

Abstract
The factors responsible for the regulation of regenerative calcium-induced calcium release (CICR) during Ca2+ spark evolution remain unclear. Cardiac ryanodine receptor (RyR) gating in rats and sheep was recorded at physiological Ca2+, Mg2+, and ATP levels and incorporated into a 3D model of the cardiac dyad, which reproduced the time course of Ca2+ sparks, Ca2+ blinks, and Ca2+ spark restitution. The termination of CICR by induction decay in the model principally arose from the steep Ca2+ dependence of RyR closed time, with the measured sarcoplasmic reticulum (SR) lumen Ca2+ dependence of RyR gating making almost no contribution. The start of CICR termination was strongly dependent on the extent of local depletion of junctional SR Ca2+, as well as the time course of local Ca2+ gradients within the junctional space. Reducing the dimensions of the dyad junction reduced Ca2+ spark amplitude by reducing the strength of regenerative feedback within CICR. A refractory period for Ca2+ spark initiation and subsequent Ca2+ spark amplitude restitution arose from 1), the extent to which the regenerative phase of CICR can be supported by the partially depleted junctional SR, and 2), the availability of releasable Ca2+ in the junctional SR. The physical organization of RyRs within the junctional space had minimal effects on Ca2+ spark amplitude when more than nine RyRs were present. Spark amplitude had a nonlinear dependence on RyR single-channel Ca2+ flux, and was approximately halved by reducing the flux from 0.6 to 0.2 pA. Although rat and sheep RyRs had quite different Ca2+ sensitivities, Ca2+ spark amplitude was hardly affected. This suggests that moderate changes in RyR gating by second-messenger systems will principally alter the spatiotemporal properties of SR release, with smaller effects on the amount released.
Introduction
In cardiac muscle, depolarization of the sarcolemma initiates Ca2+ release by activating sarcolemmal L-type Ca2+ channels, providing a local increase in cytoplasmic calcium concentration in the junctional space of the dyad (for review, see Bers (1)). This initiates [Ca2+]-dependent activation of the sarcoplasmic reticulum (SR) Ca2+ release channels, which are ryanodine receptors (RyRs) (2,3). The subsequent release of Ca2+ from the SR further increases the [Ca2+] in the junctional space and leads to regenerative RyR activation in a process known as calcium-induced calcium release (CICR) (4). In such a regenerative process, the larger SR Ca2+ flux should prevent subsequent control by the surface membrane (5) and deep-SR Ca2+-store depletion. However, the quantity of Ca2+ released from the SR has a graded dependence on the magnitude of the Ca2+ influx across the sarcolemma (5,6), and the SR releases only ∼50% of its content (see, e.g., Picht et al. (7)). The discovery of Ca2+ sparks (1,8) and local control theories (9) provided a solution to this problem by showing that graded responses can result from the spatiotemporal summation of individual Ca2+ release sites that are spatially uncoupled to limit regenerative behavior (10,11). However, local regeneration in CICR during Ca2+ sparks might still oppose reliable Ca2+ spark termination (12,13).
The termination problem in the local control mechanism has been reviewed elsewhere (12,13), and several mechanisms have been proposed to limit local regeneration. For example, it has been suggested that time-, SR-Ca2+-, and cytosolic-Ca2+-dependent RyR inactivation (4–6,14–16) would dampen regenerative behavior. However, the reported rates of inactivation are much slower than the time to peak of the Ca2+ spark (14,16,17). Stochastic RyR gating and the rapid response of local [Ca2+] to changing RyR flux may also contribute to spontaneous closure via stochastic attrition (9), although stochastic attrition on the timescale of a Ca2+ spark seems unlikely for more than ∼5–10 RyRs in a junctional cluster. The addition of inter-RyR allosteric interactions, which effectively reduces the number of RyRs in the cluster (e.g., very strong coupling will make the cluster gate as a single channel), can allow reliable termination for larger numbers of RyRs (12,18). However, direct observations of such allosteric interactions are few, and to our knowledge, such behavior has only been seen under quite nonphysiological conditions with detergent-solubilized RyRs (19).
Depletion of the SR lumen [Ca2+] may reduce the RyR release flux to the point where regenerative CICR is no longer supported (5,6,20–22), but measurements of SR [Ca2+] with fluo-5N suggest only moderate SR depletion (e.g., Picht et al. (7) and Shannon et al. (23)). We recently reported that the complex interaction of stochastically gating RyRs within a 3D spatial model could produce reliable spontaneous closure without any form of RyR inactivation (24). This closure was due to induction decay, wherein the increasing RyR closed time associated with declining RyR flux (due to junctional SR depletion) prevented closed RyRs from responding to the nearby open RyRs. Further support for the idea of induction decay comes from recent experiments in which reduced RyR conductance resulted in reduced Ca2+ spark amplitude despite the fact that there was no change in RyR gating (21). However, our previous report did not explore the generality of the result for different dyad geometries and used only gating data from rat RyRs, which seemed relatively insensitive to cytosolic Ca2+ compared to data reported elsewhere in the literature (i.e., Kd ∼ 10 μM).
Here, we expand on our previous study, examining the robustness of induction decay as a termination mechanism for both rat and sheep RyR gating in the presence of physiological levels of Mg2+ and ATP and with different dyad geometries and single-channel currents. It is important to note that the rat RyR model reproduces measured Ca2+ spark restitution (25) and that our measured RyR regulation by luminal Ca2+ has little effect on Ca2+ spark properties. The model also displays a Ca2+ spark amplitude dependence on single RyR channel current, which is similar to that recently explored using TRIS as a permeable blocking ion (21). In addition, we discuss the implications of the model for understanding the role(s) played by possible RyR gating changes and dyad restructuring.
Methods
Isolation of hearts
Healthy male adult rats (Harlan Sprague-Dawley, Indianapolis, IN) were heparinized (2000 U heparin injection BP, Weddel Pharmaceuticals, London, United Kingdom) and 15 min later anesthetized with isoflurane (Laser Animal Health, Brisbane, Australia). The hearts were rapidly removed and immediately perfused via the Langendorff method with ice-cold Krebs-Henseleit buffer (in mM, 120 NaCl, 25 NaHCO3, 10 Glucose, 5 KCl, 2 MgCl2, 1 NaH2PO4, and 2.5 CaCl2) for 2 min until the blood was washed out. Hearts were then perfused for 5 min with warmed (37°C) and oxygenated (95% O2, 5% CO2) Krebs-Henseleit buffer, after which they were snap-frozen. Sheep hearts were excised from anesthetized ewes (5% pentobartitone (IV) followed by oxygen/halothane) and rinsed in ice-cold homogenizing buffer (20 mM imidazole and 300 mM sucrose, pH 7.4 with HCl) before snap-freezing. All hearts were snap-frozen in liquid nitrogen and stored at −80°C until further use.
SR vesicle preparation
Briefly, heart tissue was homogenized in buffer containing 10 mM imidazole, 0.5 mM dithiothreitol (DTT), 3 mM sodium azide, 0.29 M sucrose, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 1 mM benzamidine, 0.5 mM phenylmethylsulfonyl fluoride, and 20 mM NaF, pH 6.9 (homogenization buffer). SR vesicles were separated by centrifugation as described in the Supporting Methods. The NaF inhibited phosphatases during the SR vesicle preparation. The whole procedure was carried out at 4°C.
Single-channel recordings
Lipid bilayers were formed as described in the Supporting Methods. Lipid bilayers separated cis (cytoplasmic) and trans (luminal) compartments. Both compartments contained 250 mM Cs+ (230 mM CsCH3O3S, 20 mM CsCl, and 10 mM TES buffer). In the trans bath, [Ca2+] was either 0.1 or 1 mM. The cis bath contained 2 mM ATP and 3 mM MgCl2 (1 mM free Mg2+), and Ca2+ was titrated between 1 and 500 μM (see Supporting Methods for details of pH and Ca 2+ buffering). The composition of the cis solution was altered by local perfusion and the composition of the trans solutions was altered by addition of aliquots of stock solutions. All experiments were performed at room temperature (21–26°C) and membrane potential of −40 mV (lumen at ground). Before analysis, the current signal was low-pass digitally filtered at 1 kHz. Channel 3 software (N. W. Laver, nic@niclaver.com) was used to detect channel events and measure open probability (Po), mean open time (τo), and mean closed time (τc).
Model geometry
The geometry of the model dyad is shown in Fig. 1. Electron micrographs suggest a large variation in dyad size and t-tubule radius (see, e.g., Franzini-Armstrong (26), Fawcett and McNutt (27), Forbes and Van Neil (28), and Forssman and Girardier (29)). In the simulations shown here, the t-tubule is a cylinder 250 nm in diameter between computational boundaries. Wrapped around its circumference, at its midpoint, is the terminal or junctional SR (j-SR), which is represented as a pancake 360 nm along the axis, 26 nm radial thickness, and 450 nm tubule circumference. RyR Ca2+ release channels were arranged in clusters with a nearest-neighbor separation of 31 nm on the j-SR membrane facing the t-tubule. The j-SR was connected to the network SR (n-SR) at its center. The n-SR was isotropically distributed throughout the computational volume to mimic a labyrinth of tubules occupying a volume fraction of 3.5% of the cytoplasmic volume. Model parameter values for dyad structure are given in the Table S1 in the Supporting Material.
Figure 1.
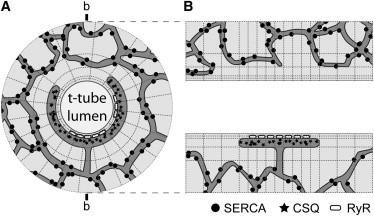
Illustration of the model geometry. (A) Cross section of the cylindrical model with a t-tubule in the center. The j-SR wraps around the t-tubule at a 15-nm radial distance (dyad cleft). RyRs are located in the j-SR membrane and release Ca2+ into the dyad cleft. The j-SR contains calsequestrin (CSQ) and is connected to the n-SR by a tubule, which allows for Ca2+ diffusion between these two compartments. The SERCA Ca2+ uptake transporters are uniformly distributed throughout the n-SR. (B) Side view of the model in the cross section indicated by b-b in A. In this view, the t-tubule lumen runs horizontally and the j-SR can be seen on one side.
Ion movements
Computational details are given in the Supporting Methods. Ca2+ diffusion and binding to Ca2+ indicators (fluo-4 and fluo-5N) in and around the dyad were calculated using Matlab (Mathworks, Natick, MA). The ion concentrations in each volume element were calculated by solving a discretized form of the electro-diffusion/transport equation that is shown here for the case of Ca2+:
| (1) |
where DCa is the diffusion coefficient for Ca2+ and Φ is the electrostatic potential arising from charged lipids on the t-tubule membrane. The term involving ∇Φ was assumed to have only radial components. JRyR represents the flow of Ca2+ through each open RyR into the adjacent volume element in the dyad cleft (with volume Vcleft), which was given by
| (2) |
where iRyR is a constant to give a 0.6-pA flux (30,31) for an open RyR with a 1 mM [Ca2+] gradient and F is Faraday’s constant. Ca2+ uptake was simulated by a simplified model for sarcoendoplasmic reticulum calcium-transport ATPase (SERCA) (32) located in the n-SR, as given by
| (3) |
where KSR,max is the maximum rate of SERCA uptake, KSERCA the cytoplasmic Ca2+ binding constant, and Vcyto the element volume. kleak sets JSERCA to zero under initial/resting conditions. KSR,max was set by reproducing the rate of decline of whole-cell [Ca2+] after setting cytoplasmic [Ca2+] to 1 μM, and the value is given in Table S3.
Jbuffer is the contribution to Ca2+ fluxes from Ca2+ reacting with a buffer, B:
| (4) |
Diffusible buffers included ATP, calmodulin, fluo-4, and fluo-5N, with diffusion coefficients described in Table S2. Troponin buffering was fixed in the cytoplasmic compartment, whereas calsequestrin was fixed in the SR compartments and concentrated mainly in the j-SR. Negatively charged lipids such as phosphatidylserine (PS) and zwitterionic phosphatidylcholine (PC) give rise to a negative net charge on the lipid bilayer and provide binding sites for cations such as Ca.2+ and K+. In the model, these effects are treated using simplifying approximations, as described previously (32) (also see Supporting Material).
The equations were solved on a cylindrical coordinate system with Neumann reflective boundaries at ±2 μm along the t-tubule and at a radius of 2 μm. Model parameters for ion diffusion and buffering are given in Table S2.
RyR gating
RyR gating was simulated using an empirical two-state (open and closed) stochastic model. The opening and closing rates were derived from two-segment linear fits to experimental transition rates (see Fig. S1), which are limited to Ca2+ levels during CICR and are not intended to describe RyR gating behavior at resting Ca2+ levels (see below). The transition rates were calculated from the reciprocal of the empirical RyR mean closed and open times, respectively. A Monte Carlo method was used to simulate stochastic transitions of each RyR, and at the computed transition times for each RyR, the integration was restarted using the new values of JRyR.
Initiation of Ca2+ sparks
Simulations were started either by 1- to 3-ms Ca2+ injection into a volume element adjacent to an RyR at a rate corresponding to 0.2 pA (to mimic an L-type Ca2+ channel opening) or by initializing one RyR in the open state (to mimic the initiation of a spontaneous Ca2+ spark). The subsequent evolution of the Ca2+ spark showed no difference between simulations triggered in these two ways (not shown).
Microscope blurring
To simulate the Ca2+ sparks and blinks as observed by confocal microscopy, the spatial distributions of fluo-4 and fluo-5N generated by the model were convolved with 3D Gaussian point-spread function. The full width at half-maximum of the Gaussian was 0.3 μm in the focal plane of the microscope (x and y directions) and 0.93 μm perpendicular to the focal plane (z direction).
Results
Fig. 2 A shows the typical pattern of RyR gating observed at various cytoplasmic [Ca2+] and in the presence of 2 mM ATP and 1 mM free Mg2+. Under these conditions, the open probability (Po) of RyRs from sheep (Fig. 2 B, open triangles) had a much weaker dependence on [Ca2+] than that of RyRs from rat at either 0.1 mM (open circles) or 1 mM (solid circles) luminal [Ca2+]. The RyR Po for both species increased to a maximum of ∼0.5 at 0.1 mM [Ca2+]. The Po for rat RyRs showed only a modest dependence on luminal Ca2+ levels, consistent with studies on sheep RyRs (33,34). For [Ca2+] between 1 and 100 μM, the mean Po varied as ∼[Ca2+]2.8 for rat (24) and ∼[Ca2+]2.1 for sheep, which was primarily due to changes in closed time (Figs. 2 C and S1). For simplicity, we used the closed-time data at 0.1 mM SR (trans) [Ca2+], because the model showed that j-SR levels dropped to about this level during the Ca2+ spark, and as shown below, allowing SR Ca2+ to influence closed time had only minor effects on Ca2+ spark properties. Any extrapolation of the empirical power function(s) describing the RyR gating seen at higher [Ca2+] to lower resting [Ca2+] predicts a resting Ca2+-spark rate that is too low and is therefore problematic. However, if the longest-closed-time data points of Fig. 2 C for rat and sheep RyRs are extrapolated to resting conditions, these data points suggest that (shorter) closed times of ∼1000 s might pertain at 0.1 μM [Ca2+]. It has been estimated that a confocal microscope samples ∼1.1 release sites per μm scanned (35). Therefore, a 100-μm scan line would record from ∼110 release sites (junctions), and with ∼20 RyRs in each junction, and assuming that most spontaneous RyR openings trigger a Ca2+ spark, the resting Ca2+ spark rate would be ∼2 s−1, in reasonable agreement with experimental observations (8,36).
Figure 2.
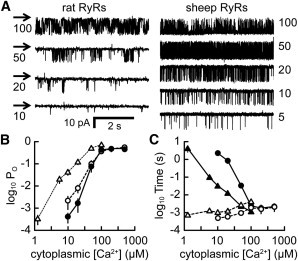
[Ca2+]-dependent RyR2 gating in lipid bilayers. Both sides of the bilayer contained (in mM) 230 CsCH3O3S and 20 CsCl (pH 7.4), with 2 ATP and 3 MgCl2 on the cytoplasmic side ([Ca2+] as indicated). (A) Channel openings at various [Ca2+] are downward deflections from the baseline (arrows). (B) Cytoplasmic [Ca2+] dependence of Po for RyRs from rat (luminal [Ca2+], 0.1 mM (solid circles) and 1 mM (open circles)) and sheep heart (luminal [Ca2+], 0.1 mM (open triangles)). Data points show the mean ± SE for n = 3–11 (solid circles), n = 4–8 (open circles), and n = 2–14 (open triangles)). Some of the rat data values have been replotted from Laver et al. (24). (C) Cytoplasmic [Ca2+] dependence of sheep RyR mean open and closed durations (0.1 mM luminal [Ca2+]). Solid symbols represent closed times and open symbols open times. Sheep RyR gating is shown by triangles and rat RyR gating, replotted from Laver et al. (24), by circles.
Morphology of simulated Ca2+ sparks and blinks
We could simulate Ca2+ sparks and associated SR Ca2+ depletion signals (Ca2+ blinks (8)) by incorporating fluo-4 and fluo-5N Ca2+ indicators into the model. Fig. 3 illustrates the time course and spatial spread of Ca2+ sparks (Fig. 3 A) and blinks (Fig. 3 B), derived from simulations of Ca2+ release by an array of 21 rat RyRs triggered by a 3 ms dihydropyridine receptor (DHPR) opening after simulated optical blurring. The simulated Ca2+ spark is similar to those observed experimentally (see also Laver et al. (24)), with the Ca2+ spark having a greater spatial width than the Ca2+ blink (Fig. 3 C). It is notable that no triggering sparklet (37) is seen; the trigger signal is essentially lost due to the small volume of the dyad cleft and the rapid activation of the RyR cluster. Although we do not have experimental records for Ca2+ blinks in rat RyRs, the simulated blinks were of comparable in amplitude, duration, and spatial extent to those reported for rabbit (minimum F/Fo = 60 ± 8%, full duration at half-maximum (FDHM) = 175 ± 7 ms (38), and 823 ± 476 nm (39). The fluo-5N fluorescence signal significantly underestimated the extent of local depletion of the j-SR due to blurring of the j-SR, t-tubule, and n-SR structures by the microscope (40). The rate of recovery of the fluo-5N fluorescence signal (time constant = 80 ms) was substantially faster than the rate of j-SR replenishment (time constant = 180 ms), which was largely due to the saturation effects in the fluo-5N signal.
Figure 3.

Simulation of Ca2+ sparks and blinks. (A) Simulated Ca2+ sparks. The simulated Ca2+ spark was generated by the array of RyRs shown in Fig. 4. (B) The corresponding simulated Ca2+ blink. Scale bars apply to both A and B. (C) Corresponding spatial profiles of spark and blink fluorescence at the times of their peak and minimum intensities, respectively. The noisy line shows the profile of an experimentally recorded Ca2+ spark (see also (23, 24) for additional comparison of model behavior to experimental data).
Ca2+ dynamics and RyR regulation during Ca2+ sparks
Fig. 4 shows an exemplar response of the model, with 21 irregularly arranged RyRs stimulated by the DHPR-trigger opening. The time courses (n = 7) of the computed average Ca2+ spark and blink signals are shown in Fig. 4 A. Mean [Ca2+] in the dyad cleft and j-SR elements is shown in Fig. 4 B. The initial rise in dyad [Ca2+] is due to the Ca2+ influx from DHPR triggering (which has no variance). Subsequent changes in [Ca2+] due to stochastic RyR gating then lead to variance in both signals. Fig. 4 C shows that shortly after initiation, the number of open RyRs increased to ∼10, which corresponds to Po ∼ 0.5, and remained at this level for ∼132 ms before the RyRs started to close; closure was complete at 22 ms. This RyR response can be understood in terms of the dyadic and j-SR [Ca2+] (Fig. 4 B). Initially, dyad [Ca2+] rose to high levels resulting from a regenerative phase of CICR that persisted for ∼15 ms. By that time, j-SR [Ca2+] had declined to ∼10% of resting levels, which reduced the Ca2+ flux via open RyRs (Fig. 4 C) and consequently reduced local dyad [Ca2+] to a point where regeneration decayed and the RyRs started to close. Once RyRs had closed, [Ca2+] in the dyad cleft equilibrated with the bulk cytoplasm and Ca2+ in the j-SR was replenished by diffusion of Ca2+ from the n-SR. The decline in Ca2+ spark fluorescence was primarily due to Ca2+ diffusion in the cytoplasm and buffering, with SERCA reuptake playing a lesser role, as previously described (41,42). In this simulation, rat RyRs were distributed in a loose cluster, as depicted in Fig. 4 D, which also shows the distribution of open RyRs and local dyad [Ca2+] at several time points. The time course of Ca2+ flux from the dyad is shown in Fig. 4 E and is in good agreement with that calculated from Ca2+ spark flux reconstructions (43). Therefore the model reproduces all of the major features of Ca2+ sparks and blinks with reasonable accuracy.
Figure 4.
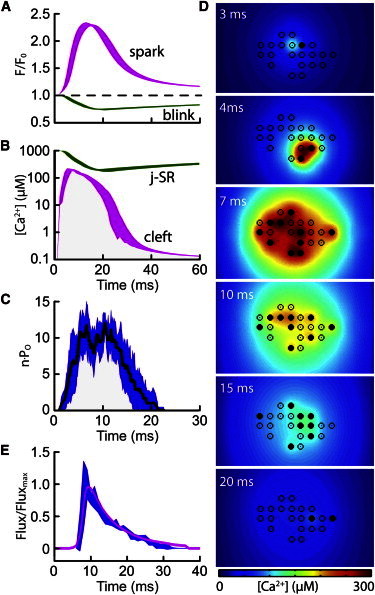
RyR cluster behavior during Ca2+ sparks. (A) Ca2+ spark and Ca2+ blink time courses for a cluster of 21 RyRs with a distribution as shown in D. (B) Spatial mean of [Ca2+] in the dyadic cleft and j-SR. (C) Numbers of open RyRs corresponding to the means in B. In A–C, line thickness shows the SD of 10 simulations. (D) Spatial distribution of [Ca2+] in the dyad at several time points during a typical simulation. RyRs are located at the circles, with solid and open circles representing open and closed RyRs, respectively. (E) Simulated time course of j-SR Ca2+ flux (wide trace) compared to the Ca2+ flux calculated from experimental Ca2+ spark profiles (line) replotted from Soeller and Cannell (43).
Termination of Ca2+ sparks
Termination of RyR activity resulted principally from the steep Ca2+ dependence of RyR closed time (Fig. 2 C). If the opening rate of RyRs is sufficiently low, the open RyR(s) that is (are) supporting local dyad [Ca2+] may close before the closed RyRs reopen to sustain CICR. We have termed this process induction decay (24), because it reflects the gradual loss of the activating Ca2+ induction process in CICR, which is responsible for the initial rapid rise in RyR Po. Thus, the termination of RyR activity depends on RyR sensitivity to [Ca2+] in the dyad cleft, as well as the time course of j-SR depletion. For rat RyRs, the minimum dyad [Ca2+] that could sustain CICR was ∼50 μM (Fig. 4 B). RyRs from sheep exhibited Po and opening rates that were ∼40-fold higher than the corresponding values for rat RyRs at 10 μM Ca2+ (Fig. 2, B and C, respectively). Fig. 5 compares the mean response of models incorporating RyRs (organized as a square cluster of 16 RyRs) from rat and sheep to a DHPR trigger. The computed time course of Ca2+ sparks (Fig. 5 A) show a similar rate of rise for rat (magenta) and sheep (blue) but a longer decay phase for sheep (Ca2+ spark parameters are given in Table 1.). The corresponding computed [Ca2+] in the dyad cleft and j-SR and the RyR Po are shown in Fig. 5, B and C, respectively. Both rat and sheep showed similar initiation of RyR activity and maximal Po. However, sheep RyR activity was sustained for ∼40 ms compared to ∼18 ms for rat RyR activity. This is because with RyRs from sheep, the minimum dyad Ca2+ level that could sustain CICR was ∼10 μM (Fig. 5 B), which was fivefold lower than the corresponding value for rat RyRs, and it took approximately twice as long for the dyad [Ca2+] to decline to this lower threshold. This different behavior may reflect physiological adaptation to the very different heart rates seen in these species (i.e., to have a shorter duration of SR release in rat RyRs). Even with more Ca2+ sensitive sheep RyRs, the combination of the limited j-SR volume and j-SR Ca2+ buffering capacity ensures that closure by induction decay occurs on the timescale of a Ca2+ spark.
Figure 5.
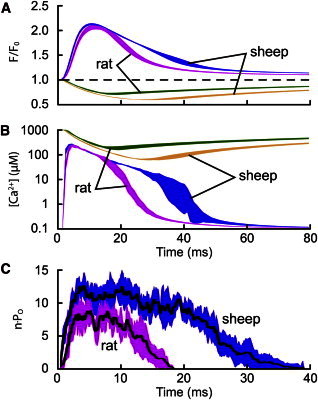
Effect of RyR Ca2+ sensitivity on Ca2+ spark activation and termination. (A) Mean time courses of Ca2+ spark and blink signals generated using a square cluster of 16 RyRs with Ca2+-activation kinetics derived from rat (n = 10) and sheep data (n = 3). (B) Corresponding spatial means of [Ca2+] in the dyadic cleft and j-SR. (C) Number of open RyRs for rat and sheep Ca2+-spark simulations, showing the mean (line) ± SD, (wide trace).
Table 1.
Morphological parameters of simulated sparks
| Condition | iRyR (pA/mM) | nRyR | Fmax/Fo | Δt(Fmax) (ms) | ΔF/Δt (ms−1) | FWHM (μm) | FDHM (ms) | n |
|---|---|---|---|---|---|---|---|---|
| Fig. 4 | 0.6 | 21 | 1.30 ± 0.03 | 14.3 | 0.15 ± 0.04 | 1.44 ± 0.03 | 19.6 ± 1.6 | 6 |
| Square RyR cluster | 0.6 | 16 | 1.04 ± 0.06 | 12.2 | 0.144 ± 0.017 | 1.40 ± 0.03 | 18.7 ± 2.0 | 17 |
| Luminal gating | 0.6 | 16 | 1.07 ± 0.02 | 12.9 | 0.141 ± 0.018 | 1.41 ± 0.03 | 19.0 ± 1.4 | 9 |
| Sheep RyRs | 0.6 | 16 | 1.07 ± 0.01 | 11.7 | 0.161 ± 0.007 | 1.42 ± 0.02 | 25.6 ± 2.1 | 3 |
| Narrower dyad | 0.6 | 16 | 0.86 ± 0.02 | 8 | 0.184 ± 0.022 | 1.07 ± 0.02 | 10.6 ± 0.9 | 4 |
| nRYR varied | 0.6 | 36 | 1.21 ± 0.01 | 12 | 0.175 ± 0.003 | 1.39 ± 0.02 | 18.8 ± 0.3 | 3 |
| 0.6 | 16 | 1.04 ± 0.06 | 12.2 | 0.144 ± 0.017 | 1.40 ± 0.03 | 18.7 ± 2.0 | 17 | |
| 0.6 | 9 | 0.95 ± 0.02 | 13.7 | 0.113 ± 0.003 | 1.42 ± 0.03 | 23.5 ± 1.4 | 3 | |
| 0.6 | 4 | 0.68 ± 0.06 | 14.3 | 0.071 ± 0.003 | 1.38 ± 0.04 | 18.4 ± 2.0 | 6 | |
| 0.6 | 2 | 0.47 ± 0.05 | 9.7 | 0.069 ± 0.003 | 1.32 ± 0.03 | 14.8 ± 1.1 | 9 | |
| iRyR varied | 0.6 | 16 | 1.04 ± 0.06 | 12.2 | 0.144 ± 0.017 | 1.40 ± 0.03 | 18.7 ± 2.0 | 17 |
| 0.4 | 16 | 0.89 ± 0.07 | 13.2 | 0.108 ± 0.035 | 1.37 ± 0.05 | 16.5 ± 1.9 | 6 | |
| 0.35 | 16 | 0.87 ± 0.07 | 12.3 | 0.107 ± 0.025 | 1.35 ± 0.03 | 15.4 ± 1.1 | 7 | |
| 0.3 | 16 | 0.83 ± 0.07 | 13.2 | 0.098 ± 0.018 | 1.36 ± 0.02 | 15.9 ± 2.2 | 10 | |
| 0.25 | 16 | 0.74 ± 0.16 | 13.9 | 0.079 ± 0.023 | 1.37 ± 0.05 | 16.0 ± 2.5 | 10 | |
| 0.2 | 16 | 0.52 ± 0.27 | 10.3 | 0.067 ± 0.022 | 1.31 ± 0.07 | 13.7 ± 3.2 | 9 | |
| 0.15 | 16 | 0.38 ± 0.14 | 9.4 | 0.057 ± 0.007 | 1.30 ± 0.07 | 12.4 ± 3.2 | 12 |
First row is for the simulation depicted in Fig. 4. Subsequent rows show how spark properties change as various factors are altered, as indicated in the first column. iRyR is the RyR single-channel Ca2+ current, nRyR the number of RyRs in the simulated cluster, Fmax/Fo the peak Ca2+ spark fluorescence change, Δt(Fmax) the time to peak of the Ca2+ spark, ΔF/Δt the rate of rise of normalized fluorescence, FWHM the width of the spark at half-maximum amplitude, and FDHM the Ca2+ spark duration measured at half-maximum amplitude. Table gives mean ± SD of n simulations.
The role of luminal regulation of RyR Po was examined using gating data derived from Fig. 2. j-SR [Ca2+] of 1 mM resulted in the RyR opening rate varying as cytoplasmic [Ca2+]1.9 compared to [Ca2+]2.8. Opening rates at intermediate j-SR [Ca2+] were given by linear interpolation. This RyR regulation by j-SR [Ca2+] in our model produced almost no change in Ca2+ spark and blink morphology (Table 1, rows 2 and 3). From these measurements, we conclude that regulation of RyR gating by Ca2+ in the j-SR may not be a major factor in determining the spatiotemporal properties of Ca2+ sparks and blinks.
Effects of RyR organization and dyad size
The role of local nanoscopic [Ca2+] gradients in maintaining regeneration in CICR is illustrated in Fig. 6 A, which shows two arrangements of 18 RyRs as either two adjacent groups or one large group with every other RyR removed. Wherever there is a gap between adjacent RyRs, CICR terminated slightly earlier, but this had little effect on the amplitude of the Ca2+ spark (Fig. 6, A and B). This paradoxical result arises from the consequences of nanoscopic [Ca2+] gradients within the dyad for determining both Ca2+ fluxes and CICR regeneration. As the RyRs become more separated, local CICR regeneration is weakened due to the spatial gradient of [Ca2+] from each open RyR resulting in adjacent RyRs experiencing lower [Ca2+] on average (leading to weakened regeneration and earlier termination). However, at the same time, the average flux associated with each RyR opening is increased due to a decreased local Ca2+ accumulation increasing the [Ca2+] gradient that determines iRyR (which offsets the former effect). Thus, nanoscopic gradients (illustrated in Figs. 4 D and 6 C) of [Ca2+] within the RyR cluster play a key role in all phases of CICR regulation, from initiation to regeneration (which allows the j-SR to release a large fraction of its total Ca2+), as well as eventual termination. The dimensions of the dyad itself also have effects on CICR termination (as well as activation probability (44)). Fig. 6 D shows that a dyad with reduced area (but the same number of RyRs) results in a smaller Ca2+ spark that is abbreviated in time course.
Figure 6.
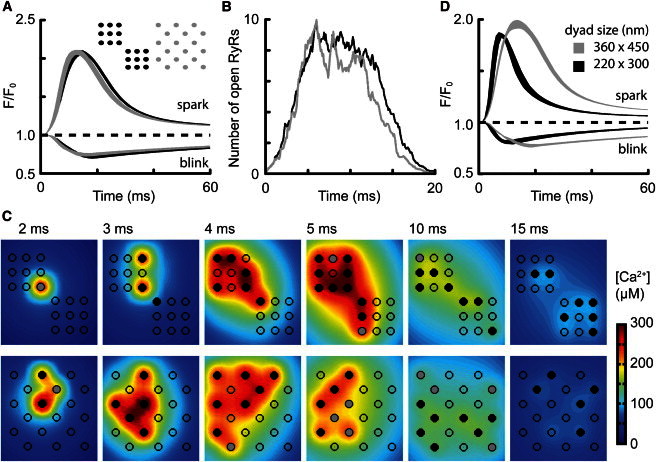
Intradyad geometric effects. (A) Ca2+ spark and blink time courses for two geometric arrangements of 18 RyRs. (Inset) Cluster organization with color key. (B) Evolution of mean Po (shown as nPo) for the two clusters indicated in A. (C) Spatial distribution of [Ca2+] in the dyad at several time points for the clusters shown in A. RyR states are depicted as described in Fig. 4 legend. (D) Effect of dyad dimensions. For a square cluster of 16 RyRs, the dyad space was set to the dimensions shown. Note that the smaller dyad results in faster Ca2+ spark termination but with a relatively small effect on amplitude.
Varying RyR cluster size between 2 and 36 RyRs revealed that Ca2+ spark properties were relatively insensitive to cluster size in clusters of more than nine RyRs (Table 1, rows 6–10). As noted previously, the shallow dependence of Ca2+ spark amplitude on the number of RyRs (24) arose from the effective diffusional resistance of the RyR open channels being less than the series diffusional resistances of the dyad cleft and SR.
Restitution of Ca2+ sparks after Ca2+ release
The time course of Ca2+ spark recovery between spontaneous Ca2+ sparks (restitution) has been used to examine SR Ca2+ depletion, as well as possible RyR inactivation (20,25,45). Although our model contained no inactivated, adapted, or refractory states, the time course of recovery of Ca2+ spark amplitude (Fig. 7 A) and probability of evoking a second Ca2+ spark (Fig. 7 B) reproduced experimental observations (25). The explanation for the apparent refractory and restitution behavior lies in the extent to which the inductive regenerative phase of CICR can be supported by the partially depleted j-SR, and this is seen by comparing the time course of restitution of j-SR [Ca2+], peak F/Fo, and nPo (Fig. 7 C).
Figure 7.
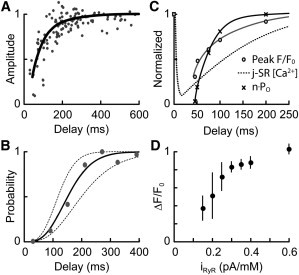
Ca2+ spark restitution and iRyR dependence. (A) Amplitude of the second Ca2+ spark (line) compared to experimental data (circles, replotted from Sobie et al. (25)). (B) Probability of triggering a second Ca2+ spark after various delays from the first. The trigger was assumed to be spontaneous RyR openings of 1 ms duration that occurred at intervals of 10 ms (left dashed line), 20 ms (solid line) and 40 ms (right dashed line). Data points (gray) are replotted from Sobie et al. (25). (C) Comparison of the time courses of j-SR [Ca2+] (dashed line), peak fluorescence (open circles) and nPo (crosses). (D) Effect of iRyR on Ca2+ spark amplitude. Sparks were simulated in a dyad containing 16 RyRs in a 4 × 4 array. The Ca2+ permeability of the RyR was varied and is expressed on the abscissa as Ca2+ current through the RyR in the presence of a 1 mM [Ca2+] difference across the SR membrane. Data show the mean ± SD for 6–17 simulations.
The close agreement of our model with the reported restitution of Ca2+ spark amplitude (Fig. 7 A) was achieved without freely adjustable parameters. Ca2+ sparks had an apparent refractory period of ∼70 ms, and the second Ca2+ spark amplitude recovered to 80% of the initial Ca2+ spark amplitude in 130 ms. The restitution in the probability of triggering a second spark also depended on both the rate of occurrence of a triggering event (a spontaneous opening of either an RyR or a DHPR) and j-SR refilling, as shown by the dotted lines in Fig. 7 B. The Ca2+ spark restitution time course was determined by the rate of refilling of the j-SR, which in turn was constrained by the model fitting the rate of recovery of Ca2+ blinks (80% in 140 ms). j-SR refilling by diffusion from the n-SR and, to a lesser extent, local Ca2+ reuptake via SERCA was transduced via the local iRyR to produce a more rapid recovery in nPo than j-SR [Ca2+] due to the power dependence of nPo on dyad [Ca2+] (Fig. 7 C). Ca2+ spark amplitude restitution also depended on the ability of the local Ca2+ flux (iRyR) to support the regenerative phase of CICR, with induction decay developing more rapidly at low j-SR [Ca2+] (see below).
Effect of RyR single-channel Ca2+ flux
Recently, the effect of reducing RyR Ca2+ conductance and current (iRyR) with impermeant ions on Ca2+ spark amplitude and frequency has been reported (21). These data provide an important additional test of the model, and Fig. 7 D shows how Ca2+ spark amplitude varied as iRyR was reduced to <0.6 pA/mM. For iRyR between 0.3 and 0.6 pA/mM, Ca2+ spark amplitude varied by 20%. However, when iRyR was reduced to <0.3 pA/mM the Ca2+ spark amplitude decreased more rapidly, reaching a ΔF/Fo of 0.4 at 0.15 pA/mM, in reasonable agreement with the data of Guo et al. (21). Reducing iRyR also hastened the onset of induction decay, resulting in a reduced time to peak for the resulting smaller-amplitude Ca2+ spark (Table 1, rows 15–17). In this regime, Ca2+ spark amplitude became more variable (as seen by the length of the error bars) as stochastic gating effects became relatively more important.
Discussion
The computer simulations presented here are based upon our best synthesis of available data on RyR gating, dyad geometry, and Ca2+ buffering. Although future work may lead to refinement of the geometry and parameters used, the model has given useful insight into the (likely) factors that determine the time course of SR Ca2+ release during Ca2+ sparks. Since the spatiotemporal evolution of Ca2+ sparks determines the rise of Ca2+ that activates contraction (35), this model can also give new insight into the factors that determine the cell-wide Ca2+ transient.
The simulations show that termination of RyR activity results from the spatiotemporal evolution of nanoscopic [Ca2+] changes within the dyad, which are not properly captured by treating the junctional space of the dyad as a single compartment (46). As the Ca2+ flux via open RyRs starts to decrease, the decline in local dyad [Ca2+] causes a decrease in the Po of neighboring RyRs, which in turn reduces the total Ca2+ flux. This decrease in flux then further decreases cluster Po (mainly via the RyR closed time), forming an inactivating process we called induction decay (24). Induction decay is essentially the reverse of the normal activating regenerative process of CICR and occurs due to the reduction in single-channel flux being unable to support regeneration of closed RyRs within the open time of adjacent RyRs. Our simulations show that the latency for induction decay to develop depends on the sensitivity of RyRs to dyad [Ca2+], as well as geometric factors such as the diameter of the dyad junction and number/organization of RyRs in the junction (Table 1 and Figs. 5 and 6).
A given dyad produces stereotyped Ca2+ sparks and blinks despite quite large stochastic variations in the number of open RyRs (compare the standard deviations in spark morphology in Fig. 4 A and Table 1 with the standard deviation in the number of open RyRs in Fig. 4 C). The reason for this behavior is that stochastic RyR gating is tempered by other, nonstochastic contributions to Ca2+ spark morphology, such as the series diffusion resistances of the dyad junction and the SR (when the RyRs have a Ca2+-release flux (iRyR) >∼0.3 pA/mM). In addition, the Ca2+ spark fluorescence is smoothed by diffusion and buffering, which act as a low-pass filter for the stochastic RyR gating. The stereotypical nature of Ca2+ sparks in these simulations is less than the Ca2+ spark variation that occurs throughout the cell (36,47) but more than that observed at a single cytoplasmic release site (48). When iRyR was <0.3 pA/mM, the resulting Ca2+ sparks showed much greater variability due to increased stochastic noise. However, reports of highly variable or even quantized spark amplitudes (49,50) could be related to peripheral superclusters (51) or multiple dyadic junctions within the recording volume (12), which are uncertain factors not considered in this model. These possibilities could be tested with the 3D stochastic modeling approach presented here, but at this time, it is unknown how variable size groups of RyRs should be combined within a supercluster fed by a single piece of extended j-SR. Nevertheless, our simulations show that the actual organization of a fixed number of closely spaced RyRs sharing a single dyadic cleft (formed by a single SR junction) has quite small effects on Ca2+ release during a Ca2+spark.
The limited effect on Ca2+ spark amplitude illustrates the remarkable resilience of dyad to moderate changes in geometry (although the geometry and RyR distribution have large effects on the ease with which CICR can be triggered (44)). The reduced diffusional resistance associated with reduced j-SR area accelerates CICR termination due to a more rapid collapse of nanoscopic CICR support, and at the same time, increases the rate at which Ca2+ leaves the dyad space (32). Reduction in the j-SR area also reduced the width of the Ca2+ spark, as the indicator signal principally arises from Ca2+ that has left the dyad cleft edges. Varying the arrangement of RyRs from uniform squares of variable numbers of RyRs to looser arrangements (see Figs. 4 and 6) had relatively small effects on the amplitude of Ca2+ sparks and did not prevent termination of CICR by induction decay.
The robust nature of CICR activation and termination and the relative constancy of Ca2+ spark amplitude demonstrated here is underscored by the relative insensitivity of the release to the number of RyRs incorporated in the dyad at n > ∼9 RyRs (Table 1, rows 6–10, and (24)). Peripheral RyR clusters appear to contain ∼14 RyRs on average (51), and more central dyads should contain a somewhat larger number of RyRs (for review, see Cannell and Kong (12)). Although extensive electron microscopic tomography data are lacking, murine junctions appear to contain ∼8–34 RyRs (52). This suggests that the dyad is not normally in a regime where Ca2+ spark amplitude is strongly influenced by the number of RyRs.
The rather limited effect of different RyR gating behavior associated with rat and sheep RyRs on Ca2+ spark amplitude might not be expected based on previous models. This result suggests that physiological modulation of RyR gating could have quite limited effects on Ca2+ spark amplitude, as the quantity released is mainly controlled by the ability of the local iRyR to support regeneration. This hypothesis is compatible with the report by Guo et al. who found that RyR phosphorylation by protein kinase A had no effect on Ca2+ spark amplitude when SR load changes were controlled (53). Also, CaMKII RyR phosphorylation increases RyR Ca2+ sensitivity (54) and prolongs the release phase of Ca2+ sparks with little effect on Ca2+ spark amplitude (53). This is in general agreement with model behavior, as shown by the Ca2+ more Ca2+-sensitive sheep RyR simulations (Fig. 5) where the onset of induction decay is delayed but Ca2+ sparks were only slightly increased in amplitude.
In connection with the above, it has been previously pointed out that a steady-state change in the amplitude of the Ca2+ transient would not be possible without a change in SR Ca2+ uptake (since time-averaged release must equal time-averaged uptake) (55). On the other hand, if pathological conditions lead to alterations in dyad geometry as a result of subcellular remodeling (such as the dyad morphing into multiple diffusionally connected subclusters of RyRs), our model shows that quite large effects on excitation-contraction coupling could result (from changes in triggering efficiency and altered Ca2+ spark termination). Such changes may help explain the paradoxical report of the development of a subpopulation of big resting Ca2+ sparks in a heart failure model without changes in SR Ca2+ content or expression levels of RyR, calsequestrin, SERCA2a, phospholamban, triadin, and junctin proteins (56). It should also be noted that there is evidence for the existence of superclusters of RyRs on the cell surface (51), so the idea that RyRs may not always be packed into a single junctional cluster (as modeled here) is not without precedent. Thus, moderate pathological changes in dyad structure or distribution of RyR should not affect the average amplitude of Ca2+ sparks in the steady state, but by altering the ability of a trigger to start CICR, they could contribute to a change in Ca2+ transient time course and/or the development of spatial nonuniformities in Ca2+, as seen in heart failure (see, e.g., Litwin et al. (57).).
Modulation of iRyR has been used recently to probe the Ca2+ spark dependence on local Ca2+ flux and SR load (21). In those experiments, chemically skinned cells were exposed to 30 mM and 60 mM Tris, which produced a 20–40% reduction in iRyR and Ca2+ spark amplitude and, importantly, also decreased Ca2+ spark time to peak (implying a more rapid onset of CICR termination). In our model, varying iRyR over the range 0.15–0.35 pA caused similar variations in the amplitude as well as the time to peak of the computed Ca2+ sparks (Fig. 7 and Table 1, rows 11–17). This forms an additional independent test of our model (note that no other parameters were changed) and adds to our confidence that this stochastic 3D model captures the fundamental behavior of the dyad and shows that induction decay dominates CICR termination (24). From their experiments, Guo et al. (21) deduced that CICR termination during a Ca2+ spark was at least partly due to the loss of ability of iRyR to support CICR, in accord with our demonstration of induction decay (24). A very recent review (58) describes induction decay as “pernicious attrition” but does not address the sufficiency and/or reliability of the falling RyR Ca2+ flux to limit CICR via the growth in RyR closed time that we show here. In any case, we view induction decay in CICR not as pernicious “pernicious,” but rather as a beneficial intrinsic component of CICR within dyads that necessarily limits SR Ca2+ release time course. We conclude that our stochastic 3D model captures the fundamental behavior of the dyad and that induction decay is sufficient to explain local termination of CICR without additional regulatory factors.
In summary, we show that a stochastic model that incorporates our current understanding of dyad structure and measurements of RyR gating in lipid bilayers reproduces the major features of Ca2+ sparks and blinks with reasonable accuracy and is able to reproduce additional properties such as the effect of changing iRyR and the time course of restitution. The key to this behavior resides in the development of induction decay, as well as the time course of Ca2+ gradients between the dyadic space and j-SR. Changing RyR gating behavior to that of more Ca2+-sensitive sheep RyRs had only small effects, suggesting that possible modulation of RyR gating by second messengers should be more effective in smaller couplons where regenerative support is weaker. Overall, the behavior of the model is reminiscent of the cluster-bomb model proposed by Stern, wherein local depletion plus stochastic attrition in a small RyR cluster limits the amount of j-SR Ca2+ released (9). However, the problem inherent in stochastic attrition (i.e., a limit to the number of RyRs that can close by this mechanism within the timescale of a Ca2+ spark) is overcome by the automatic development of induction decay within the nanoscopic cellular architecture that forms the dyad.
Acknowledgments
This work was supported by grants from the National Health and Medical Research Council and Hunter Medical Research Institute (D.L.) and the Royal Society (M.B.C.).
Supporting Material
References
- 1.Bers D.M. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 2.Meissner G., Rousseau E., Anderson K.A. Biochemical characterization of the Ca2+ release channel of skeletal and cardiac sarcoplasmic reticulum. Mol. Cell. Biochem. 1988;82:59–65. doi: 10.1007/BF00242517. [DOI] [PubMed] [Google Scholar]
- 3.Inui M., Fleischer S. Purification of Ca2+ release channel (ryanodine receptor) from heart and skeletal muscle sarcoplasmic reticulum. Methods Enzymol. 1988;157:490–505. doi: 10.1016/0076-6879(88)57098-2. [DOI] [PubMed] [Google Scholar]
- 4.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983;245:C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 5.Cannell M.B., Berlin J.R., Lederer W.J. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science. 1987;238:1419–1423. doi: 10.1126/science.2446391. [DOI] [PubMed] [Google Scholar]
- 6.Barcenas-Ruiz L., Wier W.G. Voltage dependence of intracellular [Ca2+]i transients in guinea pig ventricular myocytes. Circ. Res. 1987;61:148–154. doi: 10.1161/01.res.61.1.148. [DOI] [PubMed] [Google Scholar]
- 7.Picht E., Zima A.V., Bers D.M. Dynamic calcium movement inside cardiac sarcoplasmic reticulum during release. Circ. Res. 2011;108:847–856. doi: 10.1161/CIRCRESAHA.111.240234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng H., Lederer W.J., Cannell M.B. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 9.Stern M.D. Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannell M.B., Cheng H., Lederer W.J. The control of calcium release in heart muscle. Science. 1995;268:1045–1049. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- 11.López-López J.R., Shacklock P.S., Wier W.G. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 1995;268:1042–1045. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- 12.Cannell M.B., Kong C.H.T. Local control in cardiac E-C coupling. J. Mol. Cell. Cardiol. 2012;52:298–303. doi: 10.1016/j.yjmcc.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 13.Stern M.D., Cheng H. Putting out the fire: what terminates calcium-induced calcium release in cardiac muscle? Cell Calcium. 2004;35:591–601. doi: 10.1016/j.ceca.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 14.Lukyanenko V., Wiesner T.F., Györke S. Termination of Ca2+ release during Ca2+ sparks in rat ventricular myocytes. J. Physiol. 1998;507:667–677. doi: 10.1111/j.1469-7793.1998.667bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koh X., Srinivasan B., Levchenko A. A 3D Monte Carlo analysis of the role of dyadic space geometry in spark generation. Biophys. J. 2006;90:1999–2014. doi: 10.1529/biophysj.105.065466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valdivia H.H., Kaplan J.H., Lederer W.J. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keizer J., Levine L. Ryanodine receptor adaptation and Ca2+-induced Ca2+ release-dependent Ca2+ oscillations. Biophys. J. 1996;71:3477–3487. doi: 10.1016/S0006-3495(96)79543-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sobie E.A., Dilly K.W., Jafri M.S. Termination of cardiac Ca2+ sparks: an investigative mathematical model of calcium-induced calcium release. Biophys. J. 2002;83:59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marx S.O., Gaburjakova J., Marks A.R. Coupled gating between cardiac calcium release channels (ryanodine receptors) Circ. Res. 2001;88:1151–1158. doi: 10.1161/hh1101.091268. [DOI] [PubMed] [Google Scholar]
- 20.Ramay H.R., Liu O.Z., Sobie E.A. Recovery of cardiac calcium release is controlled by sarcoplasmic reticulum refilling and ryanodine receptor sensitivity. Cardiovasc. Res. 2011;91:598–605. doi: 10.1093/cvr/cvr143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo T., Gillespie D., Fill M. Ryanodine receptor current amplitude controls Ca2+ sparks in cardiac muscle. Circ. Res. 2012;111:28–36. doi: 10.1161/CIRCRESAHA.112.265652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zima A.V., Picht E., Blatter L.A. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ. Res. 2008;103:e105–e115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shannon T.R., Guo T., Bers D.M. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ. Res. 2003;93:40–45. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- 24.Laver D.R., Kong C.H.T., Cannell M.B. Termination of calcium-induced calcium release by induction decay: an emergent property of stochastic channel gating and molecular scale architecture. J. Mol. Cell. Cardiol. 2013;54:98–100. doi: 10.1016/j.yjmcc.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 25.Sobie E.A., Song L.-S., Lederer W.J. Local recovery of Ca2+ release in rat ventricular myocytes. J. Physiol. 2005;565:441–447. doi: 10.1113/jphysiol.2005.086496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franzini-Armstrong C. Architecture and regulation of the Ca2+ delivery system in muscle cells. Appl. Physiol. Nutr. Metab. 2009;34:323–327. doi: 10.1139/H09-017. [DOI] [PubMed] [Google Scholar]
- 27.Fawcett D.W., McNutt N.S. The ultrastructure of the cat myocardium. I. Ventricular papillary muscle. J. Cell Biol. 1969;42:1–45. doi: 10.1083/jcb.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forbes M.S., van Neil E.E. Membrane systems of guinea pig myocardium: ultrastructure and morphometric studies. Anat. Rec. 1988;222:362–379. doi: 10.1002/ar.1092220409. [DOI] [PubMed] [Google Scholar]
- 29.Forssmann W.G., Girardier L. A study of the T system in rat heart. J. Cell Biol. 1970;44:1–19. doi: 10.1083/jcb.44.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mejía-Alvarez R., Kettlun C., Fill M. Unitary Ca2+ current through cardiac ryanodine receptor channels under quasi-physiological ionic conditions. J. Gen. Physiol. 1999;113:177–186. doi: 10.1085/jgp.113.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kettlun C., González A., Fill M. Unitary Ca2+ current through mammalian cardiac and amphibian skeletal muscle ryanodine receptor channels under near-physiological ionic conditions. J. Gen. Physiol. 2003;122:407–417. doi: 10.1085/jgp.200308843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soeller C., Cannell M.B. Numerical simulation of local calcium movements during L-type calcium channel gating in the cardiac diad. Biophys. J. 1997;73:97–111. doi: 10.1016/S0006-3495(97)78051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laver D.R. Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Clin. Exp. Pharmacol. Physiol. 2007;34:889–896. doi: 10.1111/j.1440-1681.2007.04708.x. [DOI] [PubMed] [Google Scholar]
- 34.Laver D.R., Honen B.N. Luminal Mg2+, a key factor controlling RYR2-mediated Ca2+ release: cytoplasmic and luminal regulation modeled in a tetrameric channel. J. Gen. Physiol. 2008;132:429–446. doi: 10.1085/jgp.200810001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cannell M.B., Cheng H., Lederer W.J. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophys. J. 1994;67:1942–1956. doi: 10.1016/S0006-3495(94)80677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng H., Lederer M.R., Cannell M.B. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am. J. Physiol. 1996;270:C148–C159. doi: 10.1152/ajpcell.1996.270.1.C148. [DOI] [PubMed] [Google Scholar]
- 37.Wang S.Q., Song L.S., Cheng H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–596. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- 38.Zima A.V., Picht E., Bers D.M., Blatter L.A. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ. Res. 2008;103:e105–e115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brochet D.X., Yang D., Cheng H. Ca2+ blinks: rapid nanoscopic store calcium signaling. Proc. Natl. Acad. Sci. USA. 2005;102:3099–3104. doi: 10.1073/pnas.0500059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong C.H.T., Laver D.R., Cannell M.B. Extraction of sub-microscopic Ca fluxes from blurred and noisy fluorescent indicator images with a detailed model fitting approach. PLOS Comput. Biol. 2013;9:e1002931–e1002937. doi: 10.1371/journal.pcbi.1002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng H., Cannell M.B., Lederer W.J. Partial inhibition of Ca2+ current by methoxyverapamil (D600) reveals spatial nonuniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Circ. Res. 1995;76:236–241. doi: 10.1161/01.res.76.2.236. [DOI] [PubMed] [Google Scholar]
- 42.Santana L.F., Kranias E.G., Lederer W.J. Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. J. Physiol. 1997;503:21–29. doi: 10.1111/j.1469-7793.1997.021bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soeller C., Cannell M.B. Estimation of the sarcoplasmic reticulum Ca2+ release flux underlying Ca2+ sparks. Biophys. J. 2002;82:2396–2414. doi: 10.1016/S0006-3495(02)75584-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cannell M.B., Soeller C. Numerical analysis of ryanodine receptor activation by L-type channel activity in the cardiac muscle diad. Biophys. J. 1997;73:112–122. doi: 10.1016/S0006-3495(97)78052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sobie E.A., Lederer W.J. Dynamic local changes in sarcoplasmic reticulum calcium: physiological and pathophysiological roles. J. Mol. Cell. Cardiol. 2012;52:304–311. doi: 10.1016/j.yjmcc.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higgins E.R., Goel P., Sneyd J. Modelling calcium microdomains using homogenisation. J. Theor. Biol. 2007;247:623–644. doi: 10.1016/j.jtbi.2007.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng H., Song L.S., Stern M.D. Amplitude distribution of calcium sparks in confocal images: theory and studies with an automatic detection method. Biophys. J. 1999;76:606–617. doi: 10.1016/S0006-3495(99)77229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bridge J.H., Ershler P.R., Cannell M.B. Properties of Ca2+ sparks evoked by action potentials in mouse ventricular myocytes. J. Physiol. 1999;518:469–478. doi: 10.1111/j.1469-7793.1999.0469p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang S.Q., Stern M.D., Cheng H. The quantal nature of Ca2+ sparks and in situ operation of the ryanodine receptor array in cardiac cells. Proc. Natl. Acad. Sci. USA. 2004;101:3979–3984. doi: 10.1073/pnas.0306157101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shen J.-X., Wang S., Cheng H. Polymorphism of Ca2+ sparks evoked from in-focus Ca2+ release units in cardiac myocytes. Biophys. J. 2004;86:182–190. doi: 10.1016/S0006-3495(04)74095-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baddeley D., Jayasinghe I.D., Soeller C. Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc. Natl. Acad. Sci. USA. 2009;106:22275–22280. doi: 10.1073/pnas.0908971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayashi T., Martone M.E., Hoshijima M. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J. Cell Sci. 2009;122:1005–1013. doi: 10.1242/jcs.028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo T., Zhang T., Bers D.M. Ca2+/calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ. Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 54.Wehrens X.H.T., Lehnart S.E., Marks A.R. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 2004;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 55.Dibb K.M., Graham H.K., Trafford A.W. Analysis of cellular calcium fluxes in cardiac muscle to understand calcium homeostasis in the heart. Cell Calcium. 2007;42:503–512. doi: 10.1016/j.ceca.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 56.Shorofsky S.R., Aggarwal R., Balke C.W. Cellular mechanisms of altered contractility in the hypertrophied heart: big hearts, big sparks. Circ. Res. 1999;84:424–434. doi: 10.1161/01.res.84.4.424. [DOI] [PubMed] [Google Scholar]
- 57.Litwin S.E., Zhang D., Bridge J.H. Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ. Res. 2000;87:1040–1047. doi: 10.1161/01.res.87.11.1040. [DOI] [PubMed] [Google Scholar]
- 58.Gillespie D., Fill M. Pernicious attrition and inter-RyR2 CICR current control in cardiac muscle. J. Mol. Cell. Cardiol. 2013;58:53–58. doi: 10.1016/j.yjmcc.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laver D.R., O’Neill E.R., Lamb G.D. Luminal Ca2+-regulated Mg2+ inhibition of skeletal RyRs reconstituted as isolated channels or coupled clusters. J. Gen. Physiol. 2004;124:741–758. doi: 10.1085/jgp.200409092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Knollmann B.C., Chopra N., Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McLaughlin S., Mulrine N., McLaughlin A. Adsorption of divalent cations to bilayer membranes containing phosphatidylserine. J. Gen. Physiol. 1981;77:445–473. doi: 10.1085/jgp.77.4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Protasi F., Franzini-Armstrong C., Flucher B.E. Coordinated incorporation of skeletal muscle dihydropyridine receptors and ryanodine receptors in peripheral couplings of BC3H1 cells. J. Cell Biol. 1997;137:859–870. doi: 10.1083/jcb.137.4.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saito A., Inui M., Fleischer S. Ultrastructure of the calcium release channel of sarcoplasmic reticulum. J. Cell Biol. 1988;107:211–219. doi: 10.1083/jcb.107.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soeller C., Cannell M.B. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ. Res. 1999;84:266–275. doi: 10.1161/01.res.84.3.266. [DOI] [PubMed] [Google Scholar]
- 65.Reference deleted in proof.
- 66.Rizzi N., Liu N., Priori S.G. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ. Res. 2008;103:298–306. doi: 10.1161/CIRCRESAHA.108.171660. [DOI] [PubMed] [Google Scholar]
- 67.Franzini-Armstrong C., Protasi F., Ramesh V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 1999;77:1528–1539. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forbes M.S., Sperelakis N. Bridging junctional processes in couplings of skeletal, cardiac, and smooth muscle. Muscle Nerve. 1982;5:674–681. [Google Scholar]
- 69.Bers D.M. Kluwer Academic; Dordrecht, The Netherlands: 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. [Google Scholar]
- 70.Murphy R.M., Mollica J.P., Lamb G.D. Quantification of calsequestrin 2 (CSQ2) in sheep cardiac muscle and Ca2+-binding protein changes in CSQ2 knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2011;300:H595–H604. doi: 10.1152/ajpheart.00902.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ikemoto N., Nagy B., Gergely J. Studies on a metal-binding protein of the sarcoplasmic reticulum. J. Biol. Chem. 1974;249:2357–2365. [PubMed] [Google Scholar]
- 72.Wu X., Bers D.M. Sarcoplasmic reticulum and nuclear envelope are one highly interconnected Ca2+ store throughout cardiac myocyte. Circ. Res. 2006;99:283–291. doi: 10.1161/01.RES.0000233386.02708.72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.