

Abstract
The translocation of genetic material from the viral capsid to the cell is an essential part of the viral infection process. Whether the energetics of this process is driven by the energy stored within the confined nucleic acid or cellular processes pull the genome into the cell has been the subject of discussion. However, in vitro studies of genome ejection have been limited to a few head-tailed bacteriophages with a double-stranded DNA genome. Here we describe a DNA release system that operates in an archaeal virus. This virus infects an archaeon Haloarcula hispanica that was isolated from a hypersaline environment. The DNA-ejection velocity of His1, determined by single-molecule experiments, is comparable to that of bacterial viruses. We found that the ejection process is modulated by the external osmotic pressure (polyethylene glycol (PEG)) and by increased ion (Mg2+ and Na+) concentration. The observed ejection was unidirectional, randomly paused, and incomplete, which suggests that cellular processes are required to complete the DNA transfer.
Introduction
Viruses are the most abundant biological entities on our planet. They infect organisms from all three domains of life (Archaea, Bacteria, and Eukarya). It has been estimated that the world’s oceans contain ∼1030 virus particles (1). Due to their small size, viruses represent only a small fraction of the marine biomass. However, viruses have an enormous impact on cellular life in the oceans. They have been estimated to kill 20% of the microbial biomass every day, consequently affecting bio- and geochemical cycles. Furthermore, viruses cause infectious diseases (2), leading to human suffering and loss of food production.
Viruses that infect archaea were discovered in the 1970s but have recently gained more attention. To date, ∼100 archaeal viruses have been described (3,4). Since they were first discovered, archaeal viruses have captured scientists’ attention because of the diversity of their virion morphology and unique genes, which lack homologies in public databases (3). Interestingly, all of the archaeal viruses described so far have a DNA genome. Recent studies have described novel egress mechanisms for archaeal viruses that are not found in bacterial or eukaryotic viruses (5–7).
Although archaea are abundant in a variety of environments, currently isolated archaeal viruses are limited to those with extremophilic hosts (thermophiles, halophiles, and methanogens) (3,4). In contrast to bacterial viruses, most of which are head-tailed, archaeal viruses come in a variety of shapes (e.g., spindle, bottle, droplet, pleomorphic, spherical, head-tailed, or linear) (8). The most frequently encountered morphotype of the archaea-specific viruses is spindle-shaped (Fig. 1 A). These particles are commonly found in extreme environments, and so far ∼15 such viruses have been isolated (3,9–12).
Figure 1.
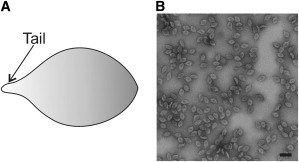
Spindle-shaped virus His1. (A) Schematic presentation of the virus particle. (B) Electron micrograph of negatively stained His1 particles. Scale bar: 100 nm.
Most of the isolated spindle-shaped viruses infect hyperthermophilic archaea, and to date only one such virus, His1, has been described for haloarchaea (Fig. 1). His1 was isolated from an Australian saltern and infects an extremely halophilic archaeon, Haloarcula hispanica (13). It contains a relatively short (14,462 bp) linear double-stranded DNA (dsDNA) genome encoding a putative B-type DNA polymerase (13,14). There are contradicting statements concerning the covalently bound genome terminal proteins (13,14); however, experimental evidence is scarce (14). Although His1 originates from a high-salinity environment, it also tolerates low salinity (13,15). The (74 × 44 nm2) His1 virion (13) is composed of one major and a few minor structural proteins, and although there is no lipid bilayer, the major capsid protein seems to be lipid modified (15). Similarities between the haloarchaeal virus His1 and other spindle-shaped viruses, which infect hyperthermophilic archaea, have led to the hypothesis that all spindle-shaped viruses with a short tail may be related and share a common ancestor (15).
Because there is practically no information on archaeal virus DNA ejection, we will briefly summarize what is known about bacteriophage DNA ejection. Early information about tight DNA packaging in bacteriophages implied that the pressure inside the capsid is high (16), and that this pressure is subsequently utilized to deliver the DNA into the cytoplasm (17,18). However, later studies indicated that the internal capsid pressure is only sufficient to initiate DNA ejection from bacteriophages (19–21), and that either host factors and viral proteins (19–24) or osmotic pressure differences between the host cytoplasm and surrounding medium (25) are needed to complete the DNA transfer. Such two-step DNA ejection has been described for several bacteriophages. Internalization of the phage T7 genome requires active pulling into the cell by RNA polymerases (20,22). Only ∼8% of the T5 genome can be injected into the host in one step. The rest of the genome is pulled in by phage-encoded proteins (26). In general, the stepwise ejection behavior of T5 is due to nicks in the DNA (21). In ϕ29, a push-pull ejection mechanism was discovered whereby 65% of the DNA is pushed by pressure and the rest is pulled in by an enzyme-assisted process (23,27).
Due to the lack of DNA ejection triggers, only a few in vitro ejection systems are available, with the most advanced being those of phages T5, λ, and SPP1 (19,25,28).
In vitro experiments have demonstrated that in bacteriophages λ and T5, DNA ejection can be suppressed by increased external osmotic pressure (29,30). The ejection is also sensitive to salt conditions. Increased magnesium concentration reduces the velocity and fraction of ejected DNA from phage λ, whereas sodium shows no such effect (31,32). Even more severe ejection suppression can be achieved by addition of spermine (32). Ejection suppression by di- and multivalent cations is explained by the fact that these cations reduce DNA repulsion forces inside the capsid (33,34). Here, we present what is to our knowledge the first in vitro observations of DNA ejection from a spindle-shaped archaeal virus. We used total internal reflection fluorescence (TIRF) microscopy to visualize the ejection. Using single-molecule experiments, we demonstrated that His1 ejection is suppressed by osmolyte-induced osmotic pressure and by increased mono- and divalent ion concentrations. The number of ejections and the ejected genome length were reduced when osmolyte or salts were added. We demonstrate that DNA ejection from His1 is directional. Our findings also support the presence of terminal proteins at both ends of the linear genome.
Materials and Methods
Particle and DNA purification
His1 virus particles were grown and purified as previously described (15). The 2× purified His1 particles were stored in His1 buffer containing 500 mM NaCl, 35 mM MgCl2, 10 mM KCl, 1 mM CaCl2, and 50 mM Tris-HCl (pH 7.2).
It has been observed that in the presence of low concentrations of detergents, His1 releases its genome without dissociating the virus particle (M.K. Pietilä, C. Hong, D.H. Bamford, and W. Chiu, unpublished data). This allows us to study this process using a single-molecule approach. DNA release and purification from the 2× purified virus particles was achieved by treatment with Nonidet P40 (0.01%; Sigma) or Triton X-100 (0.1%; Sigma) in 10-fold diluted His1 buffer for 90 min at room temperature, with or without protease treatment (proteinase K, trypsin, or bromelain at 0.5 mg/ml final concentration, at 37°C for 60 min).
For the DNA ejection experiments, we used 10-fold diluted His1 buffer containing 50 mM NaCl, 3.5 mM MgCl2, 1 mM KCl, 0.1 mM CaCl2, and 50 mM Tris-HCl (pH 7.2) (15). This buffer contains the smallest amount of salt that is sufficient for maintaining the stability of the virus particles. In control experiments, we also used 18% artificial salt water (SW; a 30% stock solution of SW contains 240 g NaCl, 30 g MgCl2⋅6H2O, 35 g MgSO4⋅7H2O, 7 g KCl, 5 ml of 1 M CaCl2⋅2H2O, and 80 ml of 1 M Tris-HCl (pH 7.2) per liter of water) and modified growth medium (MGM) containing 23% SW, 5 g peptone (Oxoid Ltd., Hampshire, UK), and 1 g Bacto yeast extract (BD, Franklin Lakes, NJ) per liter (35).
Microfluidic chamber preparation
Glass coverslips (60 × 24 × 0.17 mm3; Corning, Corning, NY) for microfluidic chambers were prepared as described previously (36). Coverslips were heated to 95°C in 0.5% Alconox detergent (Alconox, White Plains, NY) for 60 min, rinsed three times with distilled water, dried in an air stream, and used immediately to make chambers and to bind the samples. Microfluidic chambers were made as described previously (37) using microscope glass slides (75 × 25 × 1 mm3) with drilled 1.6-mm-diameter holes. We glued 0.25 mm i.d. PEEK tubing to the holes using UV-curing epoxy (NOA81; Norland Products, Cranbury, NJ). A one-channel pattern was cut into a 200-μm-thick double-stick tape spacer (Tesa AB, Kungsbacka, Sweden) that was glued to the slide. The chamber was sealed with a freshly washed and dried coverslip, and the sample was then injected.
Sample preparation
Virus particles (2× purified, A260 from 225 to 325) were diluted ∼2 × 10−4 using 10-fold diluted His1 buffer (to achieve the desired particle density on the glass) and treated with DNase I (0.0125 mg/ml; New England Biolabs, Ipswich, MA) for 20 min at 37°C (to eliminate free DNA). DNase I was heat inactivated (65°C, 15 min). The virus sample was injected into the chamber and left to settle at room temperature for 15 min. During this time, capsids were nonspecifically bound to the cleaned coverslip surface (36). The virus-containing chamber (in the microscope) was washed with 10-fold diluted His1 buffer for 5 min at a 50 μl/min flow rate (syringe pump; Harvard Apparatus, Holliston, MA). DNA ejection was triggered by injecting 10-fold diluted His1 buffer containing 0.05% of Triton X-100 (Sigma). SYBR Gold (nucleic acid gel stain, diluted 5 × 10−6, catalog No. S11494; Invitrogen/Life Technologies Ltd., Paisley, UK) was added to visualize the DNA. The flow (100 μl/min) was maintained during the experiment to fully stretch the DNA. After the ejection was triggered, the flow was switched to the buffer containing no detergent while data were collected (see Movie S1 in the Supporting Material). Fig. S1 shows a schematic image of DNA ejection in the chamber.
In the experiments with elevated osmotic pressure and ion concentrations, polyethylene glycol 300 (PEG300; Sigma-Aldrich), MgCl2, or NaCl was added at the desired concentrations. The correspondence between osmotic pressure and PEG300 concentration was calculated according to the method described by Reid and Rand (38).
Experiments with DNA
To determine the genome length and ejection directionality, we used purified His1 genome with or without protease treatment (see above). DNA was diluted in 10 mM Tris-HCl buffer (pH 8) and injected into the freshly made chamber. The length of DNA was determined by stretching it in the flow of 10-fold diluted His1 buffer, 10 mM Tris- HCl (pH 8), or 1× FastDigest (FD; Fermentas; Thermo Fisher Scientific, Waltham, MA) buffers containing SYBR gold (5 × 10−6 dilution) at 100 μl/min. In the control experiments, FD DraI enzyme (Fermentas) was used in 1× FD buffer. The DNA was diluted to 1× FD buffer and injected into the chamber, and enzyme was then added (2.5 FD units (FDU) of enzyme to 200 μl of sample). The sample was incubated for 15 min at 37°C, washed with the 10-fold diluted His1 buffer, and the fragment length was determined in the flow.
Determining ejection directionality
We determined the directionality of ejection by adding 2.5 FDU of DraI enzyme to the His1 sample (see “Sample preparation” above) after triggering the ejection, incubating the chamber at 37°C for 15 min, and determining the length of the ejected DNA fragments in the flow (DNA that has been cleaved off escapes with the flow, and only DNA fragments that are still attached to the viral particles are detectable).
Fluorescence microscopy
Experiments were carried out with a custom-built fluorescence microscope in an objective-TIRF configuration. The instrument is based on an inverted microscope (TE-2000; Nikon Instruments Melville, NY). Light from the excitation laser (Sapphire 488 50 mW; Coherent, Santa Clara, CA) is expanded ×33 with two-lens telescopes to a 23-mm-diameter beam. A mechanical shutter (99A360; Ludl Electronic Products Ltd., Hawthorne, NY) blocks the excitation light when appropriate. A plano-convex lens (LA1725-A, f = 400 mm; Thorlabs, Newton, NJ) focuses the expanded beam onto the back-focal plane of the microscope objective (CFI Apo TIRF 100× oil immersion, NA = 1.49; Nikon). The focusing lens is offset perpendicular to the optical axis to displace the excitation beam, which results in TIRF excitation at the flow-cell surface (Fig. S1). A dichroic mirror (495-Di03; Semrock, Rochester, NY) reflects the excitation light onto the optical axis of the objective. Fluorescence emission is filtered with a dedicated band-pass filter (520/35-23; Semrock) and then detected with a cooled EMCCD camera (iXon DU-897; Andor Technology, Belfast, UK). The excitation laser power was set to 5 mW, resulting in 1 mW at the sample plane. The electron-multiplying (EM) gain of the camera was 180 and the exposure time was 30 ms. The frame rate of the camera was synchronized to 15 frames per second (fps).
To collect statistics on DNA length and number of ejections, we recorded several frames in the following manner: Image frames were recorded at evenly spaced intervals because the virus particles were randomly distributed across the coverslip surface. The excitation laser was switched on and the camera was set to record continuously. After ejection was triggered, we moved the flow chamber a distance equal to the frame width along the flow direction. After translation, the image was refocused before photobleaching or photodamage occurred. This cycle was repeated for a desired number of frames.
Data analysis
Fluorescence images were analyzed in MATLAB R2011b (The MathWorks, Natick, MA). A similar analysis procedure was used for all experiments. First, we select frames of interest (stage stationary, image in focus) by manually browsing the recorded video. Next, relevant ejection events are picked by hand (corresponding to DNA lengths longer than 2 pixels, see the paragraph on uncertainty below). In the case of obtaining ejection statistics, this gives the number of ejections in each frame. Measuring the length of the ejected DNAs requires additional analysis. For the length analysis, we select nonoverlapping DNA molecules that are extended by the flow. Each frame is low-pass filtered with a Gaussian-kernel filter (σ = 1.5 pixels, window size = 3 × 3 pixels). The filtered frames are processed with an edge-detection algorithm (the Canny method (39)), which yields a binary image showing the edge contours of all ejected DNA fractions. The coordinates of the two DNA ends are determined by manually selecting the opposing ends of each edge contour. The length of each ejected DNA is computed as the Euclidean distance between its ends. Fig. S2 shows schematically the length-analysis procedure. We obtain the length time series of each single ejection event in a similar manner, and choose relevant events by browsing the video. The length of each single ejected DNA is determined as described above for each time point until photodamage destroys the molecule. Single DNA ejection events are recorded at 58 fps, which is achieved by dropping the frame size to 256 × 256 pixels. Additionally, the exposure time is set to 15 ms and the EM gain is set to 200.
We used the manual approach because it allowed us to analyze ejection events that slightly overlapped in space or that featured low fluorescence intensity. We tested a simple, fully automated thresholding-based detection algorithm, but since it failed with even slightly overlapping events (data not shown), we preferred the semiautomatic method.
The uncertainty of the DNA length measurement can be estimated if we assume that the edge-detection method gives the location of the DNA ends with a precision of 1 pixel. Then the uncertainty of the length is given by error propagation of the Euclidean distance equation. Assuming a pixel size of 106.7 nm (length calibrated value; data not shown), the uncertainty in length is 150.9 nm ( times the pixel size). As for systematic errors, we assume that the measured lengths are low estimates, since photocleavage takes place and shortens some of the DNA molecules. The magnification of the optical system and the diffraction-limited resolution put a limit to how short the ejected DNAs could be to enable their resolution. In practice, anything shorter than 2 pixels (213.4 nm) cannot be resolved reliably. Consequently, the measured length histograms have a cutoff length corresponding to this value. In the case of ejection statistics, it is not possible to count the number of ejections reliably when virus particles aggregate or when the tethers overlap significantly. However, one can minimize this kind of bias by collecting enough statistics. At high osmolyte concentrations, where the number of ejections is low (<10), the relative contribution of this error source might be significant.
Results and Discussion
Ejection velocity
Fig. 2 A shows DNA length traces of single ejection events triggered by the detergent and measured in 10-fold diluted His1 buffer. The ejection velocity varies as a function of ejected genome length. Initially, the DNA was ejected at a seemingly constant rate, which slowed down toward the end of the ejection. The velocity was estimated from a line fit to the linear part of the curve. The slope of the fit yielded a first-order approximation of ejection velocity. The mean ejection velocity was 48.97 ± 24.48 μm/s corresponding to 144 ± 72 kbp/s. Because this method gives only a first-order estimate of the ejection rate, it is insensitive to changes in velocity at the end of the ejection. This resulted in a wide velocity distribution. The frame rate of the camera limits the maximum resolvable velocity. If the whole genome (14,462 bp ≈ 4.9 μm) were ejected during one frame (1/58 s), the resulting velocity would be L × fps (1/s) = 290 μm/s (853 kbp/s). A higher frame rate would be needed to resolve a faster event. The timescale of the dye-binding kinetics could in principle affect the velocity measurement. However, the binding of SYBR Gold to ejected DNA from bacteriophage λ is essentially instantaneous (31). Hence, the observed velocity is detectable by the time resolution of the instrument. A comparison with similar measurements on bacteriophages shows that the measured ejection velocity for His1 exceeds that of phage λ (60 kbp/s) (36) and T5 (75 kbp/s) (21,40). The measured velocity is similar to the estimated velocity for T7 (140 kbp/s) (20). For His1, the ejection happens randomly, either monotonically or with one to three intermediate stops (Fig. 2 A). The ejection steps do not seem to correspond to well-defined intermediate stopping positions as is the case with bacteriophage T5 (21). This might suggest that the ejection steps are related to a conformational change or deformation of the capsid, which results in seemingly random stopping positions.
Figure 2.
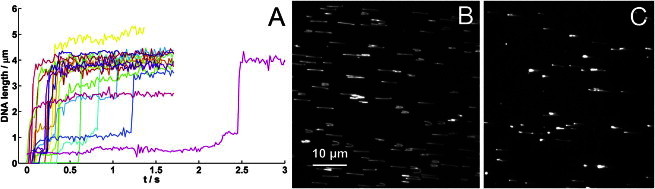
(A) Single DNA ejection events recorded at 58 fps. Linear fits to the steepest parts of the curves yield an average ejection velocity of 48.97 ± 24.48 μm/s (144 ± 72 kbp/s). (B and C) Primary fluorescence data represent purified His1 DNA nonspecifically attached to a cleaned coverslip by terminal proteins stretched out in the flow (B) and His1 DNA ejected from the virus attached to a cleaned coverslip stretched out in the flow (C).
Length of the purified genome
We observed that both ends of the DNA purified from His1 particles without protease treatment were nonspecifically bound to the etched coverslip, forming loops that were stretched by the flow (Fig. 2 B). After protease treatment, the DNA lost its ability to attach to the glass. The most efficient protease treatment was that with proteinase K (no DNA stuck to the glass), and the least effective was bromelain (a small amount (1/10 compared with the untreated sample) of DNA was found on the glass, mostly attached by one end). These experiments therefore indicated the presence of genome terminal proteins, although we did not observe different behaviors in the protease-treated and untreated DNA when analyzed by gel electrophoresis (data not shown).
The mean length of the untreated sample (5.28 ± 0.13 μm) is similar to that of the protease-treated sample (5.32 ± 0.19 μm). If DNA molecules are attached from both ends in the flow direction, the observed length could be shorter than that of a freely stretched DNA molecule. In both cases, the measured DNA length exceeded that estimated from the genome length (4.918 μm). This is attributed to lengthening of the DNA in the presence of the intercalating dye (41).
Directionality of ejection
The His1 DNA ejected from the virus particles was observed to be shorter than the total genome length (Fig. 3, A and C), indicating that complete genome ejection did not take place. Such behavior was also reported for λ and T5 (36,40,42). These observations of incomplete ejection suggest that an additional pull in the cell cytoplasm is needed to complete the DNA transfer.
Figure 3.
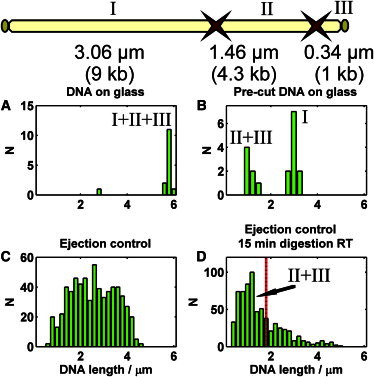
DNA length histograms for the cleavage experiment. Top: Schematic of DraI restriction sites of the His1 genome. The genome is divided into fragments I (9 kb), II (4.3 kb), and III (1 kb). (A) Undigested purified His1 genome. (B) DraI-digested purified His1 genome; the sample consists of ∼1.5 μm and ∼3 μm fragments, corresponding to 5.3 kb (II+III) and 9 kb (I), respectively. (C) Length distribution of DNA ejected from His1 particles. (D) Length distribution of DNA ejected from His1 particles and digested with DraI. Mostly short fragments of ∼1 μm are observed. Vertical line corresponds to a 5.3-kb-long fragment (II+III).
The directionality of genome ejection was resolved using a restriction enzyme cleaving His1 DNA (directionality refers to whether the left or right end of the genome comes out first; see Fig. 3 and Bath and Dyall-Smith (13)). The His1 genome has two restriction sites for DraI, dividing the genome into three parts 9 kb, 4.3 kb, and 1 kb long (from the left to the right end). In Fig. 3 the fragments are labeled I, II, and III, respectively. The control restriction analysis was done using purified His1 DNA. The digestion of this sample produced ∼1.5 μm and ∼3 μm long DNA fragments, which correspond to 5.3 kb (II+III) and 9 kb (I) fragments, respectively (Fig. 3 B; we assume that the 5.3 kb fragment is a result of incomplete digestion leaving the 4.3 kb and 1 kb fragments together). The 1 kb (∼0.3 μm) fragment was not resolvable due to the resolution limitations of the microscope.
Next, digestion was performed with DNA ejected from the virus particles. These particles were attached to the coverslip (see Materials and Methods), the ejection was triggered by detergent, and restriction enzyme was introduced into the chamber. After 15 min incubation at 37°C, the length of the ejected and digested DNA fragments was measured as previously described. The observable lengths in the digested and undigested control samples are shown in Fig. 3, C and D. When ejected DNA was digested with DraI, only short DNA fragments (∼1 μm) were observed in the chamber, indicating that the left end of the genome was ejected first and subsequently cleaved off (9 kb fragment), leaving the 5.3 kb fragment associated with the virus particle. The vertical line in Fig. 3 D corresponds to the length of a 5.3 kb fragment.
Sensitivity of DNA ejection to increased osmotic pressure
The DNA ejection efficiency of bacterial viruses depends on the osmotic pressure outside the capsid (29,43). We tested His1 for this phenomenon using PEG300 and glycerol as osmolytes. Similar to the case of phage SPP1 ejection in the presence of PEG (19), we observed two different inhibitory effects on ejection in the presence of external pressure. First, the number of ejections diminished with increasing PEG concentration (Fig. 4 A). At low PEG concentration, we observed a certain nonmonotonous behavior similar to that reported earlier for phage λ (31). Second, the length of the ejected DNA decreased up to complete inhibition of ejection. This is evident in the length histograms (Fig. 5). A wide length distribution, reaching up to the full genome length, was present in the control experiment (Fig. 5 A). Interestingly, a small amount (0.88%) of PEG300 increased the relative number of longer molecules (Fig. 5 B). Higher PEG concentrations (5–12.6%) resulted in distributions centered at shorter lengths (Fig. 5, C–F).
Figure 4.
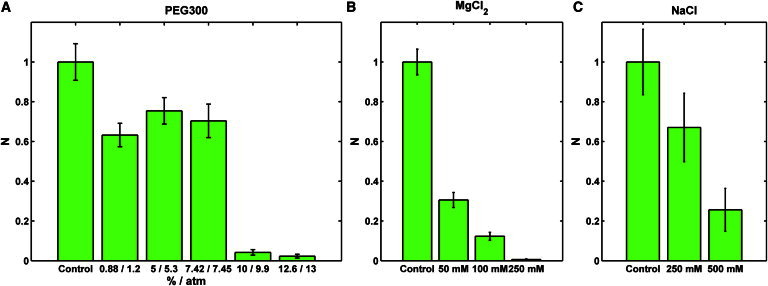
(A–C) Normalized average number of ejections per field of view in the presence of (A) PEG300 (the corresponding osmotic pressure was calculated from PEG300 concentration as described previously (38)), (B) magnesium chloride (the control sample was prepared in 10-fold diluted His1 buffer containing 3.5 mM MgCl2), and (C) sodium chloride (the control sample was prepared in 10-fold diluted His1 buffer containing 50 mM NaCl). Error bars represent 1 SD.
Figure 5.
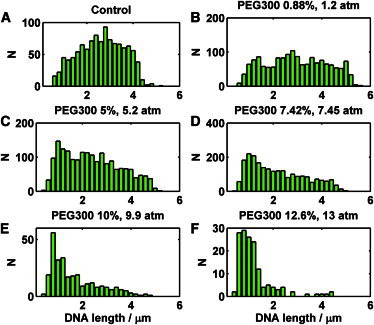
Ejected DNA length histograms in varying concentrations of PEG300. All experiments were done in 10-fold diluted His1 buffer. (A) The control experiment contained no PEG300. (B) Experiment with 0.88% PEG300 corresponding to 1.2 atm. (C) Experiment with 5% PEG300 corresponding to 5.2 atm. (D) Experiment with 7.42% PEG300 corresponding to 7.45 atm. (E) Experiment with 10% PEG300 corresponding to 9.9 atm. (F) Experiment with 12.6% PEG300 corresponding to 13 atm.
A low but approximately constant background in the experiment (there were always one or two particles per sample that were spontaneously releasing DNA) prevented us from determining the exact concentration of PEG (or the corresponding osmotic pressure) necessary to completely inhibit ejection. However, at ∼10 atm, we saw as few ejections as we did in the negative control. We saw similar behavior with glycerol as an osmolyte. In this case, the number of ejections dropped dramatically between 8.5 and 14 atm (data not shown). However, due to condensation and breakage of the ejected DNA, we cannot present quantitative data on the length of ejected DNA molecules in the presence of glycerol (Movie S2). We observed a similar effect in the presence of spermidine (data not shown). Perhaps in contrast to the case of phage λ (29), the glycerol molecules cannot penetrate the His1 capsid. Subsequently, they create an osmotic pressure difference that reduces the number of ejecting particles.
Estimating the internal capsid pressure
We estimated the internal capsid pressure of His1 using the method of Purohit et al. (43), which estimates the packaging force and pressure inside the capsid based on elastic bending and electrostatic self-repulsion of the packaged genome. Based on negative-stain electron microscopy (13), we estimate the inner space of the capsid to be approximately ellipsoidal in shape (major axis 30.4 nm, minor axes 17.7 nm). The packaged volume fraction is estimated to be ρpack = VDNA / VCapsid (43). Assuming that the DNA is a cylinder with a radius of 1 nm and length given by 0.34 nm/bp, the packaged volume fraction is ρpack = 0.495. The electrostatic parameters were F0 = 1.2 × 104 pN/nm2 and c = 0.30 nm. These values were used by Evilevitch et al. (29) (in the presence of 10 mM MgSO4). To estimate the packaging force, we first solved Eq. 1 with respect to ds (strand spacing) and R (inner radius of the packaged DNA coil) (43):
| (1) |
where kB is the Boltzmann constant, T is the temperature, and ξP is the persistence length of DNA. For an ellipsoid whose major axis is 2h and whose minor axes are Rout, the height of the packed DNA column is
| (2) |
The packaging force is
| (3) |
The internal pressure is estimated from the force needed to push a rigid rod with a radius of RDNA = 2.25 nm into the PEG solution at a force given by Eq. 3 (32). The numerical solution of Eqs. 1–3 predicts that the ejection is completely inhibited at 38.5 atm (Fig. S3 C). Our predicted pressure is considerably higher than the measured one (10 atm). The model assumes that each capsid ejects a certain fraction of its genome. It does not explain the wide distribution of ejected lengths that we observe in our experiments. Moreover, the model does not account for the presence of terminal proteins on the genome or for the unknown volume occupied by these proteins. The strand spacing ds predicted by the above computation was 2.64 nm (Fig. S3 A), which is similar to the corresponding value in dsDNA phages (∼2.7 nm) (43). The corresponding packaging force F is 15.55 pN (Fig. S3 B). Perhaps the effect of PEG on water activity inside the capsid (44) better explains the reduction in ejected DNA length. The presence of PEG in the buffer exerts osmotic pressure on the capsid wall. Because the capsid is impermeable to PEG, a hydrostatic pressure difference arises that causes water to flow out of the capsid. As a result, the electrostatic repulsion between packed DNA strands weakens, which lowers the ejection force (44). In bacterial viruses, the internal capsid pressure is estimated to be 50 atm for λ, 60 atm for φ29, and 45 atm for PRD1 (29,45). However, the osmotic pressure needed to completely suppress ejection in λ is only 20 atm (29). We estimated that the His1-packaged volume fraction is 49.5%, which is comparable to that of bacterial viruses (T7: 49%; φ29: 46%; and λ: 42% (43)).
The ejection process of a halophilic virus His1 seems to be suppressed by quite low osmotic pressure. It should be noted that it may be more efficient to trigger the ejection by virus-host protein interactions and conformational changes in vivo than in vitro.
Sensitivity of ejection to increased ion concentration
Unlike PEG300 or glycerol, mono- and divalent salts enter the capsid freely and stabilize the genome by screening the negative charge on the phosphate backbone of the DNA (32,46). We performed ejection experiments to see how the presence of mono- (Na+) and divalent (Mg2+) cations affect the ejection process. Similarly to the high osmotic pressure effect, we observed fewer ejection events when the magnesium ion concentration was increased (Fig. 4 B). Adding 50 mM of MgCl2 reduced the average length of the ejected DNA to 20% of the total genome length (Fig. 6 B). A further increase in Mg2+ concentration did not significantly change the shape of the length distribution (Fig. 6, C and D), but the number of ejections was almost completely suppressed (Fig. 4 B). The stabilizing effect of Mg2+ on encapsidated DNA is known in bacterial viruses (32,46).
Figure 6.
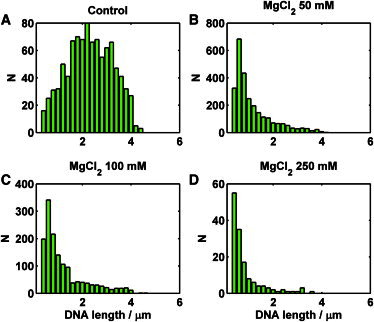
Histograms of DNA lengths ejected in the presence of magnesium chloride. (A) The control experiment was done in 10-fold diluted His1 buffer containing 3.5 mM MgCl2. (B) Modified 10-fold diluted His1 buffer containing 50 mM MgCl2. (C) Modified 10-fold diluted His1 buffer containing 100 mM MgCl2. (D) Modified 10-fold diluted His1 buffer containing 250 mM MgCl2.
The same effects (i.e., decreased number and length of ejections) were also observed with NaCl, albeit to a lesser extent. The number of ejections dropped monotonically, but a higher salt concentration (250 mM NaCl) was required to see an effect (Fig. 4 C). The mean ejected length dropped somewhat, but the distributions did not exhibit the same tendency to center around 20% of the genome length as with Mg2+ (Fig. 7). A negligible effect of sodium ions on ejection was also reported by Evilevitch et al. (32) for phage λ.
Figure 7.
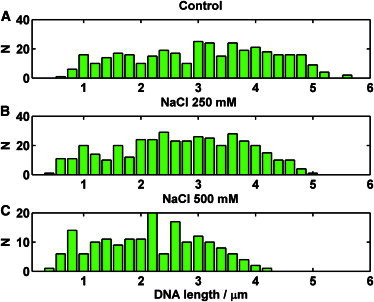
Ejected DNA length histograms in the presence of sodium chloride. (A) The control experiment was done in 10-fold diluted His1 buffer containing 50 mM NaCl. (B) Modified 10-fold diluted His1 buffer containing 250 mM NaCl. (C) Modified 10-fold diluted His1 buffer containing 500 mM NaCl.
The observation that the ejection process is not significantly hindered by high Na+ concentrations is reasonable, given that Na+ is the most abundant ion in the hypersaline environment as well as in the oceans. Moreover, haloarchaeal cells typically have high concentrations of monovalent ions (K+, Cl−) in the cytoplasm (47). Our control experiments show that the number of ejections in 18% SW (2.5 M NaCl) or 23% MGM (3.15 M NaCl) media was reduced only to 30–20% compared with 10-fold diluted His1 buffer (Fig. S4).
Conclusions
Our single-molecule experiments on the archaeal virus His1 show that the directionality of DNA ejection is fixed. The experiments also support the presence of genome terminal proteins at both ends of the linear DNA molecule. Unlike the case with most bacteriophages, His1 DNA ejection in vitro is only partial, which suggests that cellular processes are required to complete the DNA transfer. The stepwise manner of ejection and the ejection velocity in different ionic conditions require further investigation. The distribution of ejected DNA lengths is wide and centered around 50% of the total genome length. PEG300-induced osmotic pressure inhibits ejection at ∼10 atm, which agrees qualitatively with theoretical predictions. A similar effect was also observed with glycerol. Increasing the magnesium concentration drastically decreased the number of ejections and the ejected DNA length due to the stabilizing effect of charge screening. Analogous experiments with sodium ions revealed a similar behavior, but at higher ion concentrations than used for divalent ions.
Acknowledgments
We thank Päivi Hannuksela and Helin Veskiväli for excellent technical assistance.
This work was supported by Academy Professor (Academy of Finland) grants 255342 and 256518 to D.H.B. and an Academy of Finland grant (128518) to E.H. K.J.H. was supported by the Graduate School of Chemical Sensors and Microanalytical Systems. The EU ESFRI Instruct Centre for Virus Production and Purification, supported by the University of Helsinki, was used for the research.
Supporting Material
References
- 1.Suttle C.A. Marine viruses—major players in the global ecosystem. Nat. Rev. Microbiol. 2007;5:801–812. doi: 10.1038/nrmicro1750. [DOI] [PubMed] [Google Scholar]
- 2.Yang X., Yang H., Zhao G.P. Infectious disease in the genomic era. Annu. Rev. Genomics Hum. Genet. 2008;9:21–48. doi: 10.1146/annurev.genom.9.081307.164428. [DOI] [PubMed] [Google Scholar]
- 3.Pina M., Bize A., Prangishvili D. The archeoviruses. FEMS Microbiol. Rev. 2011;35:1035–1054. doi: 10.1111/j.1574-6976.2011.00280.x. [DOI] [PubMed] [Google Scholar]
- 4.Atanasova N.S., Roine E., Oksanen H.M. Global network of specific virus-host interactions in hypersaline environments. Environ. Microbiol. 2012;14:426–440. doi: 10.1111/j.1462-2920.2011.02603.x. [DOI] [PubMed] [Google Scholar]
- 5.Bize A., Karlsson E.A., Prangishvili D. A unique virus release mechanism in the Archaea. Proc. Natl. Acad. Sci. USA. 2009;106:11306–11311. doi: 10.1073/pnas.0901238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brumfield S.K., Ortmann A.C., Young M.J. Particle assembly and ultrastructural features associated with replication of the lytic archaeal virus sulfolobus turreted icosahedral virus. J. Virol. 2009;83:5964–5970. doi: 10.1128/JVI.02668-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quax T.E.F., Krupovič M., Prangishvili D. The Sulfolobus rod-shaped virus 2 encodes a prominent structural component of the unique virion release system in Archaea. Virology. 2010;404:1–4. doi: 10.1016/j.virol.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 8.Ackermann H.W., Prangishvili D. Prokaryote viruses studied by electron microscopy. Arch. Virol. 2012;157:1843–1849. doi: 10.1007/s00705-012-1383-y. [DOI] [PubMed] [Google Scholar]
- 9.Oren A., Bratbak G., Heldal M. Occurrence of virus-like particles in the Dead Sea. Extremophiles. 1997;1:143–149. doi: 10.1007/s007920050027. [DOI] [PubMed] [Google Scholar]
- 10.Sime-Ngando T., Lucas S., Prangishvili D. Diversity of virus-host systems in hypersaline Lake Retba, Senegal. Environ. Microbiol. 2011;13:1956–1972. doi: 10.1111/j.1462-2920.2010.02323.x. [DOI] [PubMed] [Google Scholar]
- 11.Rachel R., Bettstetter M., Prangishvili D. Remarkable morphological diversity of viruses and virus-like particles in hot terrestrial environments. Arch. Virol. 2002;147:2419–2429. doi: 10.1007/s00705-002-0895-2. [DOI] [PubMed] [Google Scholar]
- 12.Rice G., Stedman K., Young M.J. Viruses from extreme thermal environments. Proc. Natl. Acad. Sci. USA. 2001;98:13341–13345. doi: 10.1073/pnas.231170198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bath C., Dyall-Smith M.L. His1, an archaeal virus of the Fuselloviridae family that infects Haloarcula hispanica. J. Virol. 1998;72:9392–9395. doi: 10.1128/jvi.72.11.9392-9395.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bath C., Cukalac T., Dyall-Smith M.L. His1 and His2 are distantly related, spindle-shaped haloviruses belonging to the novel virus group, Salterprovirus. Virology. 2006;350:228–239. doi: 10.1016/j.virol.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Pietilä M.K., Atanasova N.S., Bamford D.H. Modified coat protein forms the flexible spindle-shaped virion of haloarchaeal virus His1. Environ. Microbiol. 2012 doi: 10.1111/1462-2920.12030. Oct 25. http://dx.doi.org/10.1111/1462-2920.12030 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 16.North A.C., Rich A. X-ray diffraction studies of bacterial viruses. Nature. 1961;191:1242–1245. doi: 10.1038/1911242a0. [DOI] [PubMed] [Google Scholar]
- 17.Zárybnický V. Mechanism of T-even DNA ejection. J. Theor. Biol. 1969;22:33–42. doi: 10.1016/0022-5193(69)90078-2. [DOI] [PubMed] [Google Scholar]
- 18.Hershey A.D., Chase M. Independent functions of viral protein and nucleic acid in growth of bacteriophage. J. Gen. Physiol. 1952;36:39–56. doi: 10.1085/jgp.36.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.São-José C., de Frutos M., Tavares P. Pressure built by DNA packing inside virions: enough to drive DNA ejection in vitro, largely insufficient for delivery into the bacterial cytoplasm. J. Mol. Biol. 2007;374:346–355. doi: 10.1016/j.jmb.2007.09.045. [DOI] [PubMed] [Google Scholar]
- 20.Kemp P., Gupta M., Molineux I.J. Bacteriophage T7 DNA ejection into cells is initiated by an enzyme-like mechanism. Mol. Microbiol. 2004;53:1251–1265. doi: 10.1111/j.1365-2958.2004.04204.x. [DOI] [PubMed] [Google Scholar]
- 21.de Frutos M., Letellier L., Raspaud E. DNA ejection from bacteriophage T5: analysis of the kinetics and energetics. Biophys. J. 2005;88:1364–1370. doi: 10.1529/biophysj.104.048785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molineux I.J. No syringes please, ejection of phage T7 DNA from the virion is enzyme driven. Mol. Microbiol. 2001;40:1–8. doi: 10.1046/j.1365-2958.2001.02357.x. [DOI] [PubMed] [Google Scholar]
- 23.González-Huici V., Salas M., Hermoso J.M. The push-pull mechanism of bacteriophage Ø29 DNA injection. Mol. Microbiol. 2004;52:529–540. doi: 10.1111/j.1365-2958.2004.03993.x. [DOI] [PubMed] [Google Scholar]
- 24.de Frutos M., Brasiles S., Raspaud E. Effect of spermine and DNase on DNA release from bacteriophage T5. Eur Phys J E Soft Matter. 2005;17:429–434. doi: 10.1140/epje/i2005-10019-5. [DOI] [PubMed] [Google Scholar]
- 25.Panja D., Molineux I.J. Dynamics of bacteriophage genome ejection in vitro and in vivo. Phys. Biol. 2010;7:045006. doi: 10.1088/1478-3975/7/4/045006. [DOI] [PubMed] [Google Scholar]
- 26.Lanni Y.T. First-step-transfer deoxyribonucleic acid of bacteriophage T5. Bacteriol. Rev. 1968;32:227–242. doi: 10.1128/br.32.3.227-242.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alcorlo M., González-Huici V., Salas M. The phage φ29 membrane protein p16.7, involved in DNA replication, is required for efficient ejection of the viral genome. J. Bacteriol. 2007;189:5542–5549. doi: 10.1128/JB.00402-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molineux I.J. Fifty-three years since Hershey and Chase; much ado about pressure but which pressure is it? Virology. 2006;344:221–229. doi: 10.1016/j.virol.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 29.Evilevitch A., Lavelle L., Gelbart W.M. Osmotic pressure inhibition of DNA ejection from phage. Proc. Natl. Acad. Sci. USA. 2003;100:9292–9295. doi: 10.1073/pnas.1233721100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castelnovo M., Evilevitch A. DNA ejection from bacteriophage: towards a general behavior for osmotic-suppression experiments. Eur Phys J E Soft Matter. 2007;24:9–18. doi: 10.1140/epje/i2007-10205-5. [DOI] [PubMed] [Google Scholar]
- 31.Grayson, P. 2007. The DNA ejection process in bacteriophage λ. PhD thesis. California Institute of Technology, Pasadena, CA.
- 32.Evilevitch A., Fang L.T., Knobler C.M. Effects of salt concentrations and bending energy on the extent of ejection of phage genomes. Biophys. J. 2008;94:1110–1120. doi: 10.1529/biophysj.107.115345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raspaud E., Durand D., Livolant F. Interhelical spacing in liquid crystalline spermine and spermidine-DNA precipitates. Biophys. J. 2005;88:392–403. doi: 10.1529/biophysj.104.040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raspaud E., Olvera de la Cruz M., Livolant F. Precipitation of DNA by polyamines: a polyelectrolyte behavior. Biophys. J. 1998;74:381–393. doi: 10.1016/S0006-3495(98)77795-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nuttall S.D., Dyall-Smith M.L. HF1 and HF2: novel bacteriophages of halophilic archaea. Virology. 1993;197:678–684. doi: 10.1006/viro.1993.1643. [DOI] [PubMed] [Google Scholar]
- 36.Grayson P., Han L., Phillips R. Real-time observations of single bacteriophage λ DNA ejections in vitro. Proc. Natl. Acad. Sci. USA. 2007;104:14652–14657. doi: 10.1073/pnas.0703274104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wallin A.E., Ojala H., Hæggström E. Dual-trap optical tweezers with real-time force clamp control. Rev. Sci. Instrum. 2011;82:083102. doi: 10.1063/1.3615309. [DOI] [PubMed] [Google Scholar]
- 38.Reid C., Rand R.P. Probing protein hydration and conformational states in solution. Biophys. J. 1997;72:1022–1030. doi: 10.1016/S0006-3495(97)78754-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Canny J. A computational approach to edge detection. IEEE Trans. Pattern Anal. Mach. Intell. 1986;PAMI-8:679–698. [PubMed] [Google Scholar]
- 40.Mangenot S., Hochrein M., Letellier L. Real-time imaging of DNA ejection from single phage particles. Curr. Biol. 2005;15:430–435. doi: 10.1016/j.cub.2004.12.080. [DOI] [PubMed] [Google Scholar]
- 41.Bennink M.L., Schärer O.D., Greve J. Single-molecule manipulation of double-stranded DNA using optical tweezers: interaction studies of DNA with RecA and YOYO-1. Cytometry. 1999;36:200–208. doi: 10.1002/(sici)1097-0320(19990701)36:3<200::aid-cyto9>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 42.Evilevitch A., Gober J.W., Gelbart W.M. Measurements of DNA lengths remaining in a viral capsid after osmotically suppressed partial ejection. Biophys. J. 2005;88:751–756. doi: 10.1529/biophysj.104.045088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Purohit P.K., Inamdar M.M., Phillips R. Forces during bacteriophage DNA packaging and ejection. Biophys. J. 2005;88:851–866. doi: 10.1529/biophysj.104.047134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeembaeva M., Castelnovo M., Evilevitch A. Osmotic pressure: resisting or promoting DNA ejection from phage? J. Mol. Biol. 2008;381:310–323. doi: 10.1016/j.jmb.2008.05.081. [DOI] [PubMed] [Google Scholar]
- 45.Cockburn J.J.B., Abrescia N.G.A., Stuart D.I. Membrane structure and interactions with protein and DNA in bacteriophage PRD1. Nature. 2004;432:122–125. doi: 10.1038/nature03053. [DOI] [PubMed] [Google Scholar]
- 46.Rau D.C., Lee B., Parsegian V.A. Measurement of the repulsive force between polyelectrolyte molecules in ionic solution: hydration forces between parallel DNA double helices. Proc. Natl. Acad. Sci. USA. 1984;81:2621–2625. doi: 10.1073/pnas.81.9.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oren A. The order Halobacteriales. In: Dworkin M., Falkow S., Rosenberg E., Schleifer K.-H., Stackebrandt E., editors. The Prokaryotes. A Handbook on the Biology of Bacteria. Springer; New York: 2006. pp. 113–164. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.