

Abstract
Habitat loss and fragmentation are imminent threats to biological diversity worldwide and thus are fundamental issues in conservation biology. Increased isolation alone has been implicated as a driver of negative impacts in populations associated with fragmented landscapes. Genetic monitoring and the use of measures of genetic divergence have been proposed as means to detect changes in landscape connectivity. Our goal was to evaluate the sensitivity of Wright’s F st, Hedrick’ G’st, Sherwin’s MI, and Jost’s D to recent fragmentation events across a range of population sizes and sampling regimes. We constructed an individual-based model, which used a factorial design to compare effects of varying population size, presence or absence of overlapping generations, and presence or absence of population sub-structuring. Increases in population size, overlapping generations, and population sub-structuring each reduced F st, G’st, MI, and D. The signal of fragmentation was detected within two generations for all metrics. However, the magnitude of the change in each was small in all cases, and when N e was >100 individuals it was extremely small. Multi-generational sampling and population estimates are required to differentiate the signal of background divergence from changes in Fst, G’st, MI, and D associated with fragmentation. Finally, the window during which rapid change in Fst, G’st, MI, and D between generations occurs can be small, and if missed would lead to inconclusive results. For these reasons, use of F st, G’st, MI, or D for detecting and monitoring changes in connectivity is likely to prove difficult in real-world scenarios. We advocate use of genetic monitoring only in conjunction with estimates of actual movement among patches such that one could compare current movement with the genetic signature of past movement to determine there has been a change.
Introduction
Habitat loss and fragmentation are considered to be among the most imminent threats to biological diversity worldwide and thus are fundamental issues in conservation biology [1]–[4]. Fragmentation is a complex phenomenon that is simultaneously a consequence of habitat loss and a process in and of itself [5]–[7]. It is a function of the extensiveness of individual patches, distances among those patches [8]–[10], the nature of the intervening landscape [11], and how individual species are affected by each of those aspects [12]. Understanding the joint and independent effects of loss and configuration of the remaining habitat has long been a major focus of landscape ecology due to conservation implications e.g., [13]–[17].
Although the two phenomena are intertwined, when they are examined separately habitat loss has repeatedly been shown to have larger detrimental effects than fragmentation alone [5], [7], [18]–[21]. Still, increased isolation has been implicated as a driver of population extinctions [22], declining population size of interior species [13], [23], altered social behavior [24], reduced population viability [25], [26], demographic change in general [11], [27], [28], and spread of invasive species [29]. Reduced migration under lower levels of connectivity will have genetic consequences of reduced effective population size (N e) and increased rates of inbreeding and genetic drift within newly isolated habitat patches that will affect short- and long-term potential for survival [30]–[33].
Changes in landscape composition and configuration associated with the fragmentation process have been quantified and monitored using an extensive array of landscape indices [34]–[40]. Assessing the consequences of these changes for populations and processes fundamentally requires linking the structural attributes of landscape pattern with potential or actual movement of individuals among patches [8], [40]–[43]. Movement is often documented using habitat suitability, mark-recapture, radio-telemetry, experimental removal-recolonization studies [19], [41] and demographic monitoring [44]–[46]. Unfortunately, such studies can be so data- and time-intensive that there may be little practical application for conservation of most species e.g., [47], [48]. Observing physical movement of cryptic or primarily sessile organisms in which mobility is limited to particular life stages is especially challenging [49], [50].
Genetic monitoring [51] has been proposed as a minimally invasive, relatively cost-effective means of quantifying genetic effects of changes in landscape structure. Population genetic parameters may be more sensitive for detecting changes in connectivity than traditional demographic estimates that have large error components [52]. Thus, although in many cases conservation biologists are concerned about genetic diversity for its own sake, here we are interested in the potential for using genetic changes that result from fragmentation to quantify changes in the ecological process of movement.
Direct genetic methods have been developed to detect actual dispersal events [53]–[57]. However, it is more common for investigators to document fragmentation using indirect methods that quantify the amount of divergence in populations in putatively fragmented habitat. Although potentially more powerful analytical methods have been developed [58]–[61] and are being tested [62], [63], most investigators use Wright’s F st [64] and its analogues [65]–[73].
Despite its fundamental importance and strong theoretical foundations, detecting genetic effects of fragmentation in the wild has not been as straightforward as one might expect. Attempts to link indices of landscape structure to ecological and evolutionary processes have not yielded consistent relationships and many empirical investigations of fragmentation fail to detect definitive effects [74]–[76]. In particular, empirical data are often equivocal relative to predictions of the impacts of fragmentation on genetic divergence. Inconsistent relationships may result from non-monotonic relationships between many landscape metrics and landscape configuration [10] or non-linear or threshold-like population responses along the fragmentation gradient. Additionally, not all habitat that is perceived as fragmented by humans is actually fragmented from the perspective of a species of interest, thus some investigations may be trying to quantify effects of fragmentation where it actually does not exist. As mentioned above, the point at which discrete patches are functionally fragmented depends on the scale at which a species perceives and interacts with the landscape [77]–[79]. For species in patchy habitats, connectivity ultimately depends on the degree to which land cover types between discrete patches are barriers, versus filters, versus easily traversable; information that is lacking for most species. Moreover, even if movement through a landscape is impeded or precluded through anthropogenic change, long-lived individuals that pre-date the fragmentation event would provide a genetic signature of connectivity that no longer exists [74]. These issues can be addressed through careful study design in which temporal and spatial sampling scales match potential scales of fragmentation based on the biology of the focal organism.
Of greater concern is the potential that characteristics of F st-related values might make them insufficient for detecting habitat fragmentation on time scales that are relevant for conservation management. Wright’s F st and subsequent derivations have a number of specific assumptions that are almost always violated in natural systems and complicate interpretation of genetic divergence and gene flow among populations [80]–[83]. Because F st integrates over evolutionary time it is difficult to separate current from historical processes based on a single estimate of pattern alone and it may be slow to reflect changes in migration following a fragmentation event, especially if N e remains large. Additionally, the alleles that are most likely to be lost through drift are at low frequencies in populations and these alleles contribute little to F st values [84]. Slow response may also arise from the fact that when connectivity is only reduced rather than eliminated entirely, estimates of F st may remain close to zero [83]. Finally, measures of genetic structure (e.g., F st, G st, Φst) can be depressed when within-subpopulation heterozygosity or variance is high relative to among-subpopulation levels, which is common with highly diverse markers e.g., microsatellites [85]–[89]. F st–related measures calculated from such data will never approach unity regardless of the underlying patterns of allelic diversity, and they do not behave monotonically. Hedrick [86] sought to overcome the dependence of Gst (a generalization of Wright's Fst to include multiple alleles) on levels of heterozygosity by standardizing the measure against the maximum Gst possible for the observed amount of heterozygosity. The resulting statistic, G’st varies from 0–1 in a way that better reflects the underlying patterns of genetic diversity [86], but remains fundamentally based on heterozygosity. Jost [85] proposed a measure of genetic divergence based on allelic diversity (D) that varies between 0 and 1 regardless of within-population heterozygosity, and it is suggested to better reflect population differentiation. Heller and Siegismund [90] found that values of Jost’s D calculated from data in 34 published studies were ∼60% greater than the corresponding G st values, and that G’st values were ∼85% greater than G st. The increased magnitude of both G’st and Jost’s D and potential wider range of values may provide greater ability to detect recent fragmentation events. Additionally, D is expected to be more sensitive because it is calculated based on allele diversity which will decline more rapidly than heterozygosity [84]. More recently Sherwin has proposed a standardized mutual information (MI) index [91], [92] based on Shannon’s index that also varies between 0 and 1 and is independent of heterozygosity with the added property weighing all alleles according to their frequency (i.e. neither favoring rare nor common alleles).
Because we were interested in effects of fragmentation independent of habitat loss, we evaluated the ability to detect genetic effects of fragmentation with F st, G’st, MI, and D over timeframes associated with anthropogenic habitat modification (i.e., <200 years) while controlling for population size. The number of generations necessary to make such an evaluation renders the task infeasible in a field setting. Therefore, we developed an individual-based population model to simulate genetic divergence among recently fragmented populations and measured F st, G’st, MI, and D over time. Potential for detecting change in these metrics will vary based on the amount and nature of migration among populations; therefore, we simulated two severe cases of fragmentation. In the first, migration among a set of historically panmictic populations was abruptly and completely stopped. In the second, limited gene flow among populations was allowed and subsequently ceased. The first scenario provides the most ideal situation for detecting change – going from a base condition of a Wright-Fisher population to complete isolation. The second provides a more realistic starting condition in which there is a pre-existing level of divergence among populations onto which anthropogenic fragmentation is imposed. We complement a recent investigation of the effect of dispersal distance among individuals on the time required to detect an abrupt barrier to gene flow [63] by examining multiple discrete populations and by quantifying the influence of population size, overlapping generations, and sampling effort in terms of individuals and loci on ability to detect a significant change in four measures: F st, G’st, MI, and Jost’s D.
Methods
Model Description
We generated six homogeneous panmictic populations of equal size at the start of each run. Panmixia among populations was created by allowing mating at random among individuals in all populations. The model allows variation in distances among individual population pairs but for the purposes of this evaluation all populations were equally isolated. Census size maxima (N max) within populations were set to 25, 75, 100, 500, 1000, and 3000 individuals (N e was subsequently calculated) which encompasses the size ranges of populations of most plant species listed under the U.S. Endangered Species Act (Neel unpublished data), and 71% of minimum viable population estimates for plant species world wide [93]. Initial size of each population was set to 75% of the size limit for each run and the size cap was reached within one or two generations.
At initiation, individuals were assigned two alleles at each of 20 unlinked microsatellite loci. Allele size ranged between 5 and 50 repeat units. Alleles for each locus could take on any value within the given range, and were drawn from a normal distribution with parameters μ = mean of the size range of the locus and σ2 = 5. Drawing initial allele frequencies from a normal distribution allows for accurate simulation of the stepwise mutational model of microsatellite evolution throughout a simulation [94]. These starting conditions yielded between 7 and 42 alleles per locus at the start of each simulation depending on the population size. Mutations occurred every 0.004 gamete transfer events [94]. By using a stepwise mutational model of microsatellite evolution, small changes in allelic state were more likely than large changes and the direction of mutation tended toward the mean size range of each locus [94].
Individuals were simulated to be hermaphroditic, annual plants that were self-compatible, but that did not self-fertilize more than what would be expected at random, and therefore the amount of selfing depended upon population size. All individuals had an equal probability of mating each generation. Individuals from within a population had an equal probability of being a father for all individuals within that population. The proportion of individuals contributing seed to the next generation varied around a normal distribution with the parameters μ = 50% total population size and σ2 = 1. The number of seeds produced per female was drawn from a normal distribution with parameters μ = 35 and σ2 = 5 to provide stochastic variation around a likely number of seeds per plant. Each seed had a randomly selected father. When a seed bank was included in the model, those seeds not germinating entered the seed bank; otherwise, seeds that did not germinate immediately were removed. Germination potential of seeds in the seed bank decreased over time following a negative exponential function. As the size of each population approached the population size limit, the number of viable seeds produced was reduced to reflect density dependence [95].
Each cap size was run under four conditions that independently varied presence or absence of a seed bank (i.e., non-overlapping versus overlapping generations) and presence or absence of preexisting population structure prior to population isolation. To simulate absence of population structure, panmictic populations were immediately isolated to yield an abrupt fragmentation event with the highest likelihood of being detected. To more closely reflect realistic conditions, we simulated preexisting population structure by limited seed and pollen migration as described below for 500 generations prior to stopping all migration.
At least 85% of pollen grains remained within a population and 15% had some probability of moving. Probability of dispersal from a population followed a Laplace distribution (μ = 0.4, b = 0), a commonly used dispersal kernel for plants that reflects a range of common dispersal syndromes [96]–[99]. Seeds produced from matings within populations could either stay within the population in which they were generated or they could disperse. Probability of dispersal followed the same dispersal kernel described above. After the dispersal step, seeds had a 10% chance of germinating the year after they were produced and their ultimate fate depended on whether or not generations overlapped. Although the specific values for seed production, seed germination, and pollen and seed dispersal were arbitrary, they were within the range of values that have been documented for plant species [100]–[106].
Simulations with preexisting population structure ran under the above conditions for 500 generations prior to complete isolation, those that began from panmixia were immediately isolated. Following isolation in both simulation types, the model proceeded for 200 additional generations with no migration among the 6 populations. We conducted 200 independent simulations for each of the four conditions for each of the six population size caps, yielding 24 model configurations. The resulting 4,800 simulations were run on The Lattice Project, a Grid computing system [107]–[110].
During simulations, individual populations were allowed to go extinct and to be recolonized with migrants from other populations (when migration was allowed) or from the seed bank (when overlapping generations were present). At small population sizes, individual populations would frequently go extinct. When all populations went extinct, the simulation was restarted. However, extinction of all six populations occurred in only ∼1/100 cases. We determined the total number of alleles, observed (H o) and expected (H e) heterozygosity at each generation.
In simulations without overlapping generations, we calculated the inbreeding N
e at each generation as 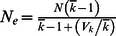 where
where 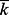 is the mean number of progeny and
is the mean number of progeny and 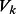 is the variance in the number of progeny at each generation [111]. In simulations with overlapping generations, N
e was calculated as N
e = T(Nb) where T is generation time defined as the average age of parents including dormancy [112] calculated following Vitalis et al. [113] and N
b is the effective number of breeders in a given year [114]. Effective population size for each population, and for each run was calculated as the harmonic mean across all generations and then averaged across simulation runs.
is the variance in the number of progeny at each generation [111]. In simulations with overlapping generations, N
e was calculated as N
e = T(Nb) where T is generation time defined as the average age of parents including dormancy [112] calculated following Vitalis et al. [113] and N
b is the effective number of breeders in a given year [114]. Effective population size for each population, and for each run was calculated as the harmonic mean across all generations and then averaged across simulation runs.
At each generation we calculated Weir and Cockerham’s [115] unbiased estimate θ, Hedrick’s G’ st [86], Sherwin’s standardized MI, and Jost’s D [85] using the estimator D est_Chao following Chao et al. [116]. We estimated the four measures from the total number of individuals using all 20 loci at each generation to provide the census or “true” estimate of θ, G’ st, MI, and D est_Chao for comparison with the subsamples of individuals and loci discussed below.
We assessed the number of generations required for θ, G’ st, MI, and D est_Chao to reach equilibrium by visually assessing asymptotic behavior. We used Fisher’s exact tests to assess whether each estimated value was significantly different from 0, assuming individuals were members of a global population and then randomly reallocated to populations while maintaining sample sizes at the realized values, and recalculating each statistic [117]. The actual value for each run was compared with the distribution of 2000 such randomizations to obtain a p-value. The number of generations after population isolation at which θ, G’ st, MI, and D est_Chao became significantly different from values at the last time-step with gene flow was tested using a one-way Dunnet multiple mean comparison test in R v2.14.1 [118]. To determine the power to detect differences we calculated the proportion of runs at each generation that was significantly different from 0. The magnitude and rate of change between consecutive generations was calculated for the first 24 generations following fragmentation for all simulations.
We sampled factorial combinations of 10, 15, and 20 loci, and 20, 30, and 50 individuals (as allowed by total maximum population sizes) at every generation over the course of each simulation run. To evaluate the effect of sample size on potential to detect fragmentation, we compared estimates of θ, G’ st, MI, and D est_Chao calculated for all factorial combinations of individuals and loci to the corresponding census value using a Tukey multiple comparison test in R. In addition, we tested estimates of each measure from all factorial combinations for significant departure from 0 using the methods described above.
Results
All Individuals and Loci
As expected, the number of alleles, H o and He tended to be higher through time in larger populations (Figure 1). Model runs with overlapping and non-overlapping generations yielded similar average allelic diversity for any given N max (2–42 alleles per locus). However, model runs with overlapping generations tended to yield higher average H o and He through time than did runs with non-overlapping generations, and differences were more pronounced at smaller N max (Figure 1).
Figure 1. Values of Na, Ho, and He for 20 loci and all individuals across all simulation conditions.

Lines from top to bottom represent the N max’s of 3000, 1000, 500, 100, 75, and 25 individuals.
In absence of overlapping generations, the harmonic mean values of N e estimates for each of the six subpopulations based on all individuals averaged over all runs were 13, 40, 52, 265, 531, 1601 individuals. These N e values represented roughly half the actual N max values of 25, 75, 100, 500, 1000, and 3000, respectively. With overlapping generations, the harmonic mean of N e estimates for each subpopulation averaged over all runs was roughly twice the N max: 43, 143, 193, 975, 1994, 5994 individuals, respectively.
As expected from theory, behavior of θ, G’ st, MI, and D est_Chao at a given time point depended on three factors: N max, presence or absence of overlapping generations, and presence or absence of population sub-structuring prior to fragmentation. Smaller N max predictably yielded larger values for any given time step (Figures 2, 3, 4, 5) except for D est_Chao when N max = 25 and generations did not overlap. For a given N max, measures were most often lower in simulations with overlapping generations than those without (Figure 2, 3, 4, 5). In simulations with population sub-structuring prior to fragmentation, θ and G’st values followed similar trajectories to those in which isolation occurred immediately after a period of panmixia (Figures 2 & 3). D est_Chao and MI values after isolation were lower when prior population sub-structuring was included whereas θ and G’st were of similar magnitude (Figures 4 & 5).
Figure 2. Average θ calculated from all individuals through time for all N max sizes.

Negative generations indicate generations with migration prior to the fragmentation event.
Figure 3. Average G’st calculated from all individuals through time for all N max sizes.

Negative generations indicate generations with migration prior to the fragmentation event.
Figure 4. Average MI calculated from all individuals through time for all N max sizes.

Negative generations indicate generations with migration prior to the fragmentation event.
Figure 5. Average Dest_Chao calculated from all individuals through time for all N max sizes.

Negative generations indicate generations with migration prior to the fragmentation event.
Across all simulations, values of G’ st, MI, and D est_Chao were generally larger than θ under the same conditions when there was no limited migration prior to isolation (Figures 2, 3, 4, 5). When population sub-structuring preceded fragmentation and generations did not overlap, the magnitudes of MI and Dest_Chao were lower than θ for N max = 25, across all 200 generations of isolation both with and without overlapping generations (Figures 4 & 5). When G’st, MI, and Dest_Chao were calculated for N max = 25 with non-overlapping generations and population sub-structuring, it had a similar rate of increase to N max = 75 and N max = 100 under the same conditions (Figure 3, 4, 5). We found two additional anomalies: a small peak in G’ st, MI, and D est_Chao existed at the start of simulations that included migration when N max ≤100 individuals (Figure 3, 4, 5).
An asymptote in values of all four measures is expected as mutation-drift equilibrium is reached [85], [86]. For θ, this asymptote was not reached during the 200 generation model runs when generations overlapped (i.e., with or without prior migration; Figure 2). For simulations without overlapping generations (with or without prior migration, θ values reached equilibrium after 60 generations when N max = 25 individuals, and approached equilibrium by the 200th generation, when N max = 75 or 100 individuals (Figure 2). G’ st, MI , and D est_Chao failed to reach equilibrium with either overlapping or non-overlapping generations when there was prior migration, but rapidly did so for N max ≤100 when isolation occurred from panmixia (Figures 3 & 5). Thus, when N max ≥500 individuals, there was no asymptote in θ, G’ st, MI, or D est_Chao values within time scales that would affect monitoring of anthropogenic landscape change, under any of the simulation conditions.
When calculated using all loci and individuals, it took two generations after cessation of gene flow for all measures to become significantly different from zero in runs starting from panmixia and from the magnitude at the final time step with migration in the runs with pre-existing structure (Table 1). For the four combinations of pre-existing structure versus panmixia and overlapping versus non-overlapping generations, the magnitude of θ, when it became significant following the fragmentation event, was between 3.4×10−4 and 0.059. The magnitude of change in G’st at the point of significance was between 0.003 and 0.30 depending on the case. At the same time point, the magnitude of change in MI was between 0.003 and 0.19 and in D est_Chao was between 0.003 and 0.47. Regardless of the simulated conditions, when N max>500 the absolute magnitude of change between generations was exceedingly small (θ<0.0033, G’st ∼0.03, MI, ∼0.001, D est_Chao<0.03).
Table 1. Difference in mean θ, G’st, MI and D values between the final migration step and 2 generations following cessation of migration for 200 runs under each set of simulation conditions based on full census of individuals and loci.
| Overlapping Generations With Migration | Non-Overlapping Generations With Migration | ||||||||
| N max | Magnitude of Difference in F st | Magnitude of Difference in G’st | Magnitude of Difference in MI | Magnitude of Difference in D | N max | Magnitude of Difference in F st | Magnitude of Difference in G’st | Magnitude of Difference in MI | Magnitude of Difference in D |
| 25 | 0.02684 | 0.04738 | 0.01904 | 0.03736 | 25 | 0.05911 | 0.1125 | 0.02398 | 0.09798 |
| 75 | 0.01032 | 0.03271 | 0.01494 | 0.02575 | 75 | 0.02019 | 0.04684 | 0.0212 | 0.02956 |
| 100 | 0.00821 | 0.02 | 0.01376 | 0.02344 | 100 | 0.01801 | 0.03811 | 0.02079 | 0.0296 |
| 500 | 0.002 | 0.01413 | 0.00645 | 0.01417 | 500 | 0.00372 | 0.02276 | 0.01062 | 0.01858 |
| 1000 | 0.00102 | 0.00493 | 0.00399 | 0.00866 | 1000 | 0.00157 | 0.01199 | 0.00607 | 0.01078 |
| 3000 | 0.00034 | 0.00309 | 0.00168 | 0.00344 | 3000 | 0.00038 | 0.00377 | 0.00203 | 0.00339 |
| From Panmixia | From Panmixia | ||||||||
| N max | Magnitude of Difference in F st | Magnitude of Difference in G’st | Magnitude of Difference in MI | Magnitude of Difference in D | N max | Magnitude of Difference in F st | Magnitude of Difference in G’st | Magnitude of Difference in MI | Magnitude of Difference in D |
| 25 | 0.04283 | 0.26758 | 0.15631 | 0.44143 | 25 | 0.04941 | 0.30642 | 0.18879 | 0.47872 |
| 75 | 0.01416 | 0.15476 | 0.06768 | 0.20209 | 75 | 0.01615 | 0.18334 | 0.08282 | 0.22671 |
| 100 | 0.01057 | 0.12654 | 0.05034 | 0.15958 | 100 | 0.01188 | 0.14867 | 0.06441 | 0.1763 |
| 500 | 0.00209 | 0.03125 | 0.0103 | 0.03541 | 500 | 0.00235 | 0.03751 | 0.01567 | 0.04035 |
| 1000 | 0.00104 | 0.01598 | 0.00695 | 0.01763 | 1000 | 0.00117 | 0.01927 | 0.00836 | 0.02004 |
| 3000 | 0.00035 | 0.00545 | 0.00256 | 0.00595 | 3000 | 0.00039 | 0.00661 | 0.00308 | 0.00674 |
We provide results for two generations because this was the point at which there was a significant difference from the last time step with migration. All differences were significant at P<0.05.
Beyond the second generation post-isolation, the magnitude of change in G’ st, MI, and D est_Chao between generations in the scenario with highest likelihood of detection (i.e., no overlap in generations and isolation occurred from panmixia) decreased sharply for a given N max (Figure 6). The decline in changes in θ between consecutive generations was more subtle, especially through generation 10 in smaller populations, and the overall magnitude of values was much lower until generation 14–16. The effect of N max was complicated in that changes between generations were reduced in both small populations and large populations, but for different reasons. The magnitude of change between generations when N max = 25 was relatively constant over time and was smaller than for N max = 75 because most populations of size 25 have already gone to fixation by the second generation, thus leaving no possibility for further divergence except through mutation. Populations with N max>75 had sufficient Ne sizes to prevent substantial divergence between subsequent generations. Populations with N max = 75 thus have the largest magnitude of change between generations until later generations when these populations also became fixed (Figure 6). Once fixation occurred within populations, the magnitude of change between generations decreased to ∼0.0005 for all measures. In the worst-case scenario for detecting change (overlapping generations and isolation from prior population sub-structure), the decline in magnitude across generations was pronounced for all four estimators and the effect of N max was more straightforward in that they declined with increasing population size. However, the average magnitude of those changes never exceeded 0.04 and most often was <0.02 (Figure 6) and thus would be unlikely to be detected in field situations. Results for the remaining two cases, 1) generations overlapped and isolation occurred from panmixia and 2) generations did not overlap and prior population structure was included were intermediate to the presented cases (data not shown).
Figure 6. Magnitude of change between consecutive sets of two-generations over the first 24 generations following termination of migration.

Bars from left to right are N max = 25, 75, 500 and 3000 with standard error. Note the scales in the upper panel differ from those in the lower panels.
Estimates from Samples
Values of θ, MI, and D est_Chao calculated from samples were statistically indistinguishable from the census estimate at all time points sampled, across all simulation conditions (Tukey multiple comparison tests not shown). Thus, for θ, MI, and D est_Chao the samples are unbiased and accurate estimates of the census values. In contrast, G’st for N max = 25 was significantly larger than the census for the first 2 generations following isolation when 20 individuals were sampled, regardless of the number of loci sampled. When N max = 75, G’st sample values were significantly larger than the census value for all generations when 20 individuals were sampled. Finally, when N max>75 values of G’st calculated from samples were significantly larger than the census value for all generations and for all sample sizes.
All sample size combinations were sufficient for detecting significant differences in θ, G’ st, MI, and D est_Chao values from 0 (when starting from panmixia), or the value prior to isolation (when prior migration was allowed) in 100% of replicates at generation 2 when Nmax<500 (as opposed to census values, which yielded significant differences by generation 2 at all N max values). When Nmax≥500, number of individuals and loci had a large effect on power to detect differences and greatly increased the time needed to reliably detect differentiation. For example, when samples of 20 individuals and 10 loci from populations with N max = 3000 with overlapping generations and isolation occurring from panmixia required 60 generations for 100% of samples to be significantly different from 0. For the same sample sizes, 18 generations were required when N max = 1000, and 12 generations were required when N max = 500. When generations did not overlap the time required for 100% of replicates to be significantly different from 0 was reduced by 50–66% (Figures 7 and 8). It took slightly longer for all samples to be significantly different from pre-isolation values when prior population structure was included (data not shown). The time required to detect a value greater than zero decreased with either larger numbers of individuals or numbers of loci (Figure 8). The addition of 10 sampled loci provided an equivalent gain to that provided by addition of 10–20 sampled individuals (Table 2).
Figure 7. Percentage of 200 replicate runs that yielded significant θ, G’st, MI, and Dest_Chao values beginning at two generations after the cessation of migration from panmixia for 20 sampled individuals and 10 sampled loci in populations overlapping generations and non-overlapping generations.

Open bars N max = 500, closed bars N max = 1000, gray bars N max = 3000.
Figure 8. Effect of number of individuals (20, 30 and 50), number of loci (10, 15, 20), and overlapping versus non-overlapping generations on the percentage of the 200 replicate runs that yielded significant Dest_Chao values 2 to 50 generations after cessation of migration for N max = 3000.

Closed bars 10 loci, open bars 15 loci, grey bars 20 loci. Data for θ, MI, and G’st are nearly identical and are not shown.
Table 2. Percentage of 200 replicate runs that yielded significant θ values 2 generations after the cessation of migration for all factorial combinations of sampled individuals and loci.
| Overlapping Generations | Non-Overlapping Generations | |||||||
| N max | Number Sampled Loci | 20 Sampled Individuals | 30 Sampled Individuals | 50 Sampled Individuals | Number Sampled Loci | 20 Sampled Individuals | 30 Sampled Individuals | 50 Sampled Individuals |
| 500 | 10 | 45 | 80.5 | 98.5 | 10 | 60.5 | 85 | 99.5 |
| 15 | 57.5 | 85.5 | 100 | 15 | 68 | 93 | 100 | |
| 20 | 73 | 96 | 100 | 20 | 85 | 98.5 | 100 | |
| 1000 | 10 | 22 | 28 | 63.5 | 10 | 25 | 37 | 73 |
| 15 | 26 | 41.5 | 81.5 | 15 | 31.5 | 49 | 87 | |
| 20 | 35.5 | 56.5 | 82.5 | 20 | 38 | 58 | 94 | |
| 3000 | 10 | 3.5 | 12.5 | 15.5 | 10 | 10.5 | 14.5 | 17.5 |
| 15 | 11.5 | 16.5 | 21 | 15 | 13.5 | 15.5 | 27 | |
| 20 | 9 | 15 | 25 | 20 | 13.5 | 11.5 | 31 | |
Discussion
Ideally, detecting changes in connectivity will provide early warning that biologically relevant habitat fragmentation has occurred so management action can be taken before the consequences become irreversible [51], [119]. The potential utility of indirect genetic methods for this purpose relies on a substantial and significant increase in genetic divergence following the end of migration relative to preexisting structure, as well the ability to detect that change under realistic field sampling conditions. We documented changes in θ, G’ st, MI, and D est_Chao of sufficient magnitude for detection (e.g., >0.05) under several combinations of population size and life history in our models. However, because the conditions under which changes in gene flow are likely to be detected by any of the measures were fairly restricted and because the values that could indicate fragmentation had occurred can also be obtained with natural subdivision. As such we suggest that these measures alone are likely to be problematic for confirming changes in landscape connectivity in time frames that will inform management.
On the positive side, all census estimates of θ, G’ st, MI, and D est_Chao were significantly different from 0 and from pre-fragmentation values within 2 generations of isolation when populations supported <500 individuals. This result is substantially more optimistic than that of Landguth et al. [63], which suggested that >100 generations were required for F st to indicate fragmentation of a continuous population of 1000 individuals divided in half by a barrier to gene flow. Because of the lag time in response of F st they recommend using Mantel’s r, which required only 1–15 generations for detection based on approaching equilibrium [63], and mention that G’st responds more similarly to Mantel’s r, but provided no corroborating data. Because they did not report the magnitude of change in the metrics or effective population sizes, further direct or detailed comparisons with our results are not possible. Our timeframe for potential detection of significant differences was also similar to that found by England et al. [62] based on changes in N e from a single population of 1000 that was abruptly isolated into 10 demes of 100. Our model differs from both Landguth et al. [63] and England et al. [62] in that we altered connectivity alone while maintaining population size whereas they simultaneously changed connectivity and population size.
Although we obtained significant differences within two generations, the magnitudes of the differences were often so small that detection in the field could be difficult. Magnitudes were largely dependent on the size and demography of the populations under investigation. In the best-case scenario for detecting change (N max = 25 with no overlap in generations and isolation occurring directly from panmixia), the magnitude of θ two generations after isolation compared with the last generation with migration increased by 0.049 resulting in an average θ value of 0.04, which would be difficult to detect as biologically significant. In the same scenario, G’st increased by an average of 0.3 (resulting in an average G’st of 0.56), MI increased by an average of 0.18 (giving a average MI of 0.31) and Jost’s D increased on average by 0.42 (yielding an average value of 0.45) greatly increasing the potential for detection relative to θ estimates. In populations with ≥500 individuals, the change in θ from prior to fragmentation to the second generation post-fragmentation was ≤0.002, which would be viewed as biologically insignificant. Although G’st, MI, and Jost’s D all had larger magnitude increases for the same scenario (Table 1), detecting the increases could still be difficult. In the most difficult circumstances for detecting change (when a seed bank was present and population sub-structuring was established prior to isolation) none of the changes in θ, G’ st, MI, nor D est_Chao exceeded 0.04 within two generations, which is well within the range of sampling error in real populations [81], [120], [121]. The lower rate of change in presence of a seedbank is likely due to the doubling of the effective population size that occurred under these conditions. In total, these results indicate that detecting change from a baseline condition in two generations will be possible only when populations are <500 individuals and only when generations do not overlap.
As a practical matter, detection of changes in genetic structure due to fragmentation presumes having samples that represent conditions prior to fragmentation for comparison. It is more likely that connectivity will be assessed only after changes in habitat amount and configuration have occurred because most often species are not studied prior to becoming of conservation concern. Despite the fact that genetic monitoring by definition requires a multi-year approach to be effective [51], few published studies of fragmentation have included such temporal sampling e.g., [122]–[127], and even these efforts have generally not extended more than a few generations. Without pre-fragmentation data, it is not possible to attribute significant values of genetic differentiation measures to anthropogenic changes because such values can result from natural subdivision of smaller populations [128]. For example, θ values at the second generation post-isolation when N max>500 individuals and there is no prior migration, were identical to cases with limited ongoing migration when N max≤100 individuals (Figure 2). Without having precise population size estimates, it would not be possible to determine whether a given θ, G’ st, MI, or D est_Chao value reflected small population size with a low level of migration or lack of migration among larger populations.
The lack of data collected prior to landscape change can be overcome by sampling from multiple demographic cohorts representing generations that originated before and after the putative fragmentation event by, for example, examining differences between adults and more recent recruits [129] or sampling across strata in a soil seed bank [130]. Several approaches can potentially overcome lack of pre-fragmentation data when sampling demographic cohorts is not possible. Chiucchi and Gibbs [128] have suggested comparing estimates of gene flow from multiple analytical approaches that reflect different time frames as a way to compare long-term and short-term levels of differentiation from a single sample. Another approach is to compare multiple populations from similar interpatch distances in different habitat matrix types in which there is strong contrast in gene flow, or in locations with versus without barriers to gene flow, or by sampling locations at varying distances from one another [131]–[133]. Alternatively, one can sample the same populations at multiple time points after landscape change and quantify the amount of change in divergence between generations. In absence of recent change, populations are expected to be at migration drift equilibrium, at which point changes between generations will be minimal (e.g, cases with limited migration, pre isolation in Figures 2, 3, 4, 5). After fragmentation that eliminates gene flow, rapid changes towards a new equilibrium are observed. The average magnitude of change across generations exceeded change seen in absence of fragmentation or in populations with substructuring due to limited migration prior to fragmentation (Figures 2, 3, 4, 5), indicating that samples at multiple time points after isolation could allow detection of fragmentation and thus provide a solution to the lack of pre-fragmentation data. However, this signature lasts only 8–10 generations (Figure 6) when populations are ≤100 individuals; beyond this point, post-isolation the rate of change between two consecutive generations is indistinguishable from that seen in populations prior to fragmentation even though the absolute values of θ, G’ st, MI, or D est_Chao were higher than they were pre-fragmentation. In populations with >100 individuals, divergence continued increasing for the 200 generations we modeled (Figure 6), thus providing a longer temporal window for detecting changes across generations. However, when N max≥500 the magnitude of change that we observed across generations may not be large enough for detecting signatures of fragmentation in field conditions especially when generations overlap and thus a time series would be inconclusive regarding any contemporary change in genetic connectivity (Figure 5).
Additionally, for all but annual species with no seedbank, the number of years required to sample across generations could be too large to provide reasonable recommendations in timeframes that are responsive to management concerns. If generation time is 5–10 years, the 10–20 years necessary to yield a clear signal of fragmentation relative to pre-fragmentation conditions or across generations post-fragmentation does not constitute an early warning. These timeframes are also too long to be suitable for documenting if management actions have successfully reestablished connectivity in an adaptive management framework [134], which requires regular and rapid assessment of the effects of management treatments. Although we did not simulate restoration of connectivity, others have found the signature of restricted gene flow (e.g. high Fst) can persist for 15–300 generations after a barrier to gene flow is removed depending on the dispersal distances [63]. A legacy of historical isolation within currently connected populations would result in misidentifying such populations as not connected by gene flow.
Should the issues surrounding sampling within the correct time window and for a sufficient length of time be overcome, the lack of power associated with sampling subsets of individuals and loci could prevent detection of changes in genetic divergence in populations of ≥500 individuals. Below that population size, sample size had no effect on the power to detect significant genetic divergence in that 100% of runs were significantly different from pre-fragmentation values. In the extreme case (N max = 3000), when generations were overlapping it took 8 generations for at least 50% of model runs to be significantly different from zero when 50 individuals and 10 loci were sampled; this time could be reduced to 6 generations if 20 loci were sampled. In contrast, when 20 individuals and 20 loci were sampled from each population, it took more than 20 generations for 50% of runs to become significantly different from zero (Figure 8). When generations were not overlapping, these times decreased to ∼4 generations for 50 individuals and ∼8 generations for 20 sampled individuals and 20 loci (Figure 8). The tradeoffs between loci and individuals were similar to those found by England et al. [62]. Given that it is often not cost effective or feasible to obtain both additional individuals and loci, it is encouraging that both options can improve estimates. It is important to note that our recommendations apply only to use of genetic data to detect a shift in genetic connectivity and are not generalizable to all types of genetic estimates. However, our results indicate the need for sample sizes for large populations that are similar to those recommended for reliable and unbiased estimates of trends in effective population sizes (a minimum of 60 individuals, sampled at least 5 years apart, and genotyped at 15 loci [135]).
In general, over the first few generations after isolation we found that D est_Chao MI, and G’st represented genetic divergence more rapidly than did Wright’s F st across all simulation conditions. This is not that surprising given that these three measures avoid biases related to high sample heterozygosity [85]–[87] in that D est_Chao and MI are calculated directly from allele frequencies and G’st controls for maximal observed heterozygosity. Although there has been disagreement surrounding the appropriateness of use of D est_Chao to the exclusion of heterozygosity-based measures [136], [137], it has been shown to behave appropriately across a wide range of allele diversities, heterozygosities, and mutation rates [85], [88], [138]. G’st and D est_Chao generally had a higher magnitude of change compared to θ, and higher overall values, except for D est_Chao when there was prior population structure occurred and N max = 25. MI was consistently lower than G’st, and D est_Chao, but was lower than θ when prior population structure was included in the model and when N max <100. The equilibrium value of MI when generations did not overlap and isolation occurred from migration was also lower than the equilibrium θ value. During the initial 70 generations when migration was occurring and N max ≤100, there was a peak in G’st, MI, and D est_Chao, which resulted from drift overwhelming migration, or from the initial increase in the number of individuals as the population cap size is reached.
Estimates of θ and G’st exceeded D est_Chao when N max was small (e.g., N max = 25) and migration was present prior to isolation. The combination of small population size and migration lead to fixation of common alleles in several populations. The pattern of fixation is what subsequently resulted in inflation of θ and G’st relative to D est_Chao. Because Wright’s F st and G’st are based on heterozygosity, which does not account for particular allelic states, identical alleles that are fixed within multiple populations do not contribute to within-population heterozygosity and thus contribute to inflation of values of θ and G’st are unable to account for the shared alleles and is therefore artificially high (Table 3). The magnitude of θ and G’st will be a function of the number of fixed alleles, but in all such cases θ and G’st are misrepresenting the underlying pattern of differentiation, and are consequently over estimating the degree of genetic differentiation relative to Dest_Chao.
Table 3. Example cases of allelic composition drawn from N max = 25, which included population sub-structuring; values calculated for θ, G’st, and Dest_Chao from these sample data.
| Fst | 0 | 1 | 1 | 1 | 1 |
| G’st | 0 | 1 | 1 | 1 | 1 |
| D | 0 | 0.5 | 0.6 | 0.8 | 1 |
| Pop 1 | A/A | A/A | A/A | A/A | A/A |
| A/A | A/A | A/A | A/A | A/A | |
| A/A | A/A | A/A | A/A | A/A | |
| Pop 2 | A/A | A/A | A/A | A/A | B/B |
| A/A | A/A | A/A | A/A | B/B | |
| A/A | A/A | A/A | A/A | B/B | |
| Pop 3 | A/A | A/A | B/B | B/B | C/C |
| A/A | A/A | B/B | B/B | C/C | |
| A/A | A/A | B/B | B/B | C/C | |
| Pop 4 | A/A | B/B | B/B | C/C | D/D |
| A/A | B/B | B/B | C/C | D/D | |
| A/A | B/B | B/B | C/C | D/D |
The fixation of common alleles removes all heterozygosity and results in inflated estimates of genetic differentiation when using F st and G’st as opposed to D.
Our results from samples show that even relatively few sampled individuals (20) or loci (10) provided unbiased estimates of θ, MI, and D est_Chao. In contrast, when population sizes were ≥100, estimates of G’st were always significantly larger than the census values regardless of the number of sampled individuals or loci. As proposed by Hedrick [86], we calculated G’st based solely on measures of heterozygosity, and did not include any adjustments for differences in population size. Without controlling for population size, it is not surprising that the values of G’st were biased relative to the census values. It is possible to calculate a corrected fixation index that accounts for bias that arises when from sampling a limited number of populations [139]. However, the method we used to calculate G’st is the formulation commonly calculated in population genetic software e.g., SMOGD [140]; GenoDive [141], is what was originally proposed by Hedrick [86], and was the formulation used in comparison of metrics conducted by Heller and Siegismund [90].
To conclude, we find that use of F st-related statistics, G’st, MI, or D for detecting and monitoring changes in connectivity among discrete populations is problematic in many real world scenarios. The conditions under which these indirect methods can best be applied include when populations support between 75 and 500 individuals, when sampling is done across multiple generations, and estimates of population size are available to allow distinguishing of the signal of background differentiation from changes associated with the loss of genetic connectivity. This multi-generation sampling must occur within the window during which rapid change in the estimators is occurring to yield conclusive results. Unfortunately, the number of years required to span a sufficient number of generations to detect a change may preclude utility. For these reasons, we caution against using indirect techniques alone for detection of fragmentation events, and advocate their use only in conjunction with direct estimates of actual movement among patches such that one could compare current movement with the genetic signature of past movement to determine that there has been a change.
Acknowledgments
The authors thank J. Pettengill, R. Burrnett, S. Ziegler, E. Lind, C. Kennedy, L. Jost, and R. Waples for helpful discussions during the early development of the model, and subsequent analyses. A. Bazinet for help in the use of the Grid computing resources and providing scripting advice. The Perlmonk online community for scripting advice.
Funding Statement
The authors have no funding or support to report.
References
- 1. Rouget M, Richardson DM, Cowling RM, Lloyd JW, Lombard AT (2003) Current patterns of habitat transformation and future threats to biodiversity in terrestrial ecosystems of the Cape Floristic Region, South Africa. Biol Conserv 112: 63–85. [Google Scholar]
- 2. Wilcove DS, Rothstein D, Dubow J, Phillips A, Losos E (1998) Quantifying threats to imperiled species in the United States. Bioscience 48: 607–615. [Google Scholar]
- 3. McKinney ML (2002) Urbanization, biodiversity, and conservation. Bioscience 52: 883–890. [Google Scholar]
- 4. Lawler JJ, Campbell SP, Guerry AD, Kolozsvary MB, O'Connor RJ, et al. (2002) The scope and treatment of threats in endangered species recovery plans. Ecol Appl 12: 663–667. [Google Scholar]
- 5. Fahrig L (2003) Effects of habitat fragmentation on biodiversity. Annu Rev Ecol Evol Syst 34: 487–515. [Google Scholar]
- 6. Saunders DA, Hobbs RJ, Margules CR (1991) Biological consequences of ecosystem fragmentation - a review. Conserv Biol 5: 18–32. [Google Scholar]
- 7. McGarigal K, McComb WC (1995) Relationships between landscape structure and breeding birds in the Oregon coast range. Ecol Monogr 65: 235–260. [Google Scholar]
- 8. Tischendorf L, Fahrig L (2000) How should we measure landscape connectivity? Landsc Ecol 15: 633–641. [Google Scholar]
- 9. Pascual-Hortal L, Saura S (2006) Comparison and development of new graph-based landscape connectivity indices: Towards the prioritization of habitat patches and corridors for conservation. Landsc Ecol 21: 959–967. [Google Scholar]
- 10. Neel MC, Mcgarigal K, Cushman SA (2004) Evaluation of class-level landscape structure metrics across gradients of area and aggregation in neutral landscapes. Landsc Ecol 19: 435–455. [Google Scholar]
- 11. Kennedy CM, Marra PP, Fagan WF, Neel MC (2010) Landscape matrix and species traits mediate responses of Neotropical resident birds to forest fragmentation in Jamaica. Ecol Monogr 80: 651–669. [Google Scholar]
- 12. Ricketts TH (2001) The matrix matters: Effective isolation in fragmented landscapes. Am Nat 158: 87–99. [DOI] [PubMed] [Google Scholar]
- 13. Bender DJ, Contreras TA, Fahrig L (1998) Habitat loss and population decline: A meta-analysis of the patch size effect. Ecology 79: 517–533. [Google Scholar]
- 14. Fahrig L, Jonsen I (1998) Effect of habitat patch characteristics on abundance and diversity of insects in an agricultural landscape. Ecosystems 1: 197–205. [Google Scholar]
- 15. Trzcinski MK, Fahrig L, Merriam G (1999) Independent effects of forest cover and fragmentation on the distribution of forest breeding birds. Ecol Appl 9: 586–593. [Google Scholar]
- 16. Fahrig L, Merriam G (1985) Habitat patch connectivity and population survival. Ecology 66: 1762–1768. [Google Scholar]
- 17. Belisle M, Clair CCS (2002) Cumulative effects of barriers on the movements of forest birds. Conserv Ecol 5: 480–495. [Google Scholar]
- 18. Brooks TM, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Rylands AB, et al. (2002) Habitat loss and extinction in the hotspots of biodiversity. Conserv Biol 16: 909–923. [Google Scholar]
- 19. Bender DJ, Tischendorf L, Fahrig L (2003) Using patch isolation metrics to predict animal movement in binary landscapes. Landsc Ecol 18: 17–39. [Google Scholar]
- 20. Fahrig L (1997) Relative effects of habitat loss and fragmentation on population extinction. J Wildl Manag 61: 603–610. [Google Scholar]
- 21. Fahrig L (2002) Effect of habitat fragmentation on the extinction threshold: A synthesis. Ecol Appl 12: 346–353. [Google Scholar]
- 22. Burkey TV, Reed DH (2006) The effects of habitat fragmentation on extinction risk: Mechanisms and synthesis. Songklanakarin J Sci Tech 28: 9–37. [Google Scholar]
- 23. Parker M, MacNally R (2002) Habitat loss and the habitat fragmentation threshold: An experimental evaluation of impacts on richness and total abundances using grassland invertebrates. Biol Conserv 105: 217–229. [Google Scholar]
- 24. Cale PG (2003) The influence of social behaviour, dispersal and landscape fragmentation on population structure in a sedentary bird. Biol Conserv 109: 237–248. [Google Scholar]
- 25. Patten MA, Wolfe DH, Shochat E, Sherrod SK (2005) Habitat fragmentation, rapid evolution and population persistence. Evol Ecol Res 7: 235–249. [Google Scholar]
- 26. Harrison S, Bruna E (1999) Habitat fragmentation and large-scale conservation: What do we know for sure? Ecography 22: 225–232. [Google Scholar]
- 27. Hovel KA, Lipcius RN (2001) Habitat fragmentation in a seagrass landscape: Patch size and complexity control blue crab survival. Ecology 82: 1814–1829. [Google Scholar]
- 28. Jules ES (1998) Habitat fragmentation and demographic change for a common plant: Trillium in old-growth forest. Ecology 79: 1645–1656. [Google Scholar]
- 29. With KA (2004) Assessing the risk of invasive spread in fragmented landscapes. Risk Anal 24: 803–815. [DOI] [PubMed] [Google Scholar]
- 30. Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10: 1500–1508. [Google Scholar]
- 31. Frankham R (1995) Conservation genetics. Annu Rev Genet 29: 305–327. [DOI] [PubMed] [Google Scholar]
- 32. Saccheri I, Kuussaari M, Kankare M, Vikman P, Fortelius W, et al. (1998) Inbreeding and extinction in a butterfly metapopulation. Nature 392: 491–494. [Google Scholar]
- 33. Westemeier RL, Brawn JD, Simpson SA, Esker TL, Jansen RW, et al. (1998) Tracking the long-term decline and recovery of an isolated population. Science 282: 1695–1698. [DOI] [PubMed] [Google Scholar]
- 34. Hargis CD, Bissonette JA, David JL (1998) The behavior of landscape metrics commonly used in the study of habitat fragmentation. Landsc Ecol 13: 167–186. [Google Scholar]
- 35. Saura S, Martinez-Millan J (2001) Sensitivity of landscape pattern metrics to map spatial extent. Photo Eng Remote Sens 67: 1027–1036. [Google Scholar]
- 36. Jaeger JAG (2000) Landscape division, splitting index, and effective mesh size: New measures of landscape fragmentation. Landsc Ecol 15: 115–130. [Google Scholar]
- 37. Schumaker NH (1996) Using landscape indices to predict habitat connectivity. Ecology 77: 1210–1225. [Google Scholar]
- 38. Gustafson EJ, Parker GR (1994) Using an index of habitat patch proximity for landscape design. Landsc Urban Plann 29: 117–130. [Google Scholar]
- 39.McGarigal K, Cushman SA, Neel MC, Ene E (2002) FRAGSTATS: Spatial pattern analysis program for categorical maps. Computer software program produced by the authors at the University of Massachusetts, Amherst. Available at the following web site: http://www.umass.edu/landeco/research/fragstats/fragstats.html. Accessed 2013 Apr 22.
- 40. Urban D, Keitt T (2001) Landscape connectivity: A graph-theoretic perspective. Ecology 82: 1205–1218. [Google Scholar]
- 41. Tischendorf L, Fahrig L (2000) On the usage and measurement of landscape connectivity. Oikos 90: 7–19. [Google Scholar]
- 42.Taylor PD, Fahrig L, With KA (2006) Landscape connectivity: A return to the basics. In: Crooks KR, Sanjayan M, editors. Connectivity Conservation. New York: Cambridge University Press. 29–43.
- 43. Collingham YC, Huntley B (2000) Impacts of habitat fragmentation and patch size upon migration rates. Ecol Appl 10: 131–144. [Google Scholar]
- 44. Dooley JL, Bowers MA (1998) Demographic responses to habitat fragmentation: Experimental tests at the landscape and patch scale. Ecology 79: 969–980. [Google Scholar]
- 45. Bowers MA, Dooley JL (1999) A controlled, hierarchical study of habitat fragmentation: Responses at the individual, patch, and landscape scale. Landsc Ecol 14: 381–389. [Google Scholar]
- 46. Bruna EM, Oli MK (2005) Demographic effects of habitat fragmentation on a tropical herb: Life-table response experiments. Ecology 86: 1816–1824. [Google Scholar]
- 47. Calabrese JM, Fagan WF (2004) A comparison-shopper's guide to connectivity metrics. Front Ecol Environ 2: 529–536. [Google Scholar]
- 48. Urban DL (2005) Modeling ecological processes across scales. Ecology 86: 1996–2006. [Google Scholar]
- 49. Wunsch M, Richter C (1998) The CaveCam–an endoscopic underwater videosystem for the exploration of cryptic habitats. Mar Ecol Prog Ser 169: 277–282. [Google Scholar]
- 50. Ellstrand NC (1992) Gene flow by pollen - implications for plant conservation genetics. Oikos 63: 77–86. [Google Scholar]
- 51. Schwartz MK, Luikart G, Waples RS (2007) Genetic monitoring as a promising tool for conservation and management. Trends Ecol Evol 22: 25–33. [DOI] [PubMed] [Google Scholar]
- 52. Ims RA, Andreassen HP (1999) Effects of experimental habitat fragmentation and connectivity on root vole demography. Journal of Animal Ecology 68: 839–852. [DOI] [PubMed] [Google Scholar]
- 53. Sork VL, Nason J, Campbell DR, Fernandez JF (1999) Landscape approaches to historical and contemporary gene flow in plants. Trends Ecol Evol 14: 219–224. [DOI] [PubMed] [Google Scholar]
- 54. Robledo-Arnuncio JJ, Austerlitz F, Smouse PE (2006) A new method of estimating the pollen dispersal curve independently of effective density. Genetics 173: 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Coates DJ, Atkins KA (2001) Priority setting and the conservation of Western Australia's diverse and highly endemic flora. Biol Conserv 97: 251–263. [Google Scholar]
- 56. Diniz-Filho JA, De Campos Telles MP (2002) Spatial autocorrelation analysis and the identification of operational units for conservation in continuous populations. Conserv Biol 16: 924–935. [Google Scholar]
- 57. Kendall KC, Stetz JB, Boulanger J, Macleod AC, Paetkau D, et al. (2009) Demography and genetic structure of a recovering grizzly bear population. J Wildl Manag 73: 3–17. [Google Scholar]
- 58. Kingman JFC (1982) The coalescent. Stoch Proc Appl 13: 235–248. [Google Scholar]
- 59. Pearse DE, Crandall KA (2004) Beyond Fst: Analysis of population genetic data for conservation. Conserv Genet 5: 585–602. [Google Scholar]
- 60. Slatkin M (1991) Inbreeding coefficients and coalescence times. Genet Res 58: 167–175. [DOI] [PubMed] [Google Scholar]
- 61. Kingman JFC (1982) On the genealogy of large populations. J Appl Prob 19A: 27–43. [Google Scholar]
- 62. England PR, Luikart G, Waples RS (2010) Early detection of population fragmentation using linkage disequilibrium estimation of effective population size. Conserv Genet 11: 2425–2430. [Google Scholar]
- 63. Landguth EL, Cushman SA, Schwartz MK, McKelvey KS, Murphy M, et al. (2010) Quantifying the lag time to detect barriers in landscape genetics. Mol Ecol 19: 4179–4191. [DOI] [PubMed] [Google Scholar]
- 64. Wright S (1951) The genetical structure of populations. Ann Eugen 15: 323–354. [DOI] [PubMed] [Google Scholar]
- 65. Wallace LE (2002) Examining the effects of fragmentation on genetic variation in Platanthera leucophaea (Orchidaceae): Inferences from allozyme and random amplified polymorphic DNA markers. Plant Species Biology 17: 37–49. [Google Scholar]
- 66. Young AG, Brown AHD, Zich FA (1999) Genetic structure of fragmented populations of the endangered daisy Rutidosis leptorrhynchoides . Conserv Biol 13: 256–265. [Google Scholar]
- 67. Hall P, Walker S, Bawa K (1996) Effect of forest fragmentation on genetic diversity and mating system in a tropical tree, Pithecellobium elegans . Conserv Biol 10: 757–768. [Google Scholar]
- 68. Meldgaard T, Nielsen EE, Loeschcke V (2003) Fragmentation by weirs in a riverine system: A study of genetic variation in time and space among populations of European grayling (Thymallus thymallus) in a Danish river system. Conserv Genet 4: 735–747. [Google Scholar]
- 69. Hanfling B, Durka W, Brandl R (2004) Impact of habitat fragmentation on genetic population structure of roach, Rutilus rutilus, in a riparian ecosystem. Conserv Genet 5: 247–257. [Google Scholar]
- 70. Meyer CFJ, Kalko EKV, Kerth G (2009) Small-scale fragmentation effects on local genetic diversity in two phyllostomid bats with different dispersal abilities in Panama. Biotropica 41: 95–102. [Google Scholar]
- 71. Li WX, Wang GT, Nie R (2008) Genetic variation of fish parasite populations in historically connected habitats: Undetected habitat fragmentation effect on populations of the nematode Procamallanus fulvidraconis in the catfish Pelteobagrus fulvidraco . J Parasitol 94: 643–647. [DOI] [PubMed] [Google Scholar]
- 72. Krauss J, Schmitt T, Seitz A, Steffan-Dewenter I, Tscharntke T (2004) Effects of habitat fragmentation on the genetic structure of the monophagous butterfly Polyommatus coridon along its northern range margin. Mol Ecol 13: 311–320. [DOI] [PubMed] [Google Scholar]
- 73. Matern A, Desender K, Drees C, Gaublomme E, Paill WG, et al. (2009) Genetic diversity and population structure of the endangered insect species Carabus variolosus in its western distribution range: Implications for conservation. Conserv Genet 10: 391–405. [Google Scholar]
- 74. Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11: 413–418. [DOI] [PubMed] [Google Scholar]
- 75. Wiegand T, Moloney KA, Naves J, Knauer F (1999) Finding the missing link between landscape structure and population dynamics: A spatially explicit perspective. Am Nat 154: 605–627. [DOI] [PubMed] [Google Scholar]
- 76. Wiens JA, Stenseth NC, Van Horne B, Ims RA (1993) Ecological mechanisms and landscape ecology. Oikos 66: 369–380. [Google Scholar]
- 77. Holland JD, Bert DG, Fahrig L (2004) Determining the spatial scale of species' response to habitat. Bioscience 54: 227–233. [Google Scholar]
- 78. Levin SA (1992) The problem of pattern and scale in ecology. Ecology 73: 1943–1967. [Google Scholar]
- 79.Crooks KR, Sanjayan M (2006) Connectivity conservation: Maintaining connections for nature. In: Crooks KR, Sanjayan M, editors. Connectivtiy Conservation. New York: Cambridge University Press. 1–19.
- 80. Bossart JL, Prowell DP (1998) Genetic estimates of population structure and gene flow: Limitations, lessons and new directions. Trends Ecol Evol 13: 202–206. [DOI] [PubMed] [Google Scholar]
- 81. Whitlock MC, McCauley DE (1999) Indirect measures of gene flow and migration: Fst not equal 1/(4Nm+1). Heredity 82: 117–125. [DOI] [PubMed] [Google Scholar]
- 82. Whitlock MC (1992) Temporal fluctuations in demographic parameters and the genetic variance among populations. Evolution 46: 608–615. [DOI] [PubMed] [Google Scholar]
- 83. Neigel JE (2002) Is Fst Obsolete? Conserv Genet 3: 167–173. [Google Scholar]
- 84. Allendorf FW (1986) Genetic drift and the loss of alleles versus heterozygosity. Zoo Biol 5: 181–190. [Google Scholar]
- 85. Jost L (2008) Gst and its relatives do not measure differentiation. Mol Ecol 17: 4015–4026. [DOI] [PubMed] [Google Scholar]
- 86. Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59: 1633–1638. [PubMed] [Google Scholar]
- 87. Meirmans PG (2006) Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 60: 2399–2402. [PubMed] [Google Scholar]
- 88. Gerlach G, Jueterbock A, Kraemer P, Deppermann J, Harmand P (2010) Calculations of population differentiation based on Gst and D: Forget Gst but not all of statistics! Mol Ecol. 19: 3845–3852. [DOI] [PubMed] [Google Scholar]
- 89. Meirmans PG, Hedrick PW (2011) Assessing population structure: FST and related measures. Mol Ecol Resour 11: 5–18. [DOI] [PubMed] [Google Scholar]
- 90. Heller R, Siegismund HR (2009) Relationship between three measures of genetic differentiation Gst, Dest and G'st: How wrong have we been? Mol Ecol 18: 2080–2083. [DOI] [PubMed] [Google Scholar]
- 91. Sherwin WB, Jabot F, Rush R, Rossetto M (2006) Measurement of biological information with applications from genes to landscapes. Mol Ecol 15: 2857–2869. [DOI] [PubMed] [Google Scholar]
- 92. Sherwin WB (2010) Entropy and information approaches to genetic diversity and its expression: Genomic geography. Entropy 12: 1765–1798. [Google Scholar]
- 93. Traill LW, Bradshaw CJA, Brook BW (2007) Minimum viable population size: A meta-analysis of 30 years of published estimates. Biol Conserv 139: 159–166. [Google Scholar]
- 94. Whittaker JC, Harbord RM, Boxall N, Mackay I, Dawson G, et al. (2003) Likelihood-based estimation of microsatellite mutation rates. Genetics 164: 781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Silander JA, Pacala SW (1985) Neighborhood predictors of plant performance. Oecologia 66: 256–263. [DOI] [PubMed] [Google Scholar]
- 96. Bullock JA, Moy IL (2004) Plants as seed traps: Inter-specific interference with dispersal. Acta Oecol 25: 35–41. [Google Scholar]
- 97. Neubert MG, Caswell H (2000) Demography and dispersal: Calculation and sensitivity analysis of invasion speed for structured populations. Ecology 81: 1613–1628. [Google Scholar]
- 98. Bullock JM, Shea K, Skarpaas O (2006) Measuring plant dispersal: An introduction to field methods and experimental design. Plant Ecol 186: 217–234. [Google Scholar]
- 99. Bullock JM, Moy IL, Coulson SJ, Clarke RT (2003) Habitat-specific dispersal: Environmental effects on the mechanisms and patterns of seed movement in a grassland herb Rhinanthus minor . Ecography 26: 692–704. [Google Scholar]
- 100. Kalamees R, Zobel M (1997) The seed bank in an Estonian calcareous grassland: Comparison of different successional stages. Folia Geobot Phytotx 32: 1–14. [Google Scholar]
- 101. Kahmen S, Poschlod P (2008) Does germination success differ with respect to seed mass and germination season? Experimental testing of plant functional trait responses to grassland management. Ann Bot-London 101: 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kelly D (1989) Demography of short-lived plants in chalk grassland. III. Population stability. J Ecol 77: 785–798. [Google Scholar]
- 103. Zammit C, Zedler PH (1990) Seed yield, seed size and germination behavior in the annual Pogogyne abramsii . Oecologia 84: 24–28. [DOI] [PubMed] [Google Scholar]
- 104. Schiller JR, Zedler PH, Black CH (2000) The effect of density-dependent insect visits, flowering phenology, and plant size on seed set of the endangered vernal pool plant Pogogyne abramsii (Lamiaceae) in natural compared to created vernal pools. Wetlands 20: 386–396. [Google Scholar]
- 105. Fox LR, Steele HN, Holl KD, Fusari MH (2006) Contrasting demographies and persistence of rare annual plants in highly variable environments. Plant Ecol 183: 157–170. [Google Scholar]
- 106. Weekley CW, Menges ES, Quintana-Ascencio PF (2007) Seedling emergence and survival of Warea carteri (Brassicaceae), an endangered annual herb of the Florida scrub. Can J Bot 85: 621–628. [Google Scholar]
- 107. Myers DS, Cummings MP (2003) Necessity is the mother of invention: A simple Grid computing system using commodity tools. J Para Dist Comp 63: 578–589. [Google Scholar]
- 108.Bazinet AL, Cummings MP (2008) The Lattice Project: A Grid research and production environment combining multiple Grid computing models. In: Weber MHW, editor. Distributed & Grid Computing - Science Made Transparent for Everyone Principles, Applications and Supporting Communities Tectum. Marburg: Rechenkraft.net. 71–85.
- 109. Bazinet AL, Myers DS, Fuetsch J, Cummings MP (2007) Grid services base library: A high-level, procedural application program interface for writing Globus-based Grid services. Fut Gen Comp Syst 22: 517–522. [Google Scholar]
- 110.Myers DS, Bazinet AL, Cummings MP (2008) Expanding the reach of Grid computing: Combining Globus - and BOINC-based systems. In: Talbi E-G, Zomaya A, editors. Grids for Bioinformatics and Computational Biology, Wiley Book Series on Parallel and Distributed Computing. New York, New York: John Wiley & Sons. 71–85.
- 111.Kimura M, Crow JF (1963) Measurement of effective population number. Evolution 17: 279-&.
- 112. Nunney L (2002) The effective size of annual plant populations: The interaction of a seed bank with fluctuating population size in maintaining genetic variation. Am Nat 160: 195–204. [DOI] [PubMed] [Google Scholar]
- 113. Vitalis R, Glemin S, Olivieri I (2004) When genes go to sleep: The population genetic consequences of seed dormancy and monocarpic perenniality. Am Nat 163: 295–311. [DOI] [PubMed] [Google Scholar]
- 114. Waples RS (2002) Effective size of fluctuating salmon populations. Genetics 161: 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370. [DOI] [PubMed] [Google Scholar]
- 116. Chao A, Jost L, Chiang SC, Jiang YH, Chazdon RL (2008) A two-stage probabilistic approach to multiple-community similarity indices. Biometrics 64: 1178–1186. [DOI] [PubMed] [Google Scholar]
- 117. Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 131: 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.R Development Core Team (2010) R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
- 119. Procaccini G, Olsen JL, Reusch TBH (2007) Contribution of genetics and genomics to seagrass biology and conservation. J Exp Mar Biol Ecol 350: 234–259. [Google Scholar]
- 120. Hamrick JL, Godt MJW (1996) Effects of life history traits on genetic diversity in plant species. Proc R Soc Lond B Biol Sci 351: 1291–1298. [Google Scholar]
- 121.Avise JC (2004) Molecular Markers, Natural History, and Evolution. Sunderland, Mass.: Sinauer Associates. 684 p. p.
- 122. Morris AB, Baucom RS, Cruzan MB (2002) Stratified analysis of the soil seed bank in the cedar glade endemic Astragalus bibullatus: Evidence for historical changes in genetic structure. Am J Bot 89: 29–36. [DOI] [PubMed] [Google Scholar]
- 123. Thornhill DJ, LaJeunesse TC, Kemp DW, Fitt WK, Schmidt GW (2006) Multi-year, seasonal genotypic surveys of coral-algal symbioses reveal prevalent stability or post-bleaching reversion. Mar Biol 148: 711–722. [Google Scholar]
- 124. Barrett LG, He TH, Lamont BB, Krauss SL (2005) Temporal patterns of genetic variation across a 9-year-old aerial seed bank of the shrub Banksia hookeriana (Proteaceae). Mol Ecol 14: 4169–4179. [DOI] [PubMed] [Google Scholar]
- 125. Hoffman EA, Blouin MS (2004) Historical data refute recent range contraction as cause of low genetic diversity in isolated frog populations. Mol Ecol 13: 271–276. [DOI] [PubMed] [Google Scholar]
- 126. Nussey DH, Coltman DW, Coulson T, Kruuk LEB, Donald A, et al. (2005) Rapidly declining fine-scale spatial genetic structure in female red deer. Mol Ecol 14: 3395–3405. [DOI] [PubMed] [Google Scholar]
- 127. Poulsen NA, Nielsen EE, Schierup MH, Loeschcke V, Gronkjaer P (2006) Long-term stability and effective population size in North Sea and Baltic Sea cod (Gadus morhua). Mol Ecol 15: 321–331. [DOI] [PubMed] [Google Scholar]
- 128. Chiucchi JE, Gibbs HL (2010) Similarity of contemporary and historical gene flow among highly fragmented populations of an endangered rattlesnake. Mol Ecol 19: 5345–5358. [DOI] [PubMed] [Google Scholar]
- 129. Young AG, Merriam HG (1994) Effects of forest fragmentation on the spatial genetic-structure of Acer saccharum Marsh (sugar maple) populations. Heredity 72: 201–208. [Google Scholar]
- 130. Baskauf CJ, Snapp S (1998) Population genetics of the cedar-glade endemic Astragalus bibullatus (Fabaceae) using isozymes. Ann Miss Bot Gar 85: 90–96. [Google Scholar]
- 131. Jaquiery J, Broquet T, Hirzel AH, Yearsley J, Perrin N (2011) Inferring landscape effects on dispersal from genetic distances: How far can we go? Mol Ecol 20: 692–705. [DOI] [PubMed] [Google Scholar]
- 132. Balkenhol N, Waits LP, Dezzani RJ (2009) Statistical approaches in landscape genetics: An evaluation of methods for linking landscape and genetic data. Ecography 32: 818–830. [Google Scholar]
- 133. Cushman SA, Landguth EL (2010) Spurious correlations and inference in landscape genetics. Mol Ecol 19: 3592–3602. [DOI] [PubMed] [Google Scholar]
- 134.Walters CJ (1986) Adaptive management of renewable resources. New York: Collier Macmillan. x, 374 p. p.
- 135. Tallmon DA, Gregovich D, Waples RS, Baker CS, Jackson J, et al. (2010) When are genetic methods useful for estimating contemporary abundance and detecting population trends? Mol Ecol Resour 10: 684–692. [DOI] [PubMed] [Google Scholar]
- 136. Ryman N, Leimar O (2009) Gst is still a useful measure of genetic differentiation - a comment on Jost's D. Mol Ecol. 18: 2084–2087. [DOI] [PubMed] [Google Scholar]
- 137. Whitlock MC (2011) G'ST and D do not replace FST . Mol Ecol 20: 1083–1091. [DOI] [PubMed] [Google Scholar]
- 138. Jost L (2009) D vs. Gst: Response to Heller and Siegismund (2009) and Ryman and Leimar (2009). Mol Ecol 18: 2088–2091. [Google Scholar]
- 139. Nei M (1987) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Crawford NG (2009) SMOGD: software for the measurement of genetic diversity. Mol Ecol Resour 10: 556–557. [DOI] [PubMed] [Google Scholar]
- 141. Meirmans PG, Van Tienderen PH (2004) GENOTYPE and GENODIVE: Two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4: 792–794. [Google Scholar]