

Abstract
Tumor cell-derived molecules such as cytokines and lipid mediators play a critical role in inducing chronic inflammation in the tumor microenvironment. We found that Th17 cells were increased in the peripheral blood, spleen and tumor tissues of mammary gland tumor-bearing mice. The Th17 cell survival factor, IL-23, was also overexpressed in tumor tissues isolated from mice and human breast cancer patients. Soluble molecules secreted from breast tumor cells but not normal breast epithelial cells induced IL-23 protein secretion in DCs via induction of p19 mRNA expression. Our data further indicate that tumor-secreted PGE2 through EP2 and EP4 receptors enhanced IL-23 p19 gene transcription through binding to the cAMP-response element in the p19 promoter. Blocking PGE2 synthesis by NS398, a COX2 inhibitor, abrogated the enhancement of p19 expression both in vitro and in vivo. Furthermore, blocking PKA by H89 completely abrogated the inductive effects of tumor conditioned medium and PGE2 on p19 transcription, whereas the cAMP active analog, Forskolin mimics the PGE2 effect. Taken together, our results indicate that tumor-secreted PGE2 induces IL-23 but not IL-12 production in the tumor microenvironment leading to Th17 cell expansion. This inductive effect of PGE2 on IL-23 p19 transcription is mediated through cAMP/PKA signaling transduction pathway.
Introduction
Chronic inflammation has been associated with increased tumor incidence and linked to tumorogenesis of breast cancer as well as other cancers, probably through recruitment of leukocytes secreting a variety of cytokines, chemokines and angiogenic factors that promote tumor growth and metastasis involving a complex interaction amid tumor, stroma and inflammatory cells (1–3). Although the role for inflammatory cells including macrophages, lymphocytes and mast cells in tumor progression is well documented, the details of the cellular and molecular interplay between host and tumor progression remain to be elucidated.
Th17 cells secrete a number of cytokines including IL-17A/F, IL-6, TNF-α and IL-22 important for inflammation, and have been associated with the pathogenesis of several autoimmune diseases (4). Currently, the role of Th17 cells in tumor progression is controversial (5, 6). It has been reported that Th17 cells are increased in the tumor microenvironment during tumor progression, including breast, prostate and gastric cancers (7–9). As a signature cytokine secreted by Th17 cells, IL-17 promotes tumor growth and metastasis through angiogenic effects and induction of chronic inflammation (6). On the other hand, it has also been reported that Th17 cells and IL-17 have antitumor effects by attracting CTL and NK cell migrating into the tumor suppressing tumor growth and metastasis (5). Nevertheless, increased Th17 cells were documented in many types of solid tumors (5) but the mechanisms by which the tumor microenvironment promotes Th17 cell development in breast cancer are still largely unknown.
Initial differentiation of Th17 cells depends on IL-6 and TGF-β stimulation, whereas IL-23 is known to be essential for Th17 cell survival and expansion, and for making pathogenic Th17 cells (10–16). Therefore, overexpression of IL-23 in the tumor microenvironment could be critical for Th17 cell development during tumor progression. Indeed, it has been reported that IL-23 was overexpressed in different types of tumors, including colon, ovarian, lung and breast cancers, etc, and mice deficient in IL-23 were resistant to chemically induced papillomas (17). More recently, reports have been published demonstrating that IL-23 can promote tumor growth and metastasis through inhibition of perforin and IFN-γ production by NK cells independent of Th17 cells/IL-17 during carcinogenesis (18). In contrast to the protumor effects of IL-23, several reports have described antitumor functions of IL-23. Overexpression of IL-23 in tumor cells reduced tumor growth and metastasis through induction of CD8+ T cell responses (19, 20). It is likely that the functions of IL-23 in tumor progression are determined by the types of tumors, the immune competence of the host, and the source of IL-23 being produced. Nevertheless, IL-23 is consistently overexpressed in both human breast cancer and mouse mammary gland tumor. However, the mechanisms that trigger IL-23 overexpression in breast tumors are still largely unknown.
IL-23 is an interleukin-12 family cytokine composed of two different subunits, one shared subunit with IL-12, p40, and another unique subunit, p19 (21). IL-23 gene expression is tightly controlled at both transcriptional and post-transcriptional levels. It has been reported that NF-κB c-Rel physically binds to the p19 promoter and induces IL-23 p19 gene expression. Conversely, deletion of c-Rel abolished IL-23 p19 expression in both dendritic cells and macrophages (22, 23). In addition, Sma- and Mad-related protein (SMAD)-3, activating transcription factor (ATF)-2, and activating protein-1 (AP-1) also play important roles in control of IL-23 p19 gene expression (24). Silencing of SMAD-3 and ATF-2 expression by shRNA reduced p19 promoter activity and protein expression in RAW264.7 cells infected with Theiler’s murine encephalomyelitis virus or treated with poly (I–C) (25). MAPKs including p38, JNK and ERK have been shown to be involved in LPS-induced IL-23 p19 gene transcription (24). So far, most studies have focused on the transcriptional regulation of p19 expression and little is known about how IL-23 is post-transcriptionally regulated in spite of the fact that IL-23 p19 mRNA has a long 3′UTR containing multiple putative adenosine/uridine-rich elements (26). Our recent work showed that IFN-γ suppresses IL-23 expression by selectively targeting p19 mRNA stability through its 3′-untranslated region (3′UTR) (27), indicating that the expression of IL-23 is controlled at both transcriptional and posttranscriptional levels.
The 4T1 tumor is an excellent mouse model for breast cancer research because its tumor development is well characterized both oncologically and immunologically (28). Tumor growth and metastatic spread of 4T1 tumor closely mimics stage IV breast cancer (29). 4T1 tumors are poorly immunogenic in that immunization with irradiated 4T1 cells provides only slight delays in tumor growth which are not sufficient to protect the animal (30). Our previous work demonstrated that administration of IL-12 to 4T1 tumor-bearing mice resulted in a substantial reduction in tumor size and decreased spontaneous metastases to the lungs, as well as significantly prolonged survival time (31, 32). 4T1 tumor cells secrete many cytokines such as TGF-β and IL-6 as well as PGE2 that may affect the host immune response to tumors. Our recent work demonstrates that the PGE2 secreted by 4T1 tumor cells suppresses CCL5 production from activated macrophages (33). However, it is unknown whether 4T1 tumor cell-secreted PGE2 affects IL-23 and/or Th17 cells during mammary gland tumor progression.
In this study, we found that 4T1 tumor-bearing mice displayed an increase in Th17 cells in the blood, spleen and tumor. IL-23 but not IL-12 was overexpressed in the tumor microenvironment. Overproduction of IL-23 was mediated by PGE2 secreted from tumor cells. Tumor-secreted PGE2 via EP2 and EP4 receptors enhanced IL-23 p19 expression at the transcriptional level through a signaling transduction pathway involving cAMP/PKA. These results elucidate an important interaction between tumor and host cells that reduce host anti-tumor immunity. Strategies controlling IL-23 overexpression could serve as new targets for development of antitumor immunotherapy.
Material and Methods
Mice
6~8 week old female Balb/c mice (obtained from The Jackson Laboratories, Bar Harbor, ME) were housed in cages with filter tops in a laminar flow hood, fed food and filtered water ad libitum at Saint Louis University Animal Facilities in accordance with the principles of Animal Care (NIH publication number 85–23, revised 1985).
Cells
The murine macrophage cell line RAW264.7 (RAW cells hereafter) and 4T1 mammary gland tumor cells were obtained from the American Type Culture Collection (ATCC), and maintained in RPMI 1640 supplemented with 2 mM glutamine, 100 units/ml of penicillin and streptomycin and 10% FBS (Sigma, St. Louis, MO, endotoxin NMT 10.0 EU/ml). To obtain dendritic cells, FLt3L-secreted B16 melanoma cell line were cultured in complete RPMI 1640 culture medium with 10% FBS and harvested when reach confluent. Then, 5 × 106 cells in 100 μl of PBS were injected subcutaneously into C57BL/6 mice. When the size of tumor reached about 2 × 2 cm in diameter (take ~ 2 weeks), the tumor-bearing mice were sacrificed and DCs were purified from spleens using mouse CD11c positive selection kit (Stemcell technology). Tumor conditioned medium (TCM) was prepared by inoculation of log-growth phase of 4T1 tumor cells into a T175 flask at a concentration of 0.6 × 106/mlwith freshly prepared complete culture medium. 72 hrs later, the culture supernatants were collected, filtered through a 0.45 μm filter, allequoted in small tubes and stored at −20°C for future use. Normal mammary gland epithelial cell line, CommaD Bgeo and FSK4 cells isolated from mammary glands of female Balb/c mice (34), were kindly provided by Dr. Daniel Medina (Baylor College of Medicine), and maintained in DMEM supplemented with 5 ml HEPES buffer (per 500 ml), insulin (10 ug/ml), EGF (5 ng/ml) and 2% FBS.
Plasmids and reagents
Mouse IL-23 p19 promoter was generated by cloning p19 promoter (1348 bp) into pGL2-basic luciferase vector (Promega) between Kpn I and Xho I sites. Primers used for cloning p19 promoter were caggacagccagggatacacagaga for sense strand and ggcacagccaggccctg for antisense strand. The CRE-mutant IL-23 p19 promoter was cloned using Quikchange II site-directed mutagenesis Kit (Strategene). All plasmid DNA for transfection were prepared with Qiagen Endo-free Maxi-Prep kits (Qiagen Inc. Valencia, CA). LPS from Escherichia coli 0217:B8 and epidermal growth factor (EGF) were purchased from Sigma-Aldrich (St, Louis, MO). NS-398, AH6809, AH23848 and SC51322 were purchased from Cayman Chemical (MI, USA). H89 and Forskolin were purchased from Calbiochem.
Reverse transcription-PCR (RT-PCR)
Reverse-transcription (RT) reactions were carried out as described previously (35) with 1 μg total RNA for RT. The following primers were used for PCR amplification of mouse IL-23 p19 cDNA: sense: TGCACCAGCGGGACATATGAATCT; antisense: TGTTGTCCTTGAGTCCTTGTGGGT; for mouse GAPDH cDNA: sense, AACTTTGGCATTGTGGAAGG; antisense, ACACATTGGGGGTAGGAACA.
Quantitative real-time PCR (qRT-PCR)
To determine the levels of mRNA expression by quantitative real time PCR, we used a modified protocol. Briefly, cDNA samples were diluted and studied at several concentrations. Diluted cDNA was mixed with a pair of primers (10 μM) targeting mouse IL-23 p19 or GAPDH cDNA sequences as described above, and with SYBR green PCR master mix (Applied Biosystem, CA) in a 15 μl volume. PCR cycling was set as: 2 min at 50°C, 10 min at 95°C for 1 cycle, followed by 40 cycles at 15 sec at 95°C, 1 min at 60°C.
Enzyme-linked immunosorbent assays (ELISA)
Supernatants from murine Flt3L-induced dendritic cell and RAW cell cultures were harvested 24 h after stimulation and stored at −70°C. Mouse IL-23 was detected using the mouse CytoSet ELISA kit (Invitrogen) according to the manufacturer’s instructions. Mouse IL-12(p40) was detected using mouse IL-12(p40) ELISA set (BD, Pharmingen). Concentrations were calculated by regression analysis of a standard curve.
Transfection assay
Transient transfections were performed by electroporation as described previously (35).
Primary transcript measurement
To determine the primary transcript of mouse IL-23 p19 gene, cDNAs were synthesized with random primers with 1 μg total RNA generated from RAW cells treated with LPS or LPS plus tumor-conditioned medium. The primers used for primary transcript were: mouse IL-23 p19 exon2 (sense): TGTTGTCCTTGAGTCCTTGTGGGT; mouse IL-23 p19 intron1 (antisense): AAACCTTCCCAGTCCTCCAAGTGT. Conditions for PCR amplification were 94°C for 4 min 1 cycle; 94°C for 15 sec, 60°C for 30 sec, 72°C for 30 sec, 30 cycles; 72°C for 7 min.
Statistical analysis
Student t test was performed wherever applicable. Standard deviation of the mean is shown unless otherwise indicated. *: p<0.05, **: p<0.01, ***: p<0.001.
Results
Mammary gland tumor-bearing mice show increased Th17 cells
To confirm whether Th17 cells were increased during breast tumor progression, we first induced mammary gland tumors in female Balb/c mice. Nineteen days after 4T1 cell injection, we isolated PBMCs, splenocytes and tumor tissues, and measured Th1 and Th17 cells by FACS analysis. The percentages of both Th1 and Th17 cells in PBMCs were significantly increased in tumor-bearing mice compared to tumor-free mice (up-panel in Figs. 1A & B). Moreover, the percentages of Th17 cells in the spleens of tumor-bearing mice were even more markedly increased than in PBMCs, whereas Th1 cells were not increased (middle panel in Figs. 1A & C). Similar Th17 cell increases without Th1 cells were present among tumor infiltrating lymphocyte (TIL) populations (lower-panel in Figs. 1A & D). Taken together, these data suggest that during breast tumor progression there are increased Th17 cells in blood, spleen and TIL in tumor-bearing host, which in turn, may facilitate tumor growth and metastasis.
FIGURE 1.
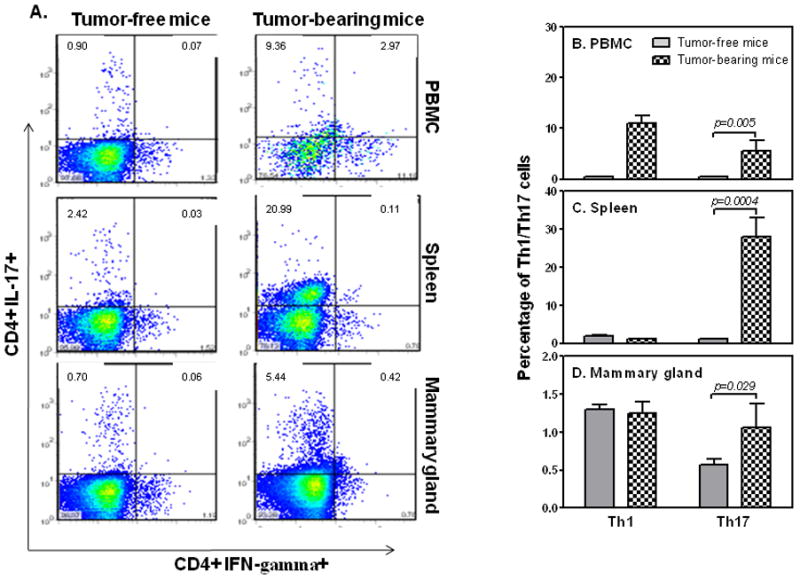
4T1 tumor-bearing mice have increased Th17 cells. 1 × 105 4T1 tumor cells were injected subcutaneously (s. c.) in the abdominal mammary gland of 6–8 week old female Balb/c mice. 19 days after 4T1 cell injection, tumor-free and tumor-bearing wild type mice were sacrificed and PBMCs, spleens and mammary glands were collected for detection of CD4+Th1 and CD4+Th17 cells by FACS analysis (A). Percentages of Th1 and Th17 cells in PBMCs (B), spleens (C), and mammary glands (D) in tumor-free and tumor-bearing mice were summarized from the data collected from five mice.
IL-23 is differentially overexpressed in tumors
The increased number of Th17 cells in tumor-bearing mice compelled us to investigate if the expression of IL-23 was also enhanced because IL-23 is a critical cytokine for Th17 cell survival and expansion. We directly isolated total RNA from breast tumors obtained from human patients and tumor-bearing mice, and measured IL-23 p19 and p40 mRNA expression as well as IL-12 p35 mRNA by quantitative real time PCR (qRT-PCR). As shown in Figure 2, IL-23 p19 (Figs. 2A&D) and p40 (Figs. 2B&E) mRNA expression were significantly increased whereas the IL-12 p35 mRNA expression (Figs. 2C&F) was only slightly increased in tumor tissues from both humans and mice, suggesting that IL-23 is differentially overexpressed in the tumor microenvironment that could facilitate the expansion and survival of Th17 cells.
FIGURE 2.
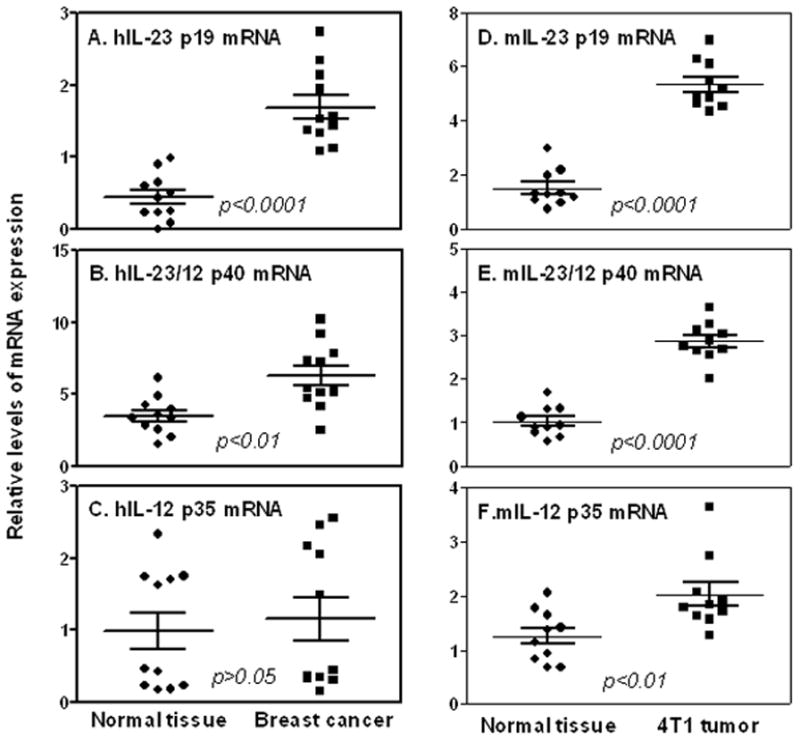
IL-23 expression is increased in breast cancer. Total RNA was extracted from tumor-surrounding tissues and breast cancer tissues from human patients (A–C) and tumor-bearing mice (D–F). Quantitative mRNA expression of human IL-23 p19 (A), human IL-12 p40 (B) and human IL-12 p35(C) in human breast tumor tissues were compared to normal adjacent tissue of the same individual (n=11). Quantitative mRNA expression of mouse IL-23 p19 (D), mouse IL-12 p40 (E) and mouse IL-12 p35 (F) in mouse normal mammary gland tissues were compared to 4T1 tumor tissues (n=10). qRT-PCR data were normalized relative to GAPDH mRNA expression levels in each respective sample and further normalized to the sample from the non-tumor surrounding tissues from the same subject, which was set as 1.
Soluble factors secreted from tumor cells increase IL-23 production
Since DCs and macrophages are major producers for IL-23 and the former are potent antigen presenting cells critical for T cell differentiation and activation, we wanted to know whether tumor-secreted factors could enhance IL-23 production from DCs. We injected subcutaneously B16 melanoma cells overexpressing Flt3-Ligand into C57B6 mice, purified the DCs from spleens 12 days after, and applied different amounts of tumor-conditioned medium (TCM) collected from 4T1 tumor cells in the presence of LPS stimulation. As we reported previously, LPS induced IL-23 production from DCs (24). Addition of TCM dose-dependently enhanced IL-23 (Fig. 3A), but suppressed IL-23/12 p40 (Fig. 3B) production in LPS-stimulated DCs. In line with protein production, IL-23 p19 mRNA expression (Fig. 3C) was enhanced whereas p40 (Fig. 3D) and p35 (Fig. 3E) mRNA expression were reduced by TCM in a dose-dependent manner in DCs stimulated with LPS. Similar IL-23 induction by TCM was also shown in activated macrophages (Data not shown). Since the p40 subunit is always produced in large quantity compared to the p19 subunit, the levels of the p19 expression determine the amount of biologically functional IL-23 production from the activated cells.
FIGURE 3.
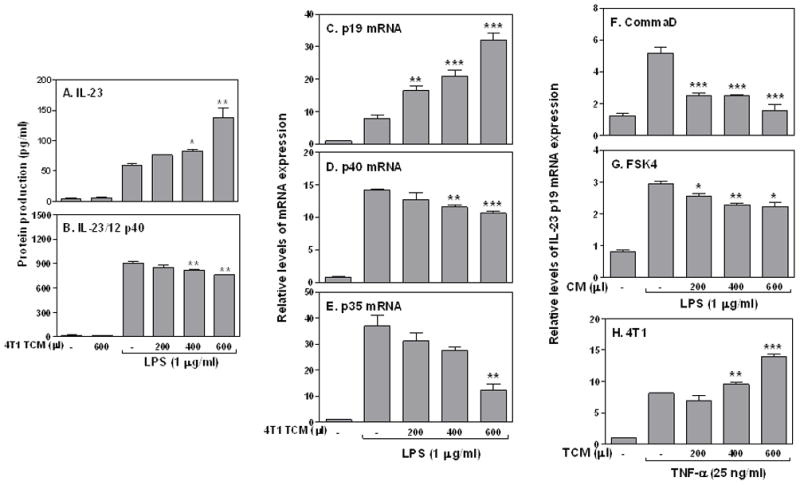
Tumor conditioned medium increases IL-23 production in DCs and macrophages. 1 × 106 Flt3 ligand-induced immature DCs were purified and seeded in each well of a 24-well culture plate. Different amounts of 4T1 TCM as indicated were added to the culture in the absence or presence of LPS (1 μg/ml) for 24 hrs to measure mouse IL-23 (A) and mouse IL-23/IL-12 p40 (B) protein levels by ELISA. (C) 3 × 106 Flt3 ligand-induced DCs were stimulated with LPS (1 μg/ml) plus different amounts of 4T1 TCM as described above for 4 h, followed by collection of total RNA to measure mouse IL-23 p19 (C), IL-23/IL-12 p40 (D) and IL-12 p35 (E) mRNA expression by qRT-PCR. The same amount of RAW cells were stimulated with LPS (1 μg/ml) in the absence or presence of different amounts of culture supernatants collected from the CommaD Bgeo (F) and FSK4 (G) cells as indicated to detect p19 mRNA expression (4 h after treatment). (H) 3 × 106 RAW cells were stimulated with TNF-α (25 ng/ml) in the absence or presence of different amounts of 4T1 TCM as indicated to detect p19 mRNA expression (8 h after treatment) by qRT-PCR. qRT-PCR data were normalized relative to GAPDH mRNA expression levels in each respective sample and further normalized to the sample from the untreated group, which was set as 1. Statistical analyses were performed between LPS or TNF-α treated group and LPS or TNF-α plus TCM or CM groups. *: p<0.05, **: p<0.01, ***: p<0.001 between two groups.
To exclude the possibility that soluble factors secreted from normal mammary gland epithelial cells may affect IL-23 induction, we collected conditioned medium (CM) from CommaD Bgeo cells and FSK4 cells (normal mammary gland epithelial cells isolated from Balb/c mice), and applied those CM to macrophages treated with LPS, followed by measurement of p19 mRNA expression. CM from CommaD (Fig. 3F) and FSK4 (Fig. 3G) cells did not induce but rather suppressed IL-23 p19 mRNA expression, indicating that soluble factors secreted from tumor cells uniquely enhanced IL-23 expression. In addition, to determine whether TCM-enhanced p19 expression happened only to TLR4 signaling, we stimulated macrophages with TNF-α in the presence of different amounts of 4T1 TCM. 4T1 TCM also enhanced TNF-α-induced p19 mRNA expression in a dose-dependent manner (Fig. 3H), indicating that soluble factors secreted from breast tumor cells can enhance IL-23 expression induced by different activation pathways.
Tumor-secreted PGE2 mediates IL-23 induction through EP2 and EP4 receptors
Our previous work indicated that 4T1 tumor cells secrete many cytokines and lipid molecules, including PGE2 (36). To determine whether PGE2 secreted by tumor cells was responsible for IL-23 induction, we collected TCM from 4T1 tumor cells treated with NS398, a specific COX2 inhibitor which can effectively block PGE2 synthesis, applied these PGE2-lacking TCM to RAW cells stimulated with LPS, and then measured p19 and p40 mRNA expression. TCM-enhanced p19 expression was almost completely abolished after blocking PGE2 synthesis in 4T1 tumor cells (Fig. 4A), whereas TCM-mediated p40 mRNA reduction was almost completely reversed (Fig. 4B), indicating that PGE2 is involved in IL-23 induction mediated by TCM. Indeed, PGE2 dose-dependently enhanced IL-23 production in DCs treated with LPS (Fig. 4C). To further verify whether tumor-secreted PGE2 mediated IL-23 induction in vivo, we injected NS398 into tumor-bearing mice, and then challenged the mice with LPS, followed by measuring p19 mRNA expression in spleens. Blocking PGE2 syntheses by NS398 significantly reduced p19 (Fig. 4D) and IL-17A (Fig. 4E) mRNA expression in tumors. The number of Th17 cells in tumors was also reduced after blocking PGE2 synthesis by NS398 (Fig. 4F). Furthermore, blocking PGE2 synthesis by NS398 in 4T1 tumor cells also completely abrogated TCM-induced p19 promoter activation (Fig. 4G). Taken together, these data indicate that tumor cell-secreted PGE2 enhances IL-23 production and Th17 cell development in the tumor microenvironment.
FIGURE 4.
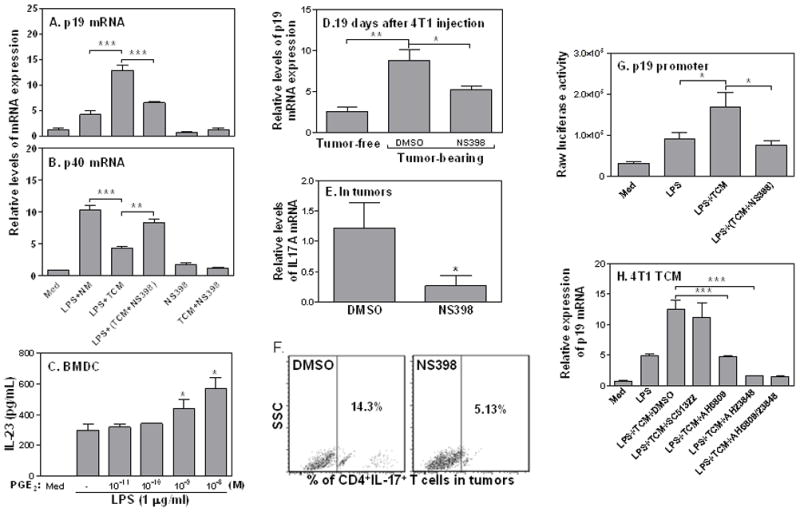
Tumor-secreted PGE2 enhances IL-23 production through EP2 and EP4 receptors. 1 × 106 RAW cells were cultured with normal media (NM), 400 μl/ml of 4T1 TCM, or 400 μl/ml of TCM collected from 4T1 cells treated with NS398 in the presence or absence of LPS (1 μg/ml) for 4 h. Note that in the fifth and sixth bar from the left, NS398 was added to the macrophage culture, not to the 4T1 culture. IL-23 p19 (A) and p40 (B) mRNA expression were measured by qRT-PCR. (C) 1 ×106 bone marrow-derived DCs were generated from bone marrow cells isolated from femurs of B6 mice under IL-4 (10 ng/ml) and GM-CSF (40 ng/ml) treatment for 6 days, and then treated with different amounts of PGE2 as indicated in the absence or presence of LPS (1 μg/ml) for 24 h. The supernatants were collected for measurement of IL-23 levels by ELISA. (D–E) 4T1 tumors were generated as described previously in Figure 1. Seven days later, NS398 (10 mg/kg) and an equal amount of DMSO were given i.p. every 3 days for total four times. 19 days post 4T1 cell injection, splenocytes were isolated and used to extract total RNA for detection of p19 (D) and IL-17A (E) mRNA expression by qRT-PCR. (F) tumor-bearing mice were treated with NS398 and DMSO as indicated above, and tumors were used to generate single cells to detect tumor-infiltrating Th17 cells by FACS analysis. (G) 5 μg of p19 promoter was transiently transfected into RAW cells by electroporation. The transfected cells were stimulated with 400 μl of 4T1 TCM or TCM collected from 4T1 cells treated with NS398 in the presence of LPS (1 μg/ml) for 7 h. The luciferase activity was measured in cell lysates. (H) 1 × 106 RAW cells were pre-treated with 10 μM SC51322 (EP1 antagonist), 3 μM AH6809 (EP2 antagonist) and 30 μM AH 23848 (EP4 antagonist) or AH6809 plus Ah23848 for 30 min, then stimulated with LPS (1 μg/ml) and/or 400 μl 4T1 TCM for 4 h to detect p19 mRNA expressionc qRT-PCR. Data shown are means plus S.D. of three-four experiments. qRT-PCR data were normalized relative to GAPDH mRNA expression levels in each respective sample and further normalized to the sample from the untreated group, which was set as 1. *: p<0.05; **: p<0.01 between two groups as indicated.
To further characterize the receptors involved in tumor PGE2-mediated IL-23 induction, we applied various PGE2 receptor antagonists to block PGE2 signaling and tested whether the inductive effects of 4T1 TCM was affected. Since there are four prostanoid receptors (EP1-EP4) involved in PGE2 signaling, we blocked these PGE2 receptors individually with SC-51322 (a selective EP1 antagonist), AH-6809 (an EP2 receptor antagonist), and AH-23848 (a selective antagonist for EP4) prior to LPS and 4T1 TCM stimulation. EP1 antagonist had no effect on TCM-mediated p19 induction whereas EP2 and EP4 antagonists each could completely abrogate TCM-induced p19 expression (Fig. 4H), demonstrating that PGE2 secreted by tumor cells enhances IL-23 induction through EP2 and EP4 receptor activation.
Tumor-secreted PGE2 promotes p19 expression at the transcriptional level through binding to the CRE site in the p19 promoter
To investigate the molecular mechanisms of TCM-mediated p19 induction, we measured primary transcript rates of the p19 gene in response to TCM in RAW cells, with primers corresponding to two different regions of the p19 gene. The use of primers specific for an intron/exon boundary region allowed us to assess the primary transcript rate of p19 inside the nucleus. As shown in Fig. 5A, p19 primary transcripts were induced by LPS within 30 min and kept increasing up to 3 hrs after LPS simulation. Addition of TCM further increased the p19 primary transcript through all time points (Fig. 5A), indicating a transcriptional enhancement of the p19 gene by TCM. PGE2 showed the same inductive effect as TCM on p19 primary transcript (Fig. 5B). To further confirm whether TCM activated p19 gene transcription, we transiently transfected mouse p19 promoter-luciferase constructs into RAW cells by electroporation, followed by stimulating the transfected cells with different amounts of TCM and LPS. Luciferase activity was measured in cell lysates. The p19 promoter activity was enhanced by TCM (Fig. 5C) and PGE2 (Fig. 5D) in a dose-dependent manner, further confirming that TCM-induced p19 gene expression is regulated at the transcriptional level.
FIGURE 5.
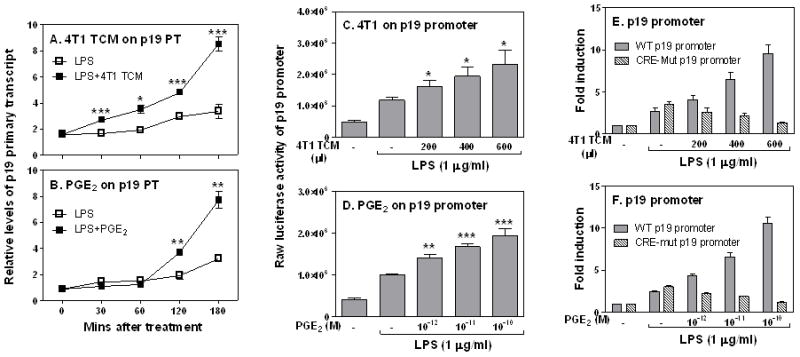
Tumor-derived PGE2 enhances IL-23 expression at the transcriptional level through binding to the CRE site in the p19 promoter. 3 × 106 RAW cells were stimulated with LPS (1 μg/ml) or LPS plus 400 μl 4T1 TCM (A), or LPS plus PGE2 (10−12 M) (B) for different times as indicated, followed by extraction of total RNA to measure p19 gene primary transcript by qRT-PCR. Data were normalized relative to GAPDH gene expression levels in each respective sample and further normalized to the results from the untreated group (Med), which was set as 1. 10 × 106 RAW cells were transiently transfected with 5 μg of mouse p19 luciferase reporter construct. The transfected cells were stimulated with different amounts of 4T1 TCM as indicated (C) or different concentration of PGE2 (D) for 7 h in the presence of LPS (1 μg/ml), followed by measurement of luciferase activity in cell lysates. 10 × 106 RAW cells were transiently transfected with 5 μg of wild type or the CRE mutant p19 luciferase reporter constructs as described above, and transfected cells were stimulated with different amounts of 4T1 TCM (E) or PGE2 (F) as indicated for 7 h in the presence of LPS (1 μg/ml), followed by measurement of luciferase activity. Luciferase data were normalized to the activity obtained with non-treated cells and represented as fold induction. All data shown are means plus SD of three-four experiments. Statistical analyses were performed between LPS treated group and LPS plus TCM treated groups. *: p<0.05, **: p<0.01, ***: p<0.001 between two groups.
To elucidate the molecular basis of TCM-mediated p19 transcriptional induction, we decided to localize the functional TCM/PGE2 response element in the p19 promoter. By searching the p19 promoter, we noticed that there is a putative cAMP response element (CRE) between −1009 and −1004. We mutated this putative CRE binding site and transiently transfected the wild type and CRE mutant into RAW cells and stimulated these cells with either TCM or PGE2 in the presence of LPS. Mutation of the CRE site in p19 promoter completely abolished responses to TCM (Fig. 5E) and PGE2 (Fig. 5F), indicating that this putative CRE binding site is critical for the responsiveness of p19 promoter to tumor-secreted PGE2 stimulation.
TCM-enhanced p19 expression is mediated through cAMP/PKA signal transduction pathway
TCM enhanced p19 transcription through the CRE site in the p19 promoter (Fig. 5F), suggesting that the inductive effect of TCM on p19 expression may be mediated by cAMP/PKA signaling pathway. To confirm this, we first blocked PKA function with the inhibitor H89 prior to 4T1 TCM and LPS stimulation, followed by measurement of p19 expression. Blocking PKA with H89 dose-dependently abolished the inductive effects of 4T1 TCM on LPS-induced p19 mRNA expression (Fig. 6A). H89 could also dose-dependently abrogate PGE2-induced p19 mRNA expression (Fig. 6B). In addition, Forskolin, a cAMP active analog, enhanced LPS-induced p19 mRNA expression (Fig. 6C) to similar levels when compared it with 4T1 TCM, further supporting the involvement of cAMP/PKA signaling in TCM-enhanced p19 expression. Furthermore, TCM-enhanced p19 promoter activation was completely abolished after blocking PKA with H89 (Fig. 6D). Taken together, these results indicate that PGE2 secreted from breast tumor cells enhances LPS-induced p19 expression through the cAMP/PKA signal transduction pathway.
FIGURE 6.
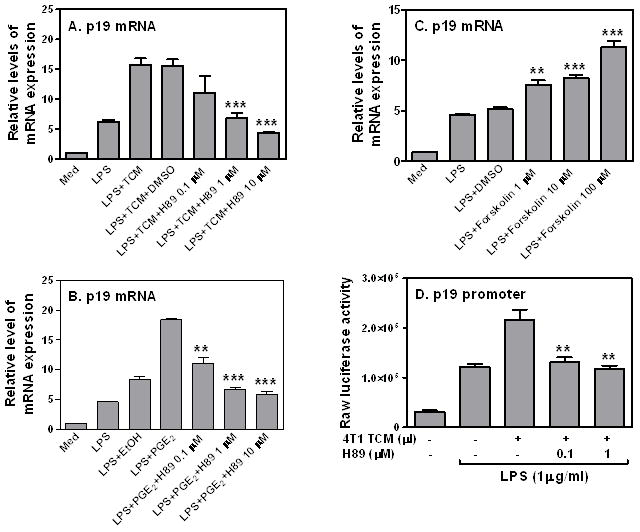
The effect of 4T1 TCM on LPS-induced p19 expression is mediated through the cAMP/PKA signaling transduction pathway. 3 × 106 RAW cells were pretreated with various amounts of H89 as indicated for 60 min, then stimulated with LPS (1 μg/ml) or LPS plus 400 μl 4T1 TCM (A) or PGE2 (10−12 M) (B) for 4 h, followed by collection of total RNA to measure p19 mRNA expression by qRT-PCR. (C) Different amounts of Forskolin as indicated were added to LPS-stimulated RAW cells for 4 hours to measure p19 mRNA expression by qRT-PCR. The same amount of dissolvent, DMSO was used as a negative control. (D) 10 × 106 RAW cells were transiently transfected with 5 μg of mouse p19 luciferase reporter construct. The transfected cells were pre-treated with different amounts of H89 as indicated for 60 min, then stimulated with LPS (1 μg/ml) or LPS plus 400 μl of 4T1 TCM for additional 7 h, followed by measurement of luciferase activity in cell lysates. qRT-PCR data were normalized relative to GAPDH mRNA expression levels in each respective sample and further normalized to the sample from the untreated group, which was set as 1. Data shown are means plus SD of 3–4 independent experiments ***: p<0.001 between two groups as indicated. Statistical analyses were performed between H89 or Forskolin treated group and LPS plus TCM and dissolvent groups.
Discussion
The link between chronic inflammation and tumorogenesis has been noted for a long time; however, the mechanisms of the causal relationships between these two events are still largely unknown. Th17 cells have been found to be directly involved in the pathogeneses of several autoimmune disorders, such as experimental autoimmune encephalomyelitis (EAE), rheumatoid arthritis (RA) and Crohn’s disease, by secreting proinflammatory cytokines which lead to accumulation of neutrophils and other immune cells causing chronic inflammation (37). The close association between Th17 cells and autoimmune inflammation motivated us to explore the relationships between chronic inflammation, Th17 cells and cancer. Recently, it has been reported that Th17 cells are increased in many solid tumors including breast cancer (5). We also found in this study that Th17 cells were increased in mammary gland tumor tissues (Figs. 1A &D). In addition, there were even greater increases of Th17 cells in spleens and blood from tumor-bearing mice (Figs. 1B&C), which has not been appreciated before. Not only the percentages but the absolute numbers of Th17 cells were also increased in tumor-bearing mice compared to tumor-free mice (Data not shown). Along with tumor progression, the percentage of Th1 cells tended to decrease and Th17 cells increased (Data not shown). The increased Th17 cells and decreased Th1 cells may contribute to tumor progression.
IL-23 induces chronic inflammation through stimulation of innate myeloid cells and stromal activation, whereas the Th17 cells are important for orchestrating inflammatory tissue destruction (18). Though initial differentiation of Th17 cells does not require IL-23, IL-23 is necessary for the maintenance and expansion of Th17 cells (37). Consistent with a previous report (17), our study confirms that IL-23 was overexpressed in breast tumor tissues. Since tumor cells secrete high amounts of TGF-β and IL-6 (36), along with the overexpression of IL-23 in the tumor microenvironment, it seems that all of these cytokines required for Th17 cell development are readily presented in the tumor microenvironment. Because the p40 subunit is a shared subunit between IL-12 and IL-23, expression of the p35 and p19 subunits determines the levels of biologically activated IL-12 and IL-23, respectively. Our results indicate that IL-23 and IL-12 expression were differentially regulated in tumors, with increased expression of the p19 subunit and little change of the p35 subunit (Figs. 2A–F). Considering the critical roles for IL-12 and IL-23 in Th1 and Th17 cell development, this differential expression may explain why there were increased Th17 cells but not Th1 cells in the tumor microenvironment.
Different from tumor tissues that expressed high levels of both p19 and p40 subunits (Figs. 2A–F), DCs and macrophages showed a reduced p40 mRNA expression in response to TCM (Figs. 3B&D). This difference in p40 expression pattern between tumor tissues and DCs/macrophages could be due to the fact that endogenous ligands in the tumor microenvironment may use different signaling pathways other than LPS/TLR4 pathway to activate p40 expression. Indeed, 4T1 tumor conditioned medium not only enhanced LPS-induced p19 mRNA expression but also increased TNF-α-stimulated p19 expression in macrophages (Fig. 3H). IL-23 p19 expression was induced only by tumor-secreted factors but not by normal mammary gland epithelial cell-secreted molecules. In contrast, molecules secreted from normal epithelial cells suppressed LPS-induced p19 expression (Figs. 3F&G), suggesting that tumor cells acquire the ability to enhance IL-23 production in the tumor microenvironment after transforming into malignant cells.
Blocking PGE2 synthesis by NS398 in 4T1 cells abolished the ability of 4T1 TCM to enhance p19 expression (Fig. 4A), indicating that tumor cell-secreted PGE2 enhances LPS-induced IL-23 production. This is consistent with previous observations that PGE2 induced IL-23 in bone marrow-derived dendritic cells (38) and with our own data (Fig. 4C). The Th17 cell-promoting effects of PGE2 are not only attributed to enhancement of IL-23 production as shown here by us and others (38) but can also be attributed to increased expression of IL-23 and IL-1 receptors. Overexpression of RORγt, IL-17 and CCR6 expression in response to PGE2 stimulation can also contribute to Th17 cell development (39). It has been recently reported that less than nanomolar concentrations of PGE2 can promote Th1 cell differentiation whereas higher concentrations of PGE2 lead to Th1 cell suppression (40). Our results also indicate the dual effects of PGE2 on IL-23 expression, with higher amounts of PGE2 suppressing and lower amounts of PGE2 enhancing IL-23 production (Data not show). Nevertheless, our study indicates, for the first time, that breast cancer cell-secreted PGE2 enhanced IL-23 production and tumor-associated Th17 cell development (Figs. 4D–F). Given that inhibition of PGE2 synthesis by NS398 in vivo only partially reduced p19 expression in tumor-bearing mice (Fig. 4D), other factors secreted by tumor cells may also be involved in activation of IL-23 during tumor development. It is of interest to investigate the endogenous molecules that induce p19 expression in tumor-bearing hosts. It appears that both EP2 and EP4 receptors are involved in TCM-mediated p19 expression. EP2 has major effects on TCM-mediated p19 induction, whereas EP4 mediates both TCM- and LPS-induced p19 expression as LPS-induced p19 mRNA expression was also completely abrogated by blocking EP4 receptor (Fig. 4H). This differential involvement of EP2 and EP4 signaling in tumor-associated IL-23 production could be used to develop potential targets for tumor immunotherapy. The transcriptional induction of the p19 subunit by TCM and PGE2 (Figs. 5A–D), suggests that the p19 subunit could be targeted for control of IL-23 production during tumor development. Identification of the CRE site in the p19 promoter essential for control of TCM-induced p19 transcription (Fig. 5E) and clarification of the cAMP/PKA pathway involved in p19 expression induced by TCM (Figs. 6A–D) will further help us to design more detailed specific molecules targeting only the p19 but not p40 gene transcription.
In conclusion, our study highlights the molecular mechanisms of breast tumor cell-secreted PGE2 in promoting IL-23 expression and Th17 cell expansion. Our findings also shed light on considering different molecules and pathways involved in promoting tumor-associated Th17 cell development as potential targets for future development of tumor treatments.
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health under Award Number CA163808 to J.L., HL098794 to M.F., and AR056729 to X.H. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We thank Dr. Daniel Medina at Baylor College of Medicine for kindly providing us the CommaD Bgeo and FSK4 cells for this study.
References
- 1.DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007;9:212. doi: 10.1186/bcr1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 4.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009 doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 5.Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol. 2010;10:248–256. doi: 10.1038/nri2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murugaiyan G, Saha B. Protumor vs antitumor functions of IL-17. J Immunol. 2009;183:4169–4175. doi: 10.4049/jimmunol.0901017. [DOI] [PubMed] [Google Scholar]
- 7.Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, Peng G. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol. 2010;184:1630–1641. doi: 10.4049/jimmunol.0902813. [DOI] [PubMed] [Google Scholar]
- 8.Derhovanessian E, Adams V, Hahnel K, Groeger A, Pandha H, Ward S, Pawelec G. Pretreatment frequency of circulating IL-17+ CD4+ T-cells, but not Tregs, correlates with clinical response to whole-cell vaccination in prostate cancer patients. Int J Cancer. 2009;125:1372–1379. doi: 10.1002/ijc.24497. [DOI] [PubMed] [Google Scholar]
- 9.Zhang B, Rong G, Wei H, Zhang M, Bi J, Ma L, Xue X, Wei G, Liu X, Fang G. The prevalence of Th17 cells in patients with gastric cancer. Biochem Biophys Res Commun. 2008;374:533–537. doi: 10.1016/j.bbrc.2008.07.060. [DOI] [PubMed] [Google Scholar]
- 10.Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 13.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 14.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 16.Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, Soumelis V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 17.Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, Basham B, McClanahan T, Kastelein RA, Oft M. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- 18.Teng MW, Andrews DM, McLaughlin N, von Scheidt B, Ngiow SF, Moller A, Hill GR, Iwakura Y, Oft M, Smyth MJ. IL-23 suppresses innate immune response independently of IL-17A during carcinogenesis and metastasis. Proc Natl Acad Sci U S A. 2010;107:8328–8333. doi: 10.1073/pnas.1003251107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang YQ, Ugai S, Shimozato O, Yu L, Kawamura K, Yamamoto H, Yamaguchi T, Saisho H, Tagawa M. Induction of systemic immunity by expression of interleukin-23 in murine colon carcinoma cells. Int J Cancer. 2003;105:820–824. doi: 10.1002/ijc.11160. [DOI] [PubMed] [Google Scholar]
- 20.Lo CH, Lee SC, Wu PY, Pan WY, Su J, Cheng CW, Roffler SR, Chiang BL, Lee CN, Wu CW, Tao MH. Antitumor and antimetastatic activity of IL-23. J Immunol. 2003;171:600–607. doi: 10.4049/jimmunol.171.2.600. [DOI] [PubMed] [Google Scholar]
- 21.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 22.Carmody RJ, Ruan Q, Liou HC, Chen YH. Essential roles of c-Rel in TLR-induced IL-23 p19 gene expression in dendritic cells. J Immunol. 2007;178:186–191. doi: 10.4049/jimmunol.178.1.186. [DOI] [PubMed] [Google Scholar]
- 23.Mise-Omata S, Kuroda E, Niikura J, Yamashita U, Obata Y, Doi TS. A proximal kappaB site in the IL-23 p19 promoter is responsible for RelA- and c-Rel-dependent transcription. J Immunol. 2007;179:6596–6603. doi: 10.4049/jimmunol.179.10.6596. [DOI] [PubMed] [Google Scholar]
- 24.Liu W, Ouyang X, Yang J, Liu J, Li Q, Gu Y, Fukata M, Lin T, He JC, Abreu M, Unkeless JC, Mayer L, Xiong H. AP-1 activated by toll-like receptors regulates expression of IL-23 p19. J Biol Chem. 2009;284:24006–24016. doi: 10.1074/jbc.M109.025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Salleeh F, Petro TM. Promoter analysis reveals critical roles for SMAD-3 and ATF-2 in expression of IL-23 p19 in macrophages. J Immunol. 2008;181:4523–4533. doi: 10.4049/jimmunol.181.7.4523. [DOI] [PubMed] [Google Scholar]
- 26.Ruhnke M, Ungefroren H, Nussler A, Martin F, Brulport M, Schormann W, Hengstler JG, Klapper W, Ulrichs K, Hutchinson JA, Soria B, Parwaresch RM, Heeckt P, Kremer B, Fandrich F. Differentiation of in vitro-modified human peripheral blood monocytes into hepatocyte-like and pancreatic islet-like cells. Gastroenterology. 2005;128:1774–1786. doi: 10.1053/j.gastro.2005.03.029. [DOI] [PubMed] [Google Scholar]
- 27.Qian X, Ning H, Zhang J, Hoft DF, Stumpo DJ, Blackshear PJ, Liu J. Posttranscriptional regulation of IL-23 expression by IFN-gamma through tristetraprolin. J Immunol. 2011;186:6454–6464. doi: 10.4049/jimmunol.1002672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pulaski BA, Ostrand-Rosenberg S. Reduction of established spontaneous mammary carcinoma metastases following immunotherapy with major histocompatibility complex class II and B7.1 cell-based tumor vaccines. Cancer Res. 1998;58:1486–1493. [PubMed] [Google Scholar]
- 29.Mi Z, Guo H, Wai PY, Gao C, Wei J, Kuo PC. Differential osteopontin expression in phenotypically distinct subclones of murine breast cancer cells mediates metastatic behavior. J Biol Chem. 2004;279:46659–46667. doi: 10.1074/jbc.M407952200. [DOI] [PubMed] [Google Scholar]
- 30.Morecki S, Yacovlev L, Slavin S. Effect of indomethacin on tumorigenicity and immunity induction in a murine model of mammary carcinoma. Int J Cancer. 1998;75:894–899. doi: 10.1002/(sici)1097-0215(19980316)75:6<894::aid-ijc12>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 31.Rakhmilevich AL, Janssen K, Hao Z, Sondel PM, Yang NS. Interleukin-12 gene therapy of a weakly immunogenic mouse mammary carcinoma results in reduction of spontaneous lung metastases via a T- cell-independent mechanism. Cancer Gene Ther. 2000;7:826–838. doi: 10.1038/sj.cgt.7700176. [DOI] [PubMed] [Google Scholar]
- 32.Shi X, Cao S, Mitsuhashi M, Xiang Z, Ma X. Genome-wide analysis of molecular changes in IL-12-induced control of mammary carcinoma via IFN-gamma-independent mechanisms. J Immunol. 2004;172:4111–4122. doi: 10.4049/jimmunol.172.7.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qian X, Zhang J, Liu J. Tumor-secreted PGE2 inhibits CCL5 production in activated macrophages through cAMP/PKA signaling pathway. J Biol Chem. 2011;286:2111–2120. doi: 10.1074/jbc.M110.154971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hollmann CA, Kittrell FS, Medina D, Butel JS. Wnt-1 and int-2 mammary oncogene effects on the beta-catenin pathway in immortalized mouse mammary epithelial cells are not sufficient for tumorigenesis. Oncogene. 2001;20:7645–7657. doi: 10.1038/sj.onc.1204980. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Qian X, Ning H, Yang J, Xiong H, Liu J. Activation of IL-27 p28 gene transcription by interferon regulatory factor 8 in cooperation with interferon regulatory factor 1. J Biol Chem. 2010;285:21269–21281. doi: 10.1074/jbc.M110.100818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitsuhashi M, Liu J, Cao S, Shi X, Ma X. Regulation of interleukin-12 gene expression and its anti-tumor activities by prostaglandin E2 derived from mammary carcinomas. J Leukoc Biol. 2004;76:322–332. doi: 10.1189/jlb.1203641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 38.Khayrullina T, Yen JH, Jing H, Ganea D. In vitro differentiation of dendritic cells in the presence of prostaglandin E2 alters the IL-12/IL-23 balance and promotes differentiation of Th17 cells. J Immunol. 2008;181:721–735. doi: 10.4049/jimmunol.181.1.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boniface K, Bak-Jensen KS, Li Y, Blumenschein WM, McGeachy MJ, McClanahan TK, McKenzie BS, Kastelein RA, Cua DJ, de Waal Malefyt R. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med. 2009;206:535–548. doi: 10.1084/jem.20082293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, Sugimoto Y, Narumiya S. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med. 2009;15:633–640. doi: 10.1038/nm.1968. [DOI] [PubMed] [Google Scholar]