in the Arf (Arf6-Q67) that is essential to hydrolysis. The AGFG consensus also uniquely lacks W14, which we predict to play a role in hydrophobic interactions with Arfs. The GIT consensus sequence is also missing a conserved aspartate homologous to D47. In fact, it is absent from all 18 GIT sequences used in our analyses. GITs also lack N1 in all but two sequences, though its predicted role in stabilization of the zinc finger might be different as a result of the location of the Arf GAP domain in GITs being at the very N-terminus of each protein. Having the set of conserved functional residues allowed us to re-examine the putative ArfGAPC2 homologs. All clearly identified ArfGAPC2 homologs share the double CXXC motifs, the catalytic R32 residue, as well as the residues homologous to W14 and D47 involved in Arf binding. Indeed of the 18 highly conserved residues, the ArfGAPC2 consensus retains 17 of them.
Thus, the ArfGAP is a very highly conserved structural domain that is predicted to have lost substantial levels of GAP activity in at least one subfamily (AGFG) and likely alternate types of regulation in at least one other (GIT). These predicted changes (including complete loss, potentially) in GAP activity or its regulation should not be confused with consequent changes in the ability to bind Arf family GTPases. Rather, we speculate that lower GAP activity with retention of binding to activated GTPases is suggestive of effector functions. The lack of highly conserved residues in the substrate binding site and other predictions are described further in the Discussion.
DISCUSSION
We have analyzed the evolutionary patterns of the ten previously identified subfamilies of proteins that contain the ArfGAP domain. This allowed us to generate novel hypotheses surrounding the origin dates for four of the ten subfamilies and update those for another five (37). We also identified a novel subfamily of Arf GAPs, dubbed ArfGAPC2, that we describe phylogenetically. We conclude that at least six subfamilies were present in the LECA and even the three more recently emerging subfamilies are older than previously predicted (37). We further examined paralog expansion, domain acquisition, and conservation of key residues, allowing us to glean both evolutionary and functional insight into Arf GAPs in ancient eukaryotes and in eukaryotes today.
With the discovery of the ArfGAPC2 proteins, we raise the total number of described subfamilies to 11. Members of the ArfGAPC2 subfamily fulfill the five-orders reciprocal best hit criterion, and share a common domain architecture: an ArfGAP domain followed by a C2 domain (Figure 3). We are confident not only that these represent a new subfamily of Arf GAPs, but that they are likely also functional, as they retain the conserved residues identified as critical for ArfGAP activity. Comparative genomics has revealed that this is an ancient, but patchy, subfamily that has been lost from opisthokonts. ArfGAPC2 is only the most recent protein subfamily found with such a distribution: for example, the recently described Adaptin 5 complex has been lost multiple times in the eukaryotic tree (11); and RabTitan, a novel Rab GTPase, displays a patchy distribution and is absent from mammals (12). This type of distribution suggests membrane-trafficking biology present in diverse eukaryotes conserved from the LECA but lost independently on more than one occasion. For those proteins not found in opisthokonts, it may be a reflection of important differences in how membrane trafficking occurs in these organisms, compared to the picture painted from studies in model organisms. Such differences might be ideally targeted for treatment against parasitic or pestilent organisms.
The three (relatively) recently arising subfamilies (ARAP, ASAP, and GIT) are involved in cell-cell communication or cell adhesion. GIT affects cell migration and focal adhesion dynamics, through interactions with PIX and paxillin, respectively (42), while ARAP is a regulator of focal adhesion dynamics and lamellipodia formation (20). It is thus intriguing that, while these subfamilies are not found broadly, they are present in the unicellular ancestors of metazoans. The choanoflagellate M. brevicollis displays the ability to attach to substrate via extracellular matrix proteins homologous to those found in humans (i.e. laminin, reeler, and ependymin domains) (43). ASAP is a regulator of actin remodeling and invadopodia formation (36). Its distribution is even more intriguing, being found in Holozoa and the apusomonad Thecamonas trahens and may be counted as an additional synapomorphy to the emerging grouping of opisthokonts and apusomonads (44, 45). Various additional proteins involved in cell communication and adhesion, once thought to be specific to metazoans, have recently been found in unicellular ancestors of animals (46, 47). Together, these data suggest that the ancestral role(s) of proteins in these three more recently arising subfamilies may be central to cell adhesion, which was potentially pre-adaptive for an eventual role in multicellularity. Based on the shared presence of triple Ank repeats, C-terminal to the ArfGAP domain, we speculate that GIT was derived from a gene duplication of either ASAP or ACAP. Similarly, it is possible that ARAP is derived from AGAP. However, these are highly speculative hypotheses to be tested when phylogenetic resolution between the subfamilies becomes feasible.
We note that the patterns of Arf GAP duplication observed in vertebrates are similar to the observed pattern of evolution for the proteins they regulate: the Arfs. Manolea et al. (48) demonstrated that the ancestral opisthokont possessed two Arf proteins, progenitor of the Class I/II Arf and a Class III Arf. The Class I/II progenitor duplicated prior to the divergence of choanoflagellates to produce a single Class I and a single Class II Arf present in all invertebrate organisms. At some point near the base of vertebrates, however, these two classes of proteins expanded; the Class I Arf underwent two duplications to produce Arf1, Arf2, and Arf3, while the Class II Arf duplicated once to produce Arf4 and Arf5. Although the pattern for the Arf GAP subfamilies in vertebrates is more complex, overall the similarity in the patterns suggests some degree of correlation and co-evolution of the Arfs and the Arf GAP subfamilies. We do not wish to imply that we have identified functional relationships. Rather, we hypothesize that the correlated duplications might be indicative of functional relationships between the sets of proteins and should serve as jumping-off points for experimental validation of Arf GAP-substrate relationships. As there is a great deal of uncertainty regarding which Arf GAPs regulate which Arf proteins in animals, the correlation in gene duplications between Arfs and Arf GAPs may prove to be important in the dissection of cellular functions for several reasons, including the incompleteness of our data on in vitro substrate specificities. The Arf GAPs may act on proteins other than Arf; prime candidates would be the Arf-like (Arl) proteins. Biochemical tests of GAP activity by several of the Arf GAPs against a few Arl proteins have yielded almost universally negative results and have led investigators to conclude there is strict specificity of Arf GAPs for the Arf proteins and not the Arls, despite their conservation of as much as >50% identity in primary sequences. A widespread problem in biochemical assays of both Arf GAP and Arf GEF activities is the dependency on the use of recombinant, isolated domains, made necessary by the size and insolubility of the full-length proteins. Differences among the Arf GAPs result in differential localization within cells, allowing for spatial regulation of Arf signaling. The presence of domains outside of the ArfGAP domain may regulate the GAP activity and provide temporal regulation to Arf signaling. These possibilities are not mutually exclusive and will be exciting to pursue experimentally in the near future. It is likely that other domains or regions outside the ArfGAP domain are capable of regulating the activities and likely specificities of Arf GAPs.
Human Arf GAPs are characterized by the possession of a broad array of domains, in addition to the ArfGAP domain. These additional domains are predicted to regulate their cellular functions as a result of modulation in the proteins’ catalytic activity, cellular location, and protein interactions (49). They also proved to be useful criteria for the definition of the ten human Arf GAP subfamilies. Our comparative analysis and re-definition of the domain structure of the Arf GAPs for the broadest taxonomic span of each subfamily revealed a clear path of domain evolution from the presumably simpler Arf GAP toolkit to the complex set of human Arf GAPs, as well as revealing gain and loss events throughout eukaryotes (Figure 2). Increasing the number of domains would increase the potential for Arf GAPs to receive a signal and act as effectors of a parallel biological pathway, up or down regulating it accordingly. Thus, these important regulators of membrane traffic could better aid in the integration of cellular systems as the descendants of LECA moved into diverse and dynamic ecological niches. This simpler complement of accessory domains in the primordial Arf GAPs is consistent with comparative analyses of other GAP families. The recently described ELMOD family (50) contains three paralogs in mammals, each only possessing the ELMOD domain, which endows the GAP activity. Similarly, analysis of the RasGAP family showed not only unexpected complexity in the LECA but a restricted complement of accessory domains restricted to the RasGAP_C, CH and C2 domains (51).
While the domain complement present in the ancient Arf GAPs was smaller than seen in the modern human array, it nonetheless consisted of several functionally distinctive modules including the ArfGAP domain, ANK repeats, and the lipid association domains BAR, PH and C2. These may provide some indication of the roles that Arf GAPs played in ancient eukaryotes and shed some light on the ancient complexity of the cell. ANK repeats function in protein-protein interaction (52). Thus, acquisition of ANK repeats might be expected to increase the scaffolding potential or number of binding partners for any protein, offering novel potential for regulation or localization. In contrast, for most PH domains associated with Arf interacting proteins, including Arf GAPs, it is more commonly involved in direct binding to phosphatidylinositides, and the binding of the PH domain to specific lipids has been proposed to function synergistically with Arf binding to recruit the protein to a specific membrane (53). It has also been suggested that removal of the PH domain can abolish Arf GAP function, even when the Arf GAP domain is present (54). As BAR domains have been shown to play a role in both the sensing and production of membrane curvature (55, 56), their presence in certain Arf GAPs suggests a role in membrane deformation analogous to that proposed for the ALPS domain in ArfGAP1 (33, 34). BAR domains also play more direct roles in modulating Arf GAP enzymatic activity through domain-domain interactions (48). C2 domains are also lipid-binding domains (57). Together the presence of these domains in the ancestral repertoire suggests that the ancestral action of Arf GAPs was likely focused on aspects of membrane biology.
In our investigations reconstructing the Arf GAP complement in the LECA, the only subfamilies for which we could not strongly infer a time point of origin based on current evidence, were AGAP and ADAP. Nonetheless, with at least six Arf GAP subfamilies inferred as present in the LECA, when current evidence suggests there was a single Arf (18), we speculate that the earliest Arf GAPs diversified and acquired new domains to facilitate their localization to distinct compartments and in so doing provided a novel source of specificity that was not yet available from the single Arf. In contrast to other Ras superfamily GTPases, which have low but readily assayable intrinsic GTPase activities, Arfs have to date been found to lack detectible intrinsic GTPase activity (58) and thus are completely dependent upon Arf GAPs to hydrolyze bound GTP and terminate signaling. Thus, this diversity of Arf GAPs may have provided the temporal resolution required for Arf signaling at distinct organelles and likely with distinct binding or accessory proteins. Later as roles were required for Arf signaling, we further speculate that new Arf genes/proteins (class I-III) were acquired and were matched by parallel diversification in the Arf GAPs. From our analyses of conserved residues within and among the Arf GAP subfamilies and the considerations above it is possible that only limited substrate specificity will be found for naked Arf GAPs. Rather, the spatial and temporal specificity in Arf signaling may lie in stable or quasi-stable complexes that are likely to include an Arf GEF, an Arf, an Arf GAP and perhaps other effectors.
We also investigated the conservation of specific residues within each subfamily as well as among the different subfamilies. Because the ArfGAP domain itself has been highly conserved throughout eukaryotic evolution, and in the majority of organisms in our analyses there was not multiple, but only a single Arf protein, we expected to find highly conserved residues critical to (i) stabilization of the ArfGAP domain fold, (ii) orientation of the catalytic arginine, and (iii) binding to the Arf GTPase substrate(s). From comparisons of conserved residues between subfamilies, we found what we predict will be functionally important differences between Arf GAP subfamilies; however, this requires validation that can only come from solving structures of additional ArfGAP domain - Arf complexes. By aligning and comparing the conserved residues in each subfamily and identifying those conserved across subfamilies we found 18 residues that were conserved in at least 80% of the subfamilies and within each subfamily. These 18 residues were identified as playing critical roles to the fold and stabilization of the ArfGAP domain and to orientation of the catalytic arginine but none were among the residues described in Ismail, et. al (28) as being in the Arf GAP-Arf binding interface, save those involved in catalysis (W14, R32, and D47). We argue that the 18 most highly conserved residues in eukaryotic Arf GAPs act in the formation and stabilization of a structure, the ArfGAP domain, required for optimal presentation of the “arginine finger” and its insertion near the gamma phosphate on the bound Arf protein. The presence of two glycines (G29 and G35), flanking this key arginine (R32), hydrophobic tryptophan (W14), and “catalytic aspartate” (D47) are each very highly conserved residues that contribute to a specific and intimate interface with the Arf and thus changes in any of these are likely to result in changes in catalytic power, substrate specificity, ability to bind Arfs, or some combination of these. This type of analysis is clearly limited by the single currently available structure for Arf·GTP-Arf GAP complex and will be even more powerful as more structural information becomes available. For example, the structure used lacked the functionally important N-terminal helix of Arf6, whose absence has been shown to have differential effects on different Arf GAP subfamilies (59).
Both comparative genomic and phylogenetic analyses of membrane traffic machinery (9, 10, 12, 13, 60, 61, 62) have revealed protein machinery consistent with an ancestor possessing multiple routes of endocytosis and exocytosis and machinery involved in dynamic interplay between the various endomembrane organelles. From the phylogenetic patterns of SNAREs, Adaptins and Rab GTPases, the Organelle Paralogy Hypothesis was derived to provide a mechanism for autogenously evolving organellar complexity (9, 10). From our results, the Arf GAPs appear to have expanded via this mechanism at the genesis stage of membrane traffic, with the Arf GTPases per se only expanding at a much later stage. Nonetheless, two very important players in membrane traffic can now be better integrated into an overall framework for autogenous organelle evolution.
MATERIALS AND METHODS
Homology Searching
Candidate Arf GAP sequences were identified in representative lineages of each of the major eukaryotic supergroups, using a combination of BLAST (63) and HMMer v2.3.2 (http://hmmer.janelia.org/) algorithms. Genome sequence was obtained via databases at the National Center for Biotechnology Information (NCBI), the Joint Genomes Institute (JGI), and individual genome projects, with specific information regarding taxon inclusion found in Table S1. Protein sequences for Acanthamoeba castellanii were translated manually, removing introns as necessary using Sequencher v4.9, such that the largest possible contiguous sequence with homology to a human Arf GAP (as assessed by BLAST) was retained.
BLASTp searches were conducted against the non-redundant (nr) database at GenBank in order to strengthen statements regarding putative absence of particular subfamilies in specific taxonomic groups. In each case, the NCBI nr-database was restricted to the broadest taxonomic grouping for the organism in question, without overlapping with that of another organism whose genome was included in this study. The functionally validated Arf GAP protein sequences from Homo sapiens and Saccharomyces cerevisiae were used as the initial queries. In order to identify any additional sequences missed by the initial BLAST searches and to avoid bias created using opisthokont queries, a Hidden Markov Model, based on the ArfGAP domain only, was built using all identified Arf GAP sequences and used to search the genomes for additional candidates.
To validate the homology assignments, all candidate Arf GAP sequences from each individual genome, identified with an E-value of <0.05, were classified using the Reciprocal Best Hit (RBH) method (38, 39). This method assigns homology based on the reciprocal retrieval of a candidate homolog and a query sequence as each other’s best scoring hit in BLAST searches. In order to increase the stringency and transparency of our analyses, we instituted additional criteria and report the limits we place on ‘best hit’ values used. For assignment of orthology to a specific subfamily, candidates had to retrieve the initial query with E-values five-orders of magnitude better (smaller) than those of the next best-scoring Arf GAP subfamily representative (hereafter referred to as the “five-orders criterion”). This provided us with a set of orthology assignments in which we have confidence and upon which we base our evolutionary conclusions. In cases that did not meet this criterion, we also assessed the relationship at a less stringent level of two orders of magnitude better than the next best subfamily representative (i.e. the “two-orders criterion”). This provides us with a set of more weakly supported hypotheses that we report, but consider clearly more speculative.
While the RBH method is widespread and standard in the field, there is inconsistency in the criteria used to define a match and because the criteria of two- and five-orders of magnitude are admittedly arbitrary, we wished to assess their accuracy, assuming that consistency of assignment corresponds to successful assignment. A series of control BLAST experiments to assess homology between Arf GAP proteins in primarily non-model organisms were carried out. BLAST searches performed were: the Naegleria gruberi Arf GAPs into the Dictyostelium discoideum genome, the Thalassiosira pseudonana Arf GAPs into the Arabidopsis thaliana genome, the Phytophthora sojae Arf GAPs into the Trichomonas vaginalis genome, and the Chlamydomonas reinhardtii Arf GAPs into the A. thaliana genome, in addition to the reciprocal of each of these experiments. In each case, both the sequences that met the five-orders criterion or the two-orders criterion were used. For each set of BLAST experiments, the positive and negative results were tallied. A result was considered positive if the query retrieved the correspondingly assigned ortholog in the target genome by the relevant criterion, e.g., the N. gruberi homolog of the “ADAP” subfamily retrieved the equivalently annotated D. discoideum sequence. For sequences identified using the five-orders criterion, 42 of 45 sequences tested (93%) returned a sequence named as being of the same subfamily at the five-orders criterion. The remaining three sequences assessed did return the appropriate ortholog, but at the two-orders criterion. For the additional 13 sequences that had been classified using the two-orders criterion, all 13 yielded positive hits at that two-orders level.
Subfamilies are deduced as present in the LECA based on their presence in at least 3 supergroups, spanning the diversity of the resolved tree of eukaryotes (6–8). Deduced losses are based on failure to identify an ortholog or a domain in two genomes of the relevant lineage.
Identification of ArfGAPC2 homologs
To search for the presence of a novel Arf GAP subfamily, within the sequences that failed to be assigned to one of the ten previously identified subfamilies (i.e. the orphan sequences), BLASTp searches using each orphan sequence were performed against the genomes encoding at least one orphan sequence. Sequences subsequently identified as having the ArfGAPC2 architecture, were similarly assessed. RBH assessment of orthology was assessed at the five-orders criterion for the reciprocal retrieval of orphans rather than Arf GAPs assigned to one of the ten previously described subfamilies. Additional phylogenetic analysis to verify the classification of ArfGAPC2 homologs versus other Arf GAP proteins sharing the same architecture was also performed, as described below.
Domain Identification
Assessment of domains present in all sequences was carried out using InterProScan at the European Molecular Biology Laboratory (EMBL) website (http://www.ebi.ac.uk/Tools/pfa/iprscan/) (64), with all 14 algorithms available for domain recognition selected. A criterion of domain presence in a minimum of 85% of the sequences of a given subfamily was set in order for a domain to be deemed conserved for that particular subfamily. This was found to be the most stringent criterion that still retained the Arf GAP domain that defines the protein family.
Phylogenetic Analysis
Tree building was restricted to only those sequences retrieved from searches of individual genomes and fulfilling the five-orders criterion described above. Analyses were performed on a dataset including all such sequences, as well as datasets for each individual subfamily and taxonomic subsets thereof, described in the results below. Table S2 provides a key corresponding datasets to figures as well as dataset size and model of sequence evolution used for analysis. All sub-datasets are available upon request and the entire dataset can be downloaded from http://www.biochem.emory.edu/labs/rkahn/arflinks.html
Sequences were aligned using MUSCLE v3.6 (65), with manual adjustment as required using MacClade v4.08. Only the ArfGAP domain could be aligned between the various proteins, and furthermore only regions of unambiguous homology were retained for analysis. Initial phylogenetic analysis was carried-out to identify long-branch sequences likely to contribute artifacts to subsequent analysis (data not shown). For each further round of analysis, datasets were re-aligned and new masks were generated, permitting greater regions of homology to be included. Model testing was carried-out using ProtTest 1.3 (66) using a Gamma rate distribution and accounting for invariant sites when necessary. Trees were built using MrBayes v3.1.2 (67) for Bayesian analysis with 1,000,000 Markov Chain Monte Carlo generations. The burn-in value was determined graphically, and all trees preceding the plateau were removed. Maximum-Likelihood bootstrap values were obtained using PhyML v2.4.4 (68) and RAxML v2.2.3 (69) using 100 pseudoreplicates.
Identification of conserved residues
For this analysis a subset of sequences, still spanning the taxonomic diversity of eukaryotes, was used. Subfamily assignments were based on the two-order criterion. The protein sequences within each subfamily were aligned using MUSCLE v3.6 and were then used to identify highly conserved residues in each subfamily. These were consistently found to cluster around the previously identified ArfGAP motif that includes a specific spacing of cysteines and the “arginine finger” (CX2CX16CX2CX4R) (24, 27). The aligned sequences were then examined to identify the most highly conserved residues within each subfamily and across them all. We empirically determined that an 80% identity cutoff was optimal as higher stringencies resulted in exclusion of so many residues as to be uninformative. We recognize the arbitrary nature of the criteria, but are confident that patterns have emerged that will be useful for investigating Arf GAP sequences that new taxon sampling will provide. This allowed the identification of specific loss of key residues in a few subfamilies as well highlighting those residues that are essentially invariant. The conserved residues were then lined up between the ten subfamilies and those residues conserved in 80% of the subfamilies were used to produce the consensus sequence.
Supplementary Material
Figure S1. Phylogenetic reconstruction of ACAP, ASAP, and AGAP with the putative T. vaginalis ASAP sequences. Noting that, as with other phylogenetic analyses, sequences that meet the five-orders criterion were used, preliminary phylogenetic analyses (data not shown) suggest that the T. vaginalis sequences met the two-orders criterion as ASAPs are also ACAPs. The T. vaginalis ASAP sequences form a moderately supported clade with T. vaginalis ACAP, suggesting that they are ACAPs, not ASAPs. For this, and all subsequent trees, the best Bayesian topology is shown. Numerical values represent Bayesian posterior probabilities/Maximum-Likelihood bootstrap values (PhyML)/Maximum-Likelihood bootstrap values (RAxML). Nodes of interest are in bold. Values for other supported nodes have been replaced by symbols: closed, dark circles = 1.00/95/95; closed, light circles = 0.95/75/75; open circles = 0.8/50/50.
Figure S2. ArfGAPC2 RBH analysis and phylogeny A) Diagram representing the reciprocal retrieval of putative ArfGAPC2 orthologs by BLASTp. Delta E-value symbols indicate the difference in orders of magnitude between the top hit (the sequence pointed to by the arrow) and the first non-orphan sequence. Thick lines indicate a difference of five orders of magnitude or greater; the dashed line indicate a difference of less than five orders of magnitude, but that retrieved another Arf GAP protein. B) Phylogenetic analysis of SMAP, ACAP, and all C2 domain-containing proteins from organisms possessing a C2 domain-containing Arf GAP. Shows C2 domain-containing sequences from Archaeplastida (A. thaliana and P. patens) and E. huxleyi sequences originally identified as SMAPs or ACAPs cluster with other ArfGAPC2 subfamily members.
Figure S3A. Phylogenetic reconstruction of ArfGAP2/3 including sequences from all supergroups with a well-supported vertebrate clade encompassing ArfGAP2 and ArfGAP3, as moderately supported independent expansions in P. patens and A. thaliana, defined by the grey boxes.
Figure S3B. Phylogenetic reconstruction of ACAP sequences. Three clades form within vertebrates (lower three boxes), although with weak to moderate support. A well-supported, independent expansion has occurred in Archaeplastida (upper box). * denote T. gondii sequences that we list here but note may be database contamination due to their lack on transcriptional support and failure to be included in a contig in the EuPath database.
Figure S3C. Phylogenetic reconstruction of AGFG. A duplication event at the base of vertebrates has produced two clades of AGFG paralogs. Two independent expansions have occurred within the Archaeplastida: one in A. thaliana and one in P. patens.
Figure S3D. Phylogenetic reconstruction of ACAP in Metazoa rooted on N. vectensis. ACAP has undergone at least two duplication events resulting in at least three paralogs in vertebrates.
Figure S3E. Phylogenetic reconstruction of ADAP in Metazoa and M. brevicollis. Grey boxes illustrate the three ADAP clades resulting from a series of duplication events.
Figure S3F. Phylogenetic reconstruction of ASAP in Metazoa. ASAP has undergone at least two duplications, producing at least three paralogs specific to vertebrates; however, an independent expansion has also occurred in Nematostella vectensis.
Figure S3G. Phylogenetic reconstruction of SMAP limited to Metazoa. SMAP has undergone a single duplication near the base of vertebrates.
Figure S3H. Phylogenetic reconstruction of ArfGAP2/3 in the Filozoa. ArfGAP2/3 has undergone a duplication event at the base of vertebrates, resulting in ArfGAP2 and ArfGAP3 clades (grey boxes).
Figure S3I. Phylogenetic analysis of AGFG sequences limited to the Filozoa. This analysis shows the well supported clades of AGFG1 and 2.
Figure S3J. Phylogenetic reconstruction of AGAP in Holozoa, rooted on Sphaeroforma arctica, depicting the two duplications that occurred to produce the three paralogs in vertebrates as outlined by the grey boxes. The H. sapiens AGAPs 4–11 are the result of an expansion of AGAP1.
Figure S4. Predictions concerning the structural roles of the most conserved residues within and among the Arf GAP families. The 18 bolded residues are conserved in at least 80% of the sequences within each subfamily are shown. The “~” symbol denotes conserved spacing in the consensus but a poorly conserved residue. Also shown is the consensus of the consensuses, i.e., those conserved residues that are present in at least 8 of the ten consensuses. The ArfGAPC2 sequences were not used to generate the consensus, but are compared below to illustrate the retention of the highly conserved residues. Numbering is from N1 to the end within these alignments and do not reflect their location within the full-length proteins. A single insertion of two residues, after D47, is present in the ARAPs (not shown), compared to other subfamilies. The structure of the ASAP3 ArfGAP domain complexed with Arf6-GDP-AlF3 ((28); PDB 3LVQ) was used to generate the following observations regarding the residues conserved across subfamilies and eukaryotic evolution: N1: pairs with backbone at N62 to help stabilize cysteine cluster fold, also effectively links two helices, may be a major global structure effect as well as local organization around cysteine cluster fold. C4, C7, C24, C27: coordinate the bound Zn2+ atom. D6: interacts with H31, to stabilize cysteine cluster. H31 is in the middle (opposite side) of the helix containing the catalytic arginine (R32), thus predicted to provide linkage between Cys-fold and R32 positioning. W14: Sandwiched between the R32 helix and loop above to form intimate surface interactions with Arf. S16: Perhaps interaction with S39 to help pin these two loops in place above more flexible region with R32. G20: No ‘function’ keeping conserved, just no space in the protein core for anything bigger, repositioning would disrupt cysteine cluster fold. G29/G35: bracket R32 in the catalytic α-helix to allow very close approach of the two surfaces – giving the R access to its target. H31: along with S39, and V41 form a hydrophobic core to the scaffold that stabilizes it; also H-bonded to D6 for further stabilization of fold. R32: catalytic “arginine finger”. S39: along with H31 form a hydrophobic core to the scaffold that stabilizes it; maybe also same as S16, above. S43: H-bond to backbone N of L483 in structure, forms a tight turn around this S side chain, positions loop and D47 at the interface. D47: “catalytic D” in that it interacts with Arf6-Q67 to assist in catalysis. G61: No space for anything else; N1 side chain is right there making a backbone interaction. N62: H-bond to the backbone.
Listing of Arf GAP sequences identified through comparative genomics. Sequences are organized first into subfamilies, then alphabetically by taxon. The Accession number is the identification number of the sequence at NCBI, JGI, or the organism’s respective genome website. Protein length reflects the length of the sequence in amino acids, found through comparative genomics. The next three columns (Accession number, Arf GAP, and E-value) indicate the accession number, and subfamily assignment of the top human Arf GAP upon reciprocal BLAST along with the E-value of that experiment. Domains of the Arf GAP sequences are given in the order provided by InterproScan. Colour coding is as follows: Sequences with white background are the sequences that meet the five-orders criterion; sequences highlighted in orange are those that meet the two-orders criterion; maroon are the sequences identified through nr-BLASTs; green are sequences that could not be classified (orphans). In the section for ArfGAPC2, sequences with grey backgrounds have a one-way BLAST with five orders of magnitude in Figure 2A; sequences with a pink background are sequences originally classified as SMAPS, but were added to the ArfGAPC2 subfamily based on the tree in Figure S2. Each subfamily is separated on the table by a double dark blue row. * denote T. gondii sequences that we list here but note may be database contamination due to their lack on transcriptional support and failure to be included in a contig in the EuPath database.
Table correlating figure with the dataset used in phylogenetic analyses, along with length of the masked alignment, and the model that best fit the dataset as determined by ProtTest v10.2 (see Materials and Methods).
Acknowledgments
FUNDING
This project was supported by an NSERC Discovery grant and an AITF New Investigator award (JBD), Czech Science Foundation grant (P305/10/0205; ME), and an NIH grant (R01-GM09158; RAK). JBD is the Canada Research Chair in Evolutionary Cell Biology.
We wish to thank P. Melançon, W. Gallin and J. Logsdon Jr., as well as members of the Dacks lab, for critical comments and discussion on the project. We also thank G. Conn for his expertise in the analysis of the Arf6-Arf GAP structure and extensive assistance in the generation of Figure 6. We would like to acknowledge the various genome projects that made their data publicly available, without which this project would not have been possible.
Abbreviations
- ANK
ankyrin repeats
- Arf
ADP-ribosylation factors
- Arl
Arf-like
- GAP
GTPase Activating Protein
- LECA
Last Eukaryotic Common Ancestor
- PH
pleckstrin homology
Footnotes
Data deposition: All alignments are available upon request.
Note: we are retaining the recommendation of the HUGO nomenclature committee’s suggestion (see Kahn, et. al 2008) and to minimize confusion, we refer to the ArfGAP domain (no space) and Arf GAP proteins (with a space).
LITERATURE CITED
- 1.Gray MW, Doolitle WF. Has the endosymbiont hypothesis been proven? Microbiological Reviews. 1982;46:1–42. doi: 10.1128/mr.46.1.1-42.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Embley TM, Martin W. Eukaryotic evolution, changes and challenges. Nature. 2006;440:623–30. doi: 10.1038/nature04546. [DOI] [PubMed] [Google Scholar]
- 3.Lane CE, Archibald JM. The eukaryotic tree of life: endosymbiosis takes its TOL. Trends in ecology & evolution. 2008;23:268–75. doi: 10.1016/j.tree.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–66. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- 5.Cai H, Reinisch K, Ferro-Novick S. Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Developmental cell. 2007;12:671–82. doi: 10.1016/j.devcel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Adl SM, Simpson AGB, Farmer MA, Andersen RA, Anderson OR, Barta JR, Bowser SS, Brugerolle G, Fensome RA, Fredericq S, James TY, Karpov S, Kugrens P, Krug J, Lane CE, Lewis LA, Lodge J, Lynn DH, Mann DG, McCourt RM, Mendoza L, Moestrup O, Mozley-Standridge SE, Nerad TA, Shearer CA, Smirnov AV, Spiegel FW, Taylor MFJR. The new higher level classification of eukaryotes with emphasis on the taxonomy of protists. The Journal of eukaryotic microbiology. 2005;52:399–451. doi: 10.1111/j.1550-7408.2005.00053.x. [DOI] [PubMed] [Google Scholar]
- 7.Walker G, Dorrell RG, Schlacht A, Dacks JB. Eukaryotic systematics: a user’s guide for cell biologists and parasitologists. Parasitology. 2011;138:1638–63. doi: 10.1017/S0031182010001708. [DOI] [PubMed] [Google Scholar]
- 8.Burki F, Okamoto N, Pombert J-F, Keeling PJ. The evolutionary history of haptophytes and cryptophytes: phylogenomic evidence for separate origins. Proceedings Biological sciences / The Royal Society. 2012;279:2246–54. doi: 10.1098/rspb.2011.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dacks JB, Field MC. Evolution of the eukaryotic membrane-trafficking system: origin, tempo and mode. Journal of cell science. 2007;120:2977–85. doi: 10.1242/jcs.013250. [DOI] [PubMed] [Google Scholar]
- 10.Dacks JB, Poon PP, Field MC. Phylogeny of endocytic components yields insight into the process of nonendosymbiotic organelle evolution. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:588–93. doi: 10.1073/pnas.0707318105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirst J, Barlow LD, Francisco GC, Sahlender DA, Seaman MNJ, Dacks JB, Robinson MS. The fifth adaptor protein complex. PLoS biology. 2011;9:e1001170. doi: 10.1371/journal.pbio.1001170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elias M, Brighouse A, Gabernet-Castello C, Field MC, Dacks JB. Sculpting the endomembrane system in deep time: high resolution phylogenetics of Rab GTPases. Journal of cell science. 2012;125:2500–8. doi: 10.1242/jcs.101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diekmann Y, Seixas E, Gouw M, Tavares-Cadete F, Seabra MC, Pereira-Leal JB. Thousands of rab GTPases for the cell biologist. PLoS computational biology. 2011;7:e1002217. doi: 10.1371/journal.pcbi.1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pereira-Leal JB, Seabra MC. Evolution of the Rab family of small GTP-binding proteins. Journal of molecular biology. 2001;313:889–901. doi: 10.1006/jmbi.2001.5072. [DOI] [PubMed] [Google Scholar]
- 15.Sanderfoot A. Increases in the number of SNARE genes parallels the rise of multicellularity among the green plants. Plant physiology. 2007;144:6–17. doi: 10.1104/pp.106.092973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dacks JB, Doolittle WF. Novel syntaxin gene sequences from Giardia, Trypanosoma and algae: implications for the ancient evolution of the eukaryotic endomembrane system. Journal of cell science. 2002;115:1635–42. doi: 10.1242/jcs.115.8.1635. [DOI] [PubMed] [Google Scholar]
- 17.Dacks JB, Doolittle WF. Molecular and phylogenetic characterization of syntaxin genes from parasitic protozoa. Molecular and Biochemical Parasitology. 2004;136:123–136. doi: 10.1016/j.molbiopara.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Kelly WG, Logsdon JM, Schurko AM, Harfe BD, Hill-Harfe KL, Kahn RA. Functional genomic analysis of the ADP-ribosylation factor family of GTPases: phylogeny among diverse eukaryotes and function in C. elegans. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2004;18:1834–50. doi: 10.1096/fj.04-2273com. [DOI] [PubMed] [Google Scholar]
- 19.D’Souza-Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nature reviews Molecular cell biology. 2006;7:347–58. doi: 10.1038/nrm1910. [DOI] [PubMed] [Google Scholar]
- 20.Inoue H, Randazzo PA. Arf GAPs and their interacting proteins. Traffic. 2007;8:1465–75. doi: 10.1111/j.1600-0854.2007.00624.x. [DOI] [PubMed] [Google Scholar]
- 21.East MP, Kahn RA. Models for the functions of Arf GAPs. Seminars in cell & developmental biology. 2011;22:3–9. doi: 10.1016/j.semcdb.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beck R, Brügger B, Wieland F. GAPs in the context of COPI: Enzymes, coat components or both? Cellular logistics. 2011;1:52–54. doi: 10.4161/cl.1.2.15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu VW. Role of ArfGAP1 in COPI vesicle biogenesis. Cellular logistics. 2011;1:55–56. doi: 10.4161/cl.1.2.15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kahn RA. GAPs: Terminator versus effector functions and the role(s) of ArfGAP1 in vesicle biogenesis. Cellular logistics. 2011;1:49–51. doi: 10.4161/cl.1.2.15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Segev N. Focusing on Arf GAPs. Cellular logistics. 2011;1:47–48. doi: 10.4161/cl.1.2.15738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kon S, Funaki T, Satake M. Putative terminator and/or effector functions of Arf GAPs in the trafficking of clathrin-coated vesicles. Cellular logistics. 2011;1:86–89. doi: 10.4161/cl.1.3.16192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cukierman E, Huber I, Rotman M, Cassel D. The ARF1 GTPase-activating protein : Zinc finger motif and golgi complex localization. Science. 1999;270:1999–2002. doi: 10.1126/science.270.5244.1999. [DOI] [PubMed] [Google Scholar]
- 28.Ismail SA, Vetter IR, Sot B, Wittinghofer A. The structure of an Arf-ArfGAP complex reveals a Ca2+ regulatory mechanism. Cell. 2010;141:812–21. doi: 10.1016/j.cell.2010.03.051. [DOI] [PubMed] [Google Scholar]
- 29.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell. 2007;130:385. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 30.Mandiyan V, Andreev J, Schlessinger J, Hubbard SR. Crystal structure of the ARF-GAP domain and ankyrin repeats of PYK2-associated protein beta. The EMBO journal. 1999;18:6890–8. doi: 10.1093/emboj/18.24.6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo R, Randazzo PA. Kinetic analysis of Arf GAP1 indicates a regulatory role for coatomer. The Journal of biological chemistry. 2008;283:21965–77. doi: 10.1074/jbc.M802268200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, Huang C, Huang K, Lee FS. Role for Gcs1p in Regulation of Arl1p at Trans-Golgi Compartments. 2005;16:4024–4033. doi: 10.1091/mbc.E05-01-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bigay J, Casella J-F, Drin G, Mesmin B, Antonny B. ArfGAP1 responds to membrane curvature through the folding of a lipid packing sensor motif. The EMBO journal. 2005;24:2244–53. doi: 10.1038/sj.emboj.7600714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mesmin B, Drin G, Levi S, Rawet M, Cassel D, Bigay J, Antonny B. Two lipid-packing sensor motifs contribute to the sensitivity of ArfGAP1 to membrane curvature. Biochemistry. 2007;46:1779–90. doi: 10.1021/bi062288w. [DOI] [PubMed] [Google Scholar]
- 35.Pryor PR, Jackson L, Gray SR, Edeling MA, Thompson A, Sanderson CM, Evans PR, Owen DJ, Luzio JP. Molecular basis for the sorting of the SNARE VAMP7 into endocytic clathrin-coated vesicles by the ArfGAP Hrb. Cell. 2008;134:817–27. doi: 10.1016/j.cell.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Randazzo PA, Inoue H, Bharti S. Arf GAPs as regulators of the actin cytoskeleton. Biology of the Cell. 2007;99:583–600. doi: 10.1042/bc20070034. [DOI] [PubMed] [Google Scholar]
- 37.Kahn RA, Bruford E, Inoue H, Logsdon JM, Nie Z, Premont RT, Randazzo PA, Satake M, Theibert AB, Zapp ML, Cassel D. Consensus nomenclature for the human ArfGAP domain-containing proteins. The Journal of cell biology. 2008;182:1039–44. doi: 10.1083/jcb.200806041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tatusov RL. A Genomic Perspective on Protein Families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 39.Bork P, Dandekar T, Diaz-Lazcoz Y, Eisenhaber F, Huynen M, Yuan Y. Predicting function: from genes to genomes and back. Journal of molecular biology. 1998;283:707–25. doi: 10.1006/jmbi.1998.2144. [DOI] [PubMed] [Google Scholar]
- 40.Choi YS, Yang J-S, Choi Y, Ryu SH, Kim S. Evolutionary conservation in multiple faces of protein interaction. Proteins. 2009;77:14–25. doi: 10.1002/prot.22410. [DOI] [PubMed] [Google Scholar]
- 41.Valencia A, Pazos F. Structural Bioinformatics. Wiley-Blackwell; 2009. Prediction of Protein-Protein Interactions from Evolutionary Information; pp. 615–632. [Google Scholar]
- 42.Hoefen RJ, Berk BC. The multifunctional GIT family of proteins. Journal of cell science. 2006;119:1469–75. doi: 10.1242/jcs.02925. [DOI] [PubMed] [Google Scholar]
- 43.King N, Westbrook MJ, Young SL, Kuo A, Abedin M, Chapman J, Fairclough S, Hellsten U, Isogai Y, Letunic I, Marr M, Pincus D, Putnam N, Rokas A, Wright KJ, Zuzow R, Dirks W, Good M, Goodstein D, Lemons D, Li W, Lyons JB, Morris A, Nichols S, Richter DJ, Salamov A, Sequencing JGI, Bork P, Lim WA, Manning G, Miller WT, McGinnis W, Shapiro H, Tjian R, Grigoriev IV, Rokhsar D. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature. 2008;451:783–8. doi: 10.1038/nature06617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim E, Simpson AGB, Graham LE. Evolutionary relationships of apusomonads inferred from taxon-rich analyses of 6 nuclear encoded genes. Molecular biology and evolution. 2006;23:2455–66. doi: 10.1093/molbev/msl120. [DOI] [PubMed] [Google Scholar]
- 45.Derelle R, Lang BF. Rooting the eukaryotic tree with mitochondrial and bacterial proteins. Molecular biology and evolution. 2012;29:1277–89. doi: 10.1093/molbev/msr295. [DOI] [PubMed] [Google Scholar]
- 46.Shalchian-Tabrizi K, Minge MA, Espelund M, Orr R, Ruden T, Jakobsen KS, Cavalier-Smith T. Multigene phylogeny of choanozoa and the origin of animals. PloS one. 2008;3:e2098. doi: 10.1371/journal.pone.0002098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sebé-pedrós A, Roger AJ, Lang FB, King N, Ruiz-trillo I. Ancient origin of the integrin-mediated adhesion and signaling machinery. Proceedings of the National Academy of Sciences. 2010;107:10142–47. doi: 10.1073/pnas.1002257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manolea F, Chun J, Chen DW, Clarke I, Summerfeldt N, Dacks JB, Melancon P. Arf3 Is Activated Uniquely at the trans-Golgi Network by Brefeldin A-inhibited Guanine Nucleotide Exchange Factors. Molecular biology of the cell. 2010;21:1836–1849. doi: 10.1091/mbc.E10-01-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jian X, Brown P, Schuck P, Gruschus JM, Balbo A, Hinshaw JE, Randazzo PA. Autoinhibition of Arf GTPase-activating protein activity by the BAR domain in ASAP1. The Journal of biological chemistry. 2009;284:1652–63. doi: 10.1074/jbc.M804218200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.East MP, Bowzard JB, Dacks J, Kahn RA. ELMO domains: evolutionary and functional characterization of a novel GTPase activating protein (GAP) domain for Arf family GTPases. The Journal of biological chemistry. 2012;287:39538–53. doi: 10.1074/jbc.M112.417477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Dam TJP, Bos JL, Snel B. Evolution of the Ras-like small GTPases and their regulators. Small GTPases. 2011;21:4–16. doi: 10.4161/sgtp.2.1.15113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cross T, Horizontally P, Bork P. Hundreds of Ankyrin-Like Repeats in Functionally Diverse Proteins : Mobile Modules o. 1993;374:363–374. doi: 10.1002/prot.340170405. [DOI] [PubMed] [Google Scholar]
- 53.Godi A, Di Campli A, Konstantakopoulos A, Di Tullio G, Alessi DR, Kular GS, Daniele T, Marra P, Lucocq JM, De Matteis MA. FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nature cell biology. 2004;6:393–404. doi: 10.1038/ncb1119. [DOI] [PubMed] [Google Scholar]
- 54.Kam JL, Miura K, Jackson TR, Gruschus J, Roller P, Stauffer S, Clark J, Aneja R, Randazzo PA. Phosphoinositide-dependent Activation of the ADP-ribosylation Factor GTPase-activating Protein ASAP1. 2000;275:9653–9663. doi: 10.1074/jbc.275.13.9653. [DOI] [PubMed] [Google Scholar]
- 55.Masuda M, Mochizuki N. Structural characteristics of BAR domain superfamily to sculpt the membrane. Seminars in cell & developmental biology. 2010;21:391–8. doi: 10.1016/j.semcdb.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 56.Field MC, Sali A, Rout MP. Evolution: On a bender--BARs, ESCRTs, COPs, and finally getting your coat. The Journal of cell biology. 2011;193:963–72. doi: 10.1083/jcb.201102042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davletov B, Sudhof T. A Single C2 Domain & from Synaptotagmin I Is Sufficient for High Affinity Ca2+/Phospholipid Binding. The Journal of biological chemistry. 1993;268:26386–26390. [PubMed] [Google Scholar]
- 58.Randazzo PA, Weiss O, Kahn RA. Preparation of recombinant ADP-ribosylation factor. Methods Enzymol. 1992;219:362–369. doi: 10.1016/0076-6879(92)19036-6. [DOI] [PubMed] [Google Scholar]
- 59.Yoon H-Y, Jacques K, Nealon B, Stauffer S, Premont RT, Randazzo PA. Differences between AGAP1, ASAP1 and Arf GAP1 in substrate recognition: interaction with the N-terminus of Arf1. Cellular signalling. 2004;16:1033–44. doi: 10.1016/j.cellsig.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 60.Pereira-Leal JB. The Ypt/Rab family and the evolution of trafficking in fungi. Traffic. 2008;9:27–38. doi: 10.1111/j.1600-0854.2007.00667.x. [DOI] [PubMed] [Google Scholar]
- 61.Koumandou VL, Dacks JB, Coulson RMR, Field MC. Control systems for membrane fusion in the ancestral eukaryote; evolution of tethering complexes and SM proteins. BMC evolutionary biology. 2007;7:29. doi: 10.1186/1471-2148-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koumandou VL, Klute MJ, Herman EK, Nunez-Miguel R, Dacks JB, Field MC. Evolutionary reconstruction of the retromer complex and its function in Trypanosoma brucei. Journal of cell science. 2011;124:1496–509. doi: 10.1242/jcs.081596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research. 1997;25:3389–402.s. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, Finn RD, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Laugraud A, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, Mistry J, Mitchell A, Mulder N, Natale D, Orengo C, Quinn AF, Selengut JD, Sigrist CJA, Thimma M, Thomas PD, Valentin F, Wilson D, Wu CH, Yeats C. InterPro: the integrative protein signature database. Nucleic acids research. 2009;37:D211–5. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–5. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 67.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 68.Guindon S, Gascuel O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Systematic Biology. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 69.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Phylogenetic reconstruction of ACAP, ASAP, and AGAP with the putative T. vaginalis ASAP sequences. Noting that, as with other phylogenetic analyses, sequences that meet the five-orders criterion were used, preliminary phylogenetic analyses (data not shown) suggest that the T. vaginalis sequences met the two-orders criterion as ASAPs are also ACAPs. The T. vaginalis ASAP sequences form a moderately supported clade with T. vaginalis ACAP, suggesting that they are ACAPs, not ASAPs. For this, and all subsequent trees, the best Bayesian topology is shown. Numerical values represent Bayesian posterior probabilities/Maximum-Likelihood bootstrap values (PhyML)/Maximum-Likelihood bootstrap values (RAxML). Nodes of interest are in bold. Values for other supported nodes have been replaced by symbols: closed, dark circles = 1.00/95/95; closed, light circles = 0.95/75/75; open circles = 0.8/50/50.
Figure S2. ArfGAPC2 RBH analysis and phylogeny A) Diagram representing the reciprocal retrieval of putative ArfGAPC2 orthologs by BLASTp. Delta E-value symbols indicate the difference in orders of magnitude between the top hit (the sequence pointed to by the arrow) and the first non-orphan sequence. Thick lines indicate a difference of five orders of magnitude or greater; the dashed line indicate a difference of less than five orders of magnitude, but that retrieved another Arf GAP protein. B) Phylogenetic analysis of SMAP, ACAP, and all C2 domain-containing proteins from organisms possessing a C2 domain-containing Arf GAP. Shows C2 domain-containing sequences from Archaeplastida (A. thaliana and P. patens) and E. huxleyi sequences originally identified as SMAPs or ACAPs cluster with other ArfGAPC2 subfamily members.
Figure S3A. Phylogenetic reconstruction of ArfGAP2/3 including sequences from all supergroups with a well-supported vertebrate clade encompassing ArfGAP2 and ArfGAP3, as moderately supported independent expansions in P. patens and A. thaliana, defined by the grey boxes.
Figure S3B. Phylogenetic reconstruction of ACAP sequences. Three clades form within vertebrates (lower three boxes), although with weak to moderate support. A well-supported, independent expansion has occurred in Archaeplastida (upper box). * denote T. gondii sequences that we list here but note may be database contamination due to their lack on transcriptional support and failure to be included in a contig in the EuPath database.
Figure S3C. Phylogenetic reconstruction of AGFG. A duplication event at the base of vertebrates has produced two clades of AGFG paralogs. Two independent expansions have occurred within the Archaeplastida: one in A. thaliana and one in P. patens.
Figure S3D. Phylogenetic reconstruction of ACAP in Metazoa rooted on N. vectensis. ACAP has undergone at least two duplication events resulting in at least three paralogs in vertebrates.
Figure S3E. Phylogenetic reconstruction of ADAP in Metazoa and M. brevicollis. Grey boxes illustrate the three ADAP clades resulting from a series of duplication events.
Figure S3F. Phylogenetic reconstruction of ASAP in Metazoa. ASAP has undergone at least two duplications, producing at least three paralogs specific to vertebrates; however, an independent expansion has also occurred in Nematostella vectensis.
Figure S3G. Phylogenetic reconstruction of SMAP limited to Metazoa. SMAP has undergone a single duplication near the base of vertebrates.
Figure S3H. Phylogenetic reconstruction of ArfGAP2/3 in the Filozoa. ArfGAP2/3 has undergone a duplication event at the base of vertebrates, resulting in ArfGAP2 and ArfGAP3 clades (grey boxes).
Figure S3I. Phylogenetic analysis of AGFG sequences limited to the Filozoa. This analysis shows the well supported clades of AGFG1 and 2.
Figure S3J. Phylogenetic reconstruction of AGAP in Holozoa, rooted on Sphaeroforma arctica, depicting the two duplications that occurred to produce the three paralogs in vertebrates as outlined by the grey boxes. The H. sapiens AGAPs 4–11 are the result of an expansion of AGAP1.
Figure S4. Predictions concerning the structural roles of the most conserved residues within and among the Arf GAP families. The 18 bolded residues are conserved in at least 80% of the sequences within each subfamily are shown. The “~” symbol denotes conserved spacing in the consensus but a poorly conserved residue. Also shown is the consensus of the consensuses, i.e., those conserved residues that are present in at least 8 of the ten consensuses. The ArfGAPC2 sequences were not used to generate the consensus, but are compared below to illustrate the retention of the highly conserved residues. Numbering is from N1 to the end within these alignments and do not reflect their location within the full-length proteins. A single insertion of two residues, after D47, is present in the ARAPs (not shown), compared to other subfamilies. The structure of the ASAP3 ArfGAP domain complexed with Arf6-GDP-AlF3 ((28); PDB 3LVQ) was used to generate the following observations regarding the residues conserved across subfamilies and eukaryotic evolution: N1: pairs with backbone at N62 to help stabilize cysteine cluster fold, also effectively links two helices, may be a major global structure effect as well as local organization around cysteine cluster fold. C4, C7, C24, C27: coordinate the bound Zn2+ atom. D6: interacts with H31, to stabilize cysteine cluster. H31 is in the middle (opposite side) of the helix containing the catalytic arginine (R32), thus predicted to provide linkage between Cys-fold and R32 positioning. W14: Sandwiched between the R32 helix and loop above to form intimate surface interactions with Arf. S16: Perhaps interaction with S39 to help pin these two loops in place above more flexible region with R32. G20: No ‘function’ keeping conserved, just no space in the protein core for anything bigger, repositioning would disrupt cysteine cluster fold. G29/G35: bracket R32 in the catalytic α-helix to allow very close approach of the two surfaces – giving the R access to its target. H31: along with S39, and V41 form a hydrophobic core to the scaffold that stabilizes it; also H-bonded to D6 for further stabilization of fold. R32: catalytic “arginine finger”. S39: along with H31 form a hydrophobic core to the scaffold that stabilizes it; maybe also same as S16, above. S43: H-bond to backbone N of L483 in structure, forms a tight turn around this S side chain, positions loop and D47 at the interface. D47: “catalytic D” in that it interacts with Arf6-Q67 to assist in catalysis. G61: No space for anything else; N1 side chain is right there making a backbone interaction. N62: H-bond to the backbone.
Listing of Arf GAP sequences identified through comparative genomics. Sequences are organized first into subfamilies, then alphabetically by taxon. The Accession number is the identification number of the sequence at NCBI, JGI, or the organism’s respective genome website. Protein length reflects the length of the sequence in amino acids, found through comparative genomics. The next three columns (Accession number, Arf GAP, and E-value) indicate the accession number, and subfamily assignment of the top human Arf GAP upon reciprocal BLAST along with the E-value of that experiment. Domains of the Arf GAP sequences are given in the order provided by InterproScan. Colour coding is as follows: Sequences with white background are the sequences that meet the five-orders criterion; sequences highlighted in orange are those that meet the two-orders criterion; maroon are the sequences identified through nr-BLASTs; green are sequences that could not be classified (orphans). In the section for ArfGAPC2, sequences with grey backgrounds have a one-way BLAST with five orders of magnitude in Figure 2A; sequences with a pink background are sequences originally classified as SMAPS, but were added to the ArfGAPC2 subfamily based on the tree in Figure S2. Each subfamily is separated on the table by a double dark blue row. * denote T. gondii sequences that we list here but note may be database contamination due to their lack on transcriptional support and failure to be included in a contig in the EuPath database.
Table correlating figure with the dataset used in phylogenetic analyses, along with length of the masked alignment, and the model that best fit the dataset as determined by ProtTest v10.2 (see Materials and Methods).