

Abstract
The intrinsically disordered region (IDR) of a protein is an important topic in molecular biology. The functional significance of IDRs typically involves gene-regulation processes and is closely related to posttranslational modifications such as phosphorylation. We previously reported that the Drosophila facilitates chromatin transcription (FACT) protein involved in chromatin remodeling contains an acidic ID fragment (AID) whose phosphorylation modulates FACT binding to nucleosomes. Here, we performed dynamic atomic force microscopy and NMR analyses to clarify how the densely phosphorylated AID masks the DNA binding interface of the high-mobility-group domain (HMG). Dynamic atomic force microscopy of the nearly intact FACT revealed that a small globule temporally appears but quickly vanishes within each mobile tail-like image, corresponding to the HMG-containing IDR. The lifespan of the globule increases upon phosphorylation. NMR analysis indicated that phosphorylation induces no ordered structure but increases the number of binding sites in AID to HMG with an adjacent basic segment, thereby retaining the robust electrostatic intramolecular interaction within FACT even in the presence of DNA. These data lead to the conclusion that the inhibitory effect of nucleosome binding is ascribed to the increase in the probability of encounter between HMG and the phosphorylated IDR.
Introduction
In recent years data derived from bioinformatics, protein physical chemistry, and tertiary structure analyses, such as x-ray crystallography and NMR, have led to the conclusion that numerous functional proteins contain regions or domains that do not form well-defined orderly three-dimensional structures but rather exist as dynamic ensembles of interconverting flexible conformers (1–3). These intrinsically disordered regions (IDRs) are ubiquitously found in eukaryotic proteins that play crucial roles in gene regulation within nuclei (4–8). It is known that these IDRs are involved in functionally important molecular recognition, where they fold into ordered conformations through binding to rigid ordered protein subunits or domains, a process that is generally termed coupled folding (9,10). Conversely, IDRs in a number of proteins do not necessarily undergo global disorder-to-order transitions upon binding. Moreover, several important regulatory interactions involve dynamic complexes in which main chains of IDRs continue to fluctuate without forming defined architectures (11,12). These binding events appear to be modulated by critical posttranslational modifications including phosphorylation, acetylation, methylation, and ubiquitination (8,11,13).
Our previous work reported that high-speed atomic force microscopy (HS-AFM) can simultaneously visualize each behavior of IDRs and rigid domains of Drosophila chromatin transcription-facilitating protein (dFACT) on a substrate surface in solution (14). This method is suitable for studying proteins involved in gene expression, such as transcriptional regulators and chromatin-remodeling factors. For example, FACT, classified as a chromatin remodeler, is a heterodimer complex that consists of SSRP1 and SPT16 subunits (Fig. 1). Both subunits contain several rigid domains and IDRs that play crucial roles in chromatin remodeling and transcriptional elongation (15–19). Thus, FACT is a suitable target for investigating the mechanistic and functional aspects of IDRs in solution through HS-AFM and NMR analyses.
Figure 1.
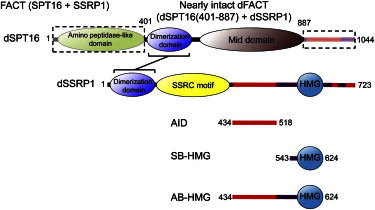
A schematic drawing of the domain organization of dSPT16 and dSSRP1, the two subunits in dFACT. The fragments used in this work are listed: AID; SB-HMG (residues 543–554), which contains a part of the BID region colored in purple (residues 519–554); and AB-HMG. These fragments jointly constitute the regulatory region of dSSRP1 binding to DNA. The heterodimeric complex, consisting of dSPT16 (residues 401–887) and the full-length dSSRP1, was used for HS-AFM measurements.
In a previous study on interactions between dFACT and nucleosomes, we found that dFACT initially binds nucleosomes and/or nucleosomal DNA via a high-mobility-group domain (HMG) and an HMG-flanking basic ID segment (BID) of the dSSRP1 subunit, which jointly forms a DNA-binding element (Fig. 1) (20). The acidic ID segment (AID), adjacent to BID, forms intramolecular interactions with both HMG and BID (Fig. 1). Extensive phosphorylation of AID strikingly increases the number of negative charges, thereby strengthening the intramolecular interactions. As a result, the binding of dFACT to nucleosomal DNA is blocked, preventing the formation of rigid structures (20). The physiological significance of this control mechanism is highlighted by rapid chromatin transactions during early embryogenesis through dephosphorylation in the maternally transmitted dSSRP1 after fertilization (20). In humans, the dephosphorylated SSRP1, including the HMG, is essential for FACT to exhibit higher binding affinity with nucleosomes (19). In contrast, the yeast FACT-HMG functions as an isolated Nhp6a/b protein (15,21,22). Thus, the phosphorylation-dependent regulation is likely to be conserved only in higher eukaryotes. Despite the importance of AID phosphorylation, the detailed blocking mechanism remains unresolved at the atomic level.
To clarify the mechanism of intramolecular interaction between the AID segment and the DNA-binding elements, we investigated the detailed effects of phosphorylation by combining NMR spectroscopy and HS-AFM. Specifically, we prepared large amounts of various fragments, which were labeled with stable isotopes for NMR in both the phosphorylated and nonphosphorylated states, whereas FACT molecules nearly intact in size were observed by HS-AFM. Thus, this study allowed us to conclude that dense phosphorylation causes an electrostatic reinforcement of the interactions between the acidic ID region and the DNA-binding elements, and that the essence of the interactions is dynamic and transient, thus lacking specific and stable binding sites in domains or fragments.
Materials and Methods
Protein preparation
The dFACT used was the same as that described in detail in our previous study (20). The fragments used in this work are schematically drawn in Fig. 1: AID, the short basic IDR plus HMG (SB-HMG), and the acidic and basic IDRs plus HMG (AB-HMG). These fragments were expressed in Escherichia coli. Details of the purification of each fragment and phosphorylation of the AID and AB-HMG fragments by casein kinase II (CK2) are described in the Supporting Material.
For the dynamic AFM experiments, the His-tagged Drosophila SPT16 (401–887) (dSPT16) and Drosophila SSRP1 (dSSRP1) were cloned into a pFastBacDual plasmid. To obtain the Ser/Thr-to-Ala mutants at the 10 phosphorylation sites (10SA; S443A, S472A, S476A, T477A, S488A, S496A, S500A, S502A, S506A, and S515A (Fig.S1 in the Supporting Material)), site-directed mutagenesis of the dFACT proteins was performed by the QuikChange method (Stratagene, La Jolla, CA). The nearly intact dFACT heterodimers (His-dSPT16 (401–887) + dSSRP1) were coexpressed as the fully phosphorylated form (wild–type (WT)) and a nonphosphorylated form (10SA) in Sf9 insect cells and purified as described previously (20).
Dynamic AFM analysis of the dFACT heterodimer
Dynamic AFM imaging experiments of the nearly intact dFACT heterodimers (Fig. 1) were carried out as described previously (14) using a laboratory-built HS-AFM (23,24). Briefly, we diluted the dFACT-WT or dFACT-10SA samples to ∼2 nM with buffer A (20 mM Tris-HCl, pH7.5, 50 mM KCl, 10 mM MgCl2, and 0.5% glycerol (vol/vol)). The diluted samples were used within 3 h. A droplet (2 μL) of a diluted sample was deposited on a freshly cleaved mica surface (∼1 mm in diameter and <0.05 mm in thickness), which had been glued onto a glass stage (2 mm in diameter and 2 mm in height). After incubation for 3 min, molecules that were not attached to the mica surface were removed by rinsing with ∼20 μL of buffer A. The sample surface was not allowed to dry. Subsequently, the sample stage was immersed in a liquid cell filled with ∼60 μL of buffer A in which a small cantilever had been fixed. Imaging was carried out by HS-AFM in tapping mode. Details of the HS-AFM imaging experiments were summarized in a recent work (25). For image analysis, we applied three filters in the following order. First, a low-pass filter to remove spike noise; second, a flattening filter to make the xy-plane flat; third, a line-to-line base collection filter to minimize the base height difference between lines.
NMR spectroscopy
The backbone 1H/13C/15N resonance assignments of AID and phosphorylated AID fragments were performed with a standard set of triple resonance experiments on a DMX600 spectrometer (Bruker, Billerica, MA). (BMRB entry 11511) Details are available in the Supporting Material.
NMR titration experiments
The interactions between the isolated AID and SB-HMG fragments were performed in two ways: one experiment used nonlabeled AID titrated into 15N-labeled SB-HMG, and the other experiment involved titrating nonlabeled SB-HMG into 15N-labeled AID. To explore how phosphorylation of AID influenced its interaction with binding partners, a phosphorylated AID fragment was also used in the titration experiments. The chemical-shift changes observed in a series of two-dimensional (2D) 1H-15N heteronuclear single-quantum coherence (HSQC) spectra during the titration were analyzed by numerical curve fitting to elucidate the number of effective binding sites, n, and the dissociation constants, KD, according to a previously published procedure (26). In the calculations, we applied a global fitting procedure that incorporates all the chemical-shift-change profiles for the residues under study, with n and KD as global adjustables (27). Monte Carlo simulation was applied to estimate the statistical errors for n and KD, assuming an uncertainty for each peak position on a 1H-15N HSQC spectrum of 0.002 ppm (1H) and 0.02 ppm (15N), respectively, and using 64 synthetic data sets with Gaussian noise (28). Experimental details in the titration experiments between the isolated AID and SB-HMG fragments are described in the Supporting Material.
NMR analysis on the interaction between AB-HMG and double-stranded DNA
We carried out NMR spectral analysis for the 15N-labeled nonphosphorylated and phosphorylated AB-HMG fragments in the absence and presence of DNA (Fig. 1). The limited solubility and the severe spectral overlap in the spectra of AB-HMG did not permit complete assignment of the resonances arising from the backbone nuclei. Only a limited number of resonances were assigned for AB-HMG. We used double-stranded DNA (dsDNA) with the sequence 5′-d(CGCGATATCGCG)2-3′. The reaction mixture including the dsDNA was passed through a NAP-10 column to exchange the buffer to 50 mM Tris-HCl, pH 6.6, the buffer used for the NMR experiments.
Results
Molecular features of phosphorylated and nonphosphorylated dFACT heterodimers visualized by HS-AFM
The wild-type dFACT (dFACT-WT) is spontaneously phosphorylated in Sf9 insect cells and is expressed as the fully phosphorylated form at 10 phosphorylation sites. In contrast, a dFACT mutant containing Ser/Thr-Ala substitutions at each of these sites, dFACT-10SA, is expressed as the nonphosphorylated form, as described previously (20). The molecular features of dFACT-WT (the phosphorylated form) and dFACT-10SA (the nonphosphorylated form) attached to mica surfaces in the buffer solution were directly visualized by HS-AFM (Fig. 2, A and B, respectively; also see Movies S1 and S2). At first glance, it appears that both molecules consist of a large globular domain and a long tail region. The large globular domain appears to be well adsorbed onto the mica surface, and its position does not change significantly over time. In contrast, the long tail region exhibits rapid fluctuations because of thermal agitations, indicating that this region has less affinity to the mica surface. These molecular features are consistent with the results of our previous study (14). Considering our previous data (14), we can assign the large globular domain (termed GD1, see Fig. 2 C) to the structured domain consisting of dSPT16 (401−887) and dSSRP1 (1−404) (Fig. 1). The long tail region can be assigned to the IDR connected to the HMG (Fig. 1).
Figure 2.
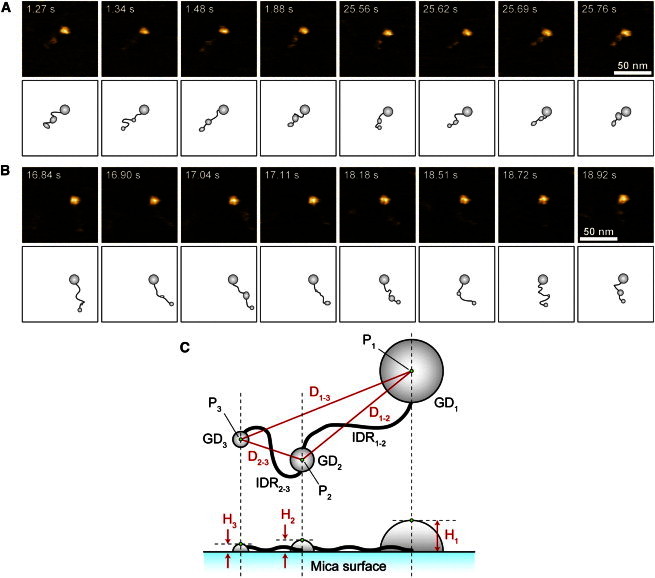
Typical HS-AFM images showing the molecular features of dFACT-WT (A) and dFACT-10SA (B). These HS-AFM images were clipped from the movie files (see Movie S1 for dFACT-WT and Movie S2 for dFACT-10SA). Every image was taken at 67.08 ms/frame (∼15 frames/s). The time from the beginning of the clip is indicated at the upper left of each image. Scanning area, 100 × 100 nm2 with 80 × 80 pixel; Z-scale, 4.0 nm. The observed molecular features of dFACT are schematized according to the definitions in C. These schematics were drawn freehand by tracing the AFM images by visual estimation. (C) Top-view (upper) and side-view (lower) schematics simply represent the characteristics of dFACT observed by HS-AFM. Gray-colored ellipses and black-colored thick solid lines represent the GDs and the IDRs, respectively. The symbols used for image analysis are also depicted. For every image, we selected three points (P1–P3) (green dots) representing the peak heights (Hi) in their respective GDs. Using these points, a distance from the highest point of one GD to that of another was determined. For example, the distance from P1 of GD1 to P2 of GD2 was expressed as D1–2. In some images, the appearance of GD2 or GD3 was unclear; there was no point with distinct heights around the area usually seen. In this case, P2 and P3 with the highest height around the middle and the end of the IDR were selected.
With careful observations, we noticed that the IDR has two small globular domains, which were connected with IDR1–2 and IDR2–3 (Fig. 2 C). One of the small globular domains (GD3 (see Fig. 2 C)) was consistently observed at the end of the IDR and its appearance is not substantially different between the two constructs. In our previous study, this small globular domain was not observed, although the sample was prepared using the E. coli expression system. Furthermore, the amino acid sequence indicates no tertiary structure at the end of the IDR. Taken together, we postulate that unknown posttranslational modifications would induce a globular domain at the end of the IDR.
On the other hand, the other small globular domain (GD2 (Fig. 2 C)) appears temporally around the middle of the IDR but shortly vanishes. In other words, we observed that the height of GD2 changes over time. In addition, the figure of GD2 appears to be retained for a longer period in dFACT-WT than in dFACT-10SA, whereas the length of the IDR appears to be slightly shorter in dFACT-WT than in dFACT-10SA. Notably, the distance between GD1 and GD2 appears to be much shorter in dFACT-WT; however, the distance between GD2 and GD3 is similar in both constructs. Similar tendencies were observed for most of the other protein molecules examined in each construct.
Quantitative analysis of distinct HS-AFM images between phosphorylated and nonphosphorylated dFACT
To quantitatively evaluate minute variations of the above-mentioned molecular features, we performed image analysis. Because the IDRs are highly flexible and can adopt similar molecular features at an arbitrary time, successive AFM images must be analyzed without skipping images to maintain arbitrariness of data sampling. To identify IDRs that contribute to changes in molecular features, a straightforward approach is required for contour length analysis of IDRs. However, considering the spatiotemporal resolution of the HS-AFM, it is currently not possible to apply contour-length analysis to successive AFM images without skipping images. As an alternative, we created a simple schematic representation of the molecular features of dFACT observed by HS-AFM (Fig. 2 C). In this schematic, the heights of the three globular domains (H1–H3) and the end-to-end distances between the two globular domains (D1–2, D2–3, and D1–3) were obtained from a single AFM image. This approach allowed us to analyze almost all AFM images, even when the AFM images had poor signal/noise (S/N) ratios. The end-to-end distance can be used to adequately evaluate the length of a biological polymer on a surface, as described previously (29), and our data therefore support our interpretation. The analysis was performed on three typical molecules for the phosphorylated (dFACT-WT) and nonphosphorylated (dFACT-10SA) constructs, and the results are summarized in Table 1. We believe that the three molecules selected lie within the population as a whole of each construct, because most of the molecules observed displayed similar molecular features. For example, we can see four molecules of dFACT-10SA simultaneously (Movie S3), all of which appear to have similar molecular features. Note that the parameters described herein are not likely to have reached the most probable values, because we analyzed 4586–5770 images of only three molecules for each construct. However, we believe that these parameters will be convincing for our conclusions because they represent well the molecular features of each construct. The details are described below.
Table 1.
Summary of the AFM image analysis
| Parameter | dFACT-WT | dFACT-10SA |
|---|---|---|
| H1 | 3.9 ± 0.3 nm | 3.9 ± 0.3 nm |
| H3 | 1.6 ± 0.3 nm | 1.6 ± 0.4 nm |
| D1–2 | 15 ± 5 nm | 17 ± 5 nm |
| D2–3 | 10 ± 3 nm | 11 ± 4 nm |
| D1–3 | 21 ± 5 nm | 23 ± 7 nm |
| D1–2/(D1–2 + D2–3) | 0.59 ± 0.11 | 0.62 ± 0.11 |
| Number of molecules analyzed | 3 | 3 |
| Number of frames analyzed | 5770 | 4586 |
Heights and distances are shown by mean ± SD. Units of areas are arbitrary. The accuracy of the HS-AFM measurement for the z-direction (i.e., background noise) was 0.15 nm. The standard deviations of the Gaussian fits were therefore restricted to >0.15 nm. Note that we did not apply Gaussian fittings to the H2 distributions because the results obtained had no statistical relevance.
The distributions of H1 appeared to be similar for both constructs, and the mean height was 3.9 nm (Fig. 3, A and D, and Table 1), indicating no significant difference in the GD1 for the two constructs. This height is the same as that obtained in our previous study (14). Although the distributions of H3 appeared to differ, the mean height was 1.6 nm, with similar standard deviations for both constructs (Fig. 3, C and F, and Table 1), indicating that the GD3 domain may not differ between the two constructs. In contrast, distributions for H2 were notably higher for dFACT-WT than for dFACT-10SA (Fig. 3, B and E). Two peaks could be seen in the H2 distribution of dFACT-10SA around 1.1 nm and 1.7 nm (Fig. 3 E). The peak around 1.1 nm was higher and wider than that around 1.7 nm, and the ratio of the areas of the two peaks was ∼2:1 (low/high) (Fig. 3 E). Because we observed at least two physical states in GD2 (i.e., a lower-height and a higher-height state), this result indicates that the GD2 of dFACT-10SA tends toward a lower-height state than a higher one. On the other hand, the H2 distribution of dFACT-WT appears to be a combination of a large peak around 1.6 nm and a small peak around 1.1 nm because a small shoulder around 1.1 nm could be seen in the distribution (Fig. 3 B). The area of the large peak was more than twice as wide as that of the small peak, indicating that the GD2 of dFACT-WT tends toward a higher-height state than a lower one. Consequently, these results suggest that the GD2 of dFACT-WT resides in the higher-height state much longer than does that of dFACT-10SA. Considering the two height values seen in the H2 distributions (i.e., 1.1 nm for the lower-height state and 1.6–1.7 nm for the higher-height state), the height of the HMG on one side corresponded to ∼1 nm (30) and the height of the IDR was determined to be 0.4–0.6 nm. These data collectively suggest that the lower-height state (H2 of ∼1.1 nm) and higher-height state (H2 of 1.6–1.7 nm) represent the HMG alone and the HMG associated with the IDR, respectively.
Figure 3.
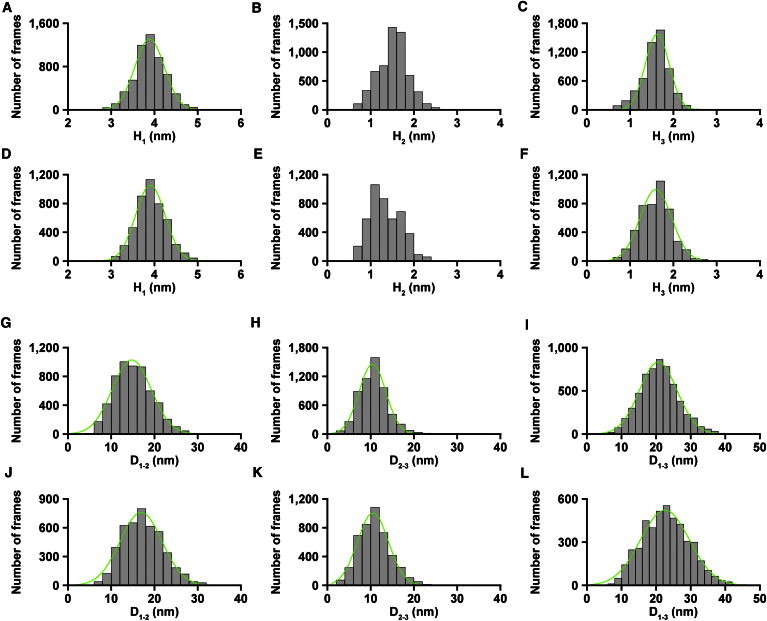
Height distributions of three GDs and distributions of distance between two GDs of dFACT. Height distributions (H1–H3) are shown for dFACT-WT (A–C) and dFACT-10SA (D–F). Distance distributions (D1–2, D2–3 and D1–3) are shown for dFACT-WT (G–I) and dFACT-10SA (J–L). Green lines represent single-Gaussian fitting. Note that we did not apply Gaussian fittings to the H2 distributions because the results obtained had no statistical relevance. A summary of the analysis is presented in Table 1. These results were obtained from analysis applied to three molecules for each construct.
The distributions of D2–3 were similar between dFACT-WT and dFACT-10SA (Fig. 3, H and K), and the mean length of D2–3 showed almost the same values (Table 1). The length of IDR2–3 did not change in response to phosphorylation. Meanwhile, the mean length of D1–2 of dFACT-WT (15 ± 5 nm) was shorter by ∼2 nm than that of dFACT-10SA (17 ± 5 nm) (Fig. 3, G and J, and Table 1). Therefore, the shorter state of IDR1–2 within dFACT-WT (the phosphorylated form) is generated by the interaction between HMG and IDR1–2, which contains the phosphorylated AID, but not that between HMG and IDR2–3. The mean length of D1–3 of dFACT-WT (21 ± 5 nm) was also shorter by ∼2 nm than that of dFACT-10SA (23 ± 7 nm) (Fig. 3, I and L, Table1). This difference can be ascribed to the difference in the D1-2 for the two constructs because we observed no difference greater than 2 nm in D2-3.
To estimate the mean position of the GD2 in the long tail domain, the ratios of D1–2/(D1–2 + D2–3) for dFACT-WT (0.59 ± 0.11) and dFACT-10SA (0.62 ± 0.11) were determined (Fig. 4), indicating that phosphorylation of IDR1–2 shifts the mean position of GD2 to GD1 but not to GD3. Combined with the results in Fig. 3, these results demonstrate that the height change for GD2 and the length changes for D1–2 in dFACT-WT result from the interaction between the HMG and IDR1–2, but not between the HMG and IDR2–3. This observation is in good agreement with our previous model (20).
Figure 4.
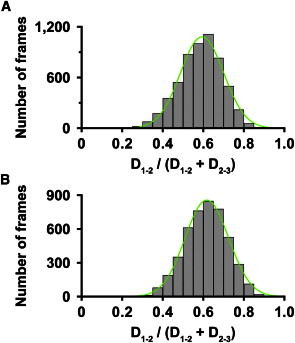
Plots showing the ratio of D1–2/(D1–2 + D2–3), which indicate the mean positions of GD2 in the tail regions of dFACT-WT (A) and dFACT-10SA (B). Green lines represent single-Gaussian fitting. A summary of the analysis is presented in Table 1. These results were obtained from analysis applied to three molecules for each construct.
To summarize the HS-AFM data, GD2 temporally appears and then vanishes in both constructs. However, it is important that the lifetime of the higher-height state in dFACT-WT is much longer than that in dFACT-10SA. In addition, dFACT-WT seems to have a shorter IDR than dFACT-10SA. These data indicate that the higher-height state of GD2 originates from the interaction between the HMG and IDR1–2, which contains the phosphorylated AID (Fig. 1). Analysis by HS-AFM is highly suitable for directly observing the dynamic behavior of large protein complexes on the surface of a substrate in solution. The findings described here should be analyzed by NMR, which can provide detailed local and atomic information on protein-protein interactions.
Structural characterization of the phosphorylated AID fragment
The fragments used in the NMR analysis are schematically drawn in Fig. 1. These fragments were expressed in E. coli and were phosphorylated by CK2 in vitro (Fig. S2). The limited signal dispersion of the isolated AID fragment in the 2D 1H-15N HSQC spectrum is typical of a disordered protein (Fig. 5 A). The 15N{1H} heteronuclear nuclear Overhauser effect (NOE) values for most residues of the AID were negative, indicating that the fragment is essentially in a disordered state. Residues around 453 showed positive heteronuclear NOE values. This region is rich in hydrophobic amino acids and is therefore likely to have restricted local backbone motions; although the motional properties do not seem functionally relevant (Fig. 5 C). CK2 successfully phosphorylated all nine canonical phosphoacceptor serines within the AID, as revealed from high-field NMR chemical shift changes in both the 1H and 15N dimensions (Fig. 5 B); T477 was not phosphorylated in this sample preparation, presumably due to the lower activity of CK2 toward Thr residues (31). The phosphorylation of AID did not induce any structural changes, because the increase in the resonance dispersion in the 2D 1H-15N HSQC spectrum (data not shown) was negligible. The 15N{1H} heteronuclear NOE profile for the phosphorylated AID (pAID) also indicated that it is in a disordered state (Fig. 5 C). Nonetheless, the heteronuclear NOE values of pAID did show an overall small but significant increase in the size of the NOE values when compared with the data for the nonphosphorylated form (Fig. 5 C). Presumably, electrostatic repulsion among phosphate groups may induce some fragment stiffness.
Figure 5.
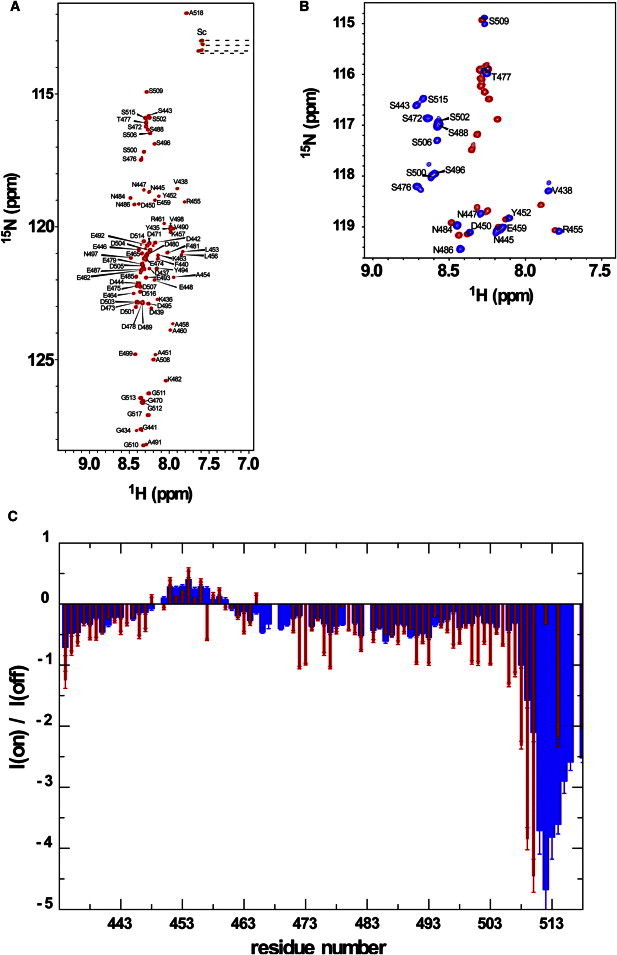
NMR spectral characterization of AID fragments. (A) Backbone resonance assignments of 15N-labeled AID in the 2D 1H-15N HSQC spectrum. (B) Spectral comparison in a region of the 2D 1H-15N HSQC spectra for nonphosphorylated (red) and phosphorylated AID (blue). The backbone amide signals of the phosphorylated Ser shows an upfield shift in both the 1H and 15N dimensions. (C) Comparison of the 15N{1H} heteronuclear NOE profiles between nonphosphorylated (red) and phosphorylated (blue) AID fragments.
Interactions between the isolated AID and SB-HMG fragments
To elucidate the binding modes between isolated fragments, both the AID and pAID were titrated to the 15N-labeled SB-HMG (Fig. 6, A and B), which contains a short basic IDR and the HMG (Fig. 1). The normalized chemical-shift changes observed in the 2D 1H-15N HSQC spectra of 15N-labeled SB-HMG in the titration (Fig. 6, A and B) are plotted versus the residue numbers (Fig. 6, C and D). The comparison between the two profiles showed that AID and pAID share similar binding sites on SB-HMG, irrespective of phosphorylation. Although they do have contact with the DNA binding surface of the HMG, both fragments primarily bind to the BID, because the residues in the BID showed greater chemical shifts than those in the HMG (Fig. 6, C and D). Furthermore, we found in our previous work that the BID is essential for intramolecular interaction with the AID (20). The magnitudes of the chemical shift changes by the addition of pAID are overall greater than those observed for AID binding, indicating that pAID is likely to have higher affinity to SB-HMG. That is, the increase in the population of the bound form of SB-HMG with pAID causes greater spectral changes relative to the free-state spectrum (27).
Figure 6.
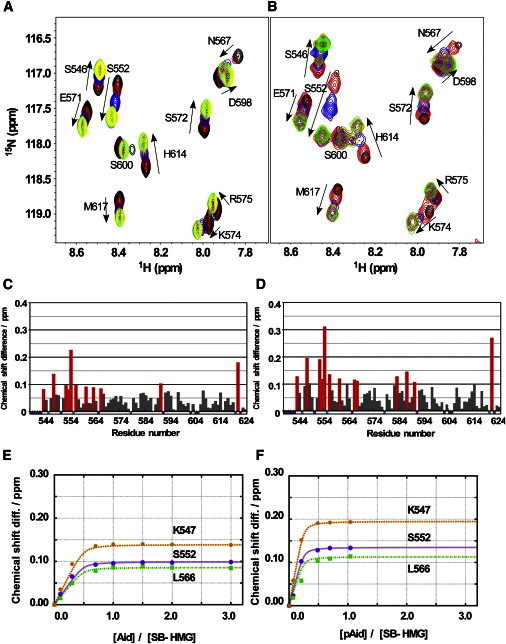
NMR titration experiments using the 15N-labeled SB-HMG fragment. (A) Chemical-shift changes in the titration of nonphosphorylated AID fragment (AID) to 15N-labeled SB-HMG; AID/SB-HMG molar ratios were 0.0 (black), 0.3 (red), 0.7 (blue), 1.0 (orange), 1.5 (pink), 2.0 (green), and 3.0 (yellow). (B) The corresponding spectral changes observed for phosphorylated AID (pAID); pAID/SB-HMG ratios were 0.00 (black), 0.07 (red), 0.21 (blue), 0.49 (orange), 0.70 (pink), and 1.04 (green). (C and D) Histograms of the chemical-shift differences in SB-HMG upon binding with AID (C) and pAID (D). Chemical-shift differences are plotted against residue numbers of SB-HMG. Red bars indicate that the chemical-shift differences are over the average plus one standard deviation. Short bars in cyan and purple along the x axis represent prolines and the residues for which assignment information was missing. Chemical-shift changes are shown for the representative residues in SB-HMG in the titrations with AID (E) and pAID (F). The numerically determined dissociation constant, KD, and number of binding sites, n, for the AID and pAID titration experiments were KD = 9.7 ± 0.3 μM, n = 2.0 ± 0.1 and KD = 1.01 ± 0.02 μM, n = 3.9 ± 0.1, respectively. Values for KD and n were determined by global fitting using the residues marked by red bars in C and D (see Supporting Material).
The numerical fittings to the chemical shift changes against the molar ratios of AID or pAID to SB-HMG showed that phosphorylation alters their binding modes to SB-HMG (Fig. 6, E and F) (26,32). To determine the KD and n values from the experiments by using 15N-labeled SB-HMG, we included in our calculation 11 residues for AID and 30 residues for pAID, showing that the significant changes deviated more than the mean + 1 SD (Fig. 6, C and D, red bars). The number of SB-HMG-binding sites on AID was estimated at n = 2.0 ± 0.1 with a dissociation constant of KD = 9.7 ± 0.3 μM, assuming that all the binding sites on AID have equivalent affinities for SB-HMG. In the case of SB-HMG binding to pAID, the number of binding sites was estimated at n = 3.9 ± 0.1 with KD = 1.01 ± 0.02 μM. This indicates that phosphorylation of AID doubles its number of SB-HMG-binding sites in addition to enhancing its affinity for SB-HMG. In agreement with these data from NMR, isothermal calorimetry (ITC) experiments using the SB-HMG and AID or pAID showed that the number of binding sites on AID approximately doubles, depending upon phosphorylation (data not shown).
The SB-HMG titration to 15N-labeled AID or pAID was also examined. The spectral changes in the 2D 1H-15N HSQC spectra for AID and pAID are shown in Fig. 7, A and B, respectively. The 20 residues in AID showed significant spectral changes (chemical-shift changes deviating by more than the mean + 1 SD of the entire data) upon binding to SB-HMG. It is thus likely that these residues are involved in binding to SB-HMG (Fig. 7 E). The residues are classified into two groups according to their affinity (nine high-affinity residues (Fig. 7 C, left, and Fig. 7 E, blue bars) and 11 low-affinity residues (Fig. 7 C, right, and Fig. 7 E, red bars)). The global fitting calculation, using the chemical shift profiles for the nine high-affinity residues gave a KD value of 3.7 ± 0.1 μM, whereas the KD value for the 11 low-affinity residues was 43 ± 1 μM. It should be noted that one-to-one binding was assumed in these calculations. The residues showed linear spectral changes according to SB-HMG concentration (Fig. 7 A), indicating that the high- and low-affinity residues do not form individual binding sites. If residues with different affinities form individual binding sites, the chemical-shift-change profiles should become nonunidirectional (33). Therefore, the binding surfaces on AID for SB-HMG may contain both high- and low-affinity residues. In fact, the residues of 15N-labeled SB-HMG (Fig. 6 C, red bars) showed a slightly lower affinity for AID (KD = 9.7 ± 0.3 μM) than did the high-affinity residues in AID (KD = 3.7 ± 0.1 μM). These results imply that the high- and low-affinity residues of AID do not interact with SB-HMG in an individual manner but form multiple complexes in a dynamic equilibrium (i.e., a dynamic complex) (34).
Figure 7.
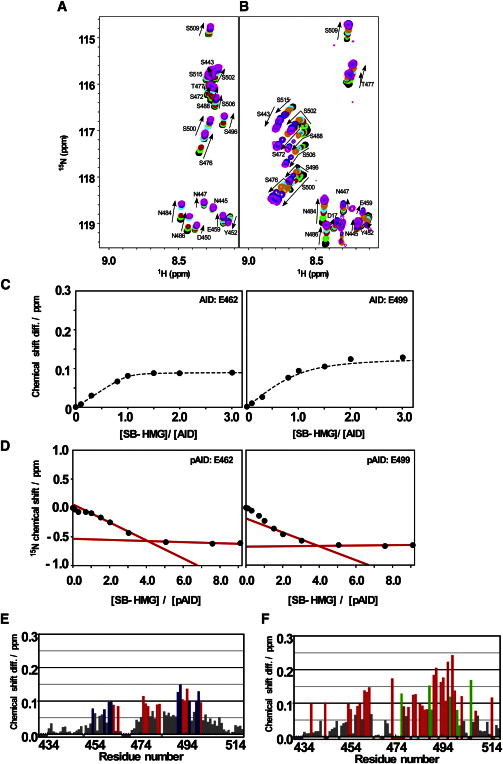
Spectral changes on 15N-labeled AID and pAID in the titration with the unlabeled SB-HMG. (A) Spectral changes induced by the titration of AID; SB-HMG/AID ratios were 0.0 (black), 0.1 (green), 0.3 (red), 0.7 (cyan), 1.0 (yellow), 1.5 (purple), 2.0 (blue), and 3.0 (pink). (B) Spectral changes by pAID; SB-HMG/pAID ratios were 0.0 (black), 0.3 (green), 1.0 (red), 2.0 (cyan), 3.0 (yellow), 5.0 (purple), 7.0 (blue), and 9.0 (pink). (C) Chemical-shift-change profiles for representative residues in AID, showing the high (left) and low (right) affinities to SB-HMG. (D) The 15N chemical-shift changes observed for representative residues in pAID according to the titration. Spectral changes determined that the maximal binding number of SB-HMG to pAID is 4. (E and F) Histograms of the chemical-shift differences in AID (E) and pAID (F) upon binding with SB-HMG. Chemical-shift differences are plotted against residue numbers for AID and pAID. Residues with a mean + 1 SD are colored. Residues in AID with blue bars have higher affinity (KD = 3.7 ± 0.1 μM) than those with red bars (KD = 43 ± 1 μM). The residues in pAID with green bars showed kinked titration traces, as in B. Short down bars in cyan and purple along the x axis indicate prolines and unassigned residues, respectively. Short down bars in red indicate phosphorylated Ser residues. Short down orange bars are residues whose signals were incompletely traced due to severe signal overlap during the titration.
The titration experiments using 15N-labled pAID showed that pAID could accommodate up to four SB-HMG molecules (Fig. 7, B and D), which is consistent with the result from the analysis using 15N-labeled SB-HMG (n = 3.9 ± 0.1). Some residues in pAID showed nonunidirectional spectral changes in the titration of SB-HMG (Fig. 7 B). These residues are localized in the C-terminal half, which is rich in phosphoserine and acidic residues (Fig. 7 F, green bars). Similar to the nonlinear spectral changes in multiple ligands binding to the protein (27,35), it is likely that multiple SB-HMGs bind to the phosphor-Ser-rich region of pAID. The nonunidirectional spectral changes may therefore be caused by the expansion of the phosphor-Ser-rich region in response to the multiple bindings of SB-HMG.
Taken together, these data allowed us to conclude that nonphosphorylated AID contains binding surfaces that can accommodate up to two SB-HMG molecules, whereas phosphorylation of AID expands the surface by a factor of 2. The binding surfaces expanded by phosphorylation should increase the intramolecular encounter probability between the AID and BID-HMG segments in the dSSRP1 subunit.
Changes of intramolecular interactions between AID and BID-HMG in response to phosphorylation and dsDNA binding
To explore how AID forms intramolecular interactions with BID-HMG, we compared the 2D 1H-15N HSQC spectra of an AB-HMG fragment and the isolated AID (Fig. S3). The limited solubility (<0.1 mM) and the longer IDR of AB-HMG hampered assignment of the resonances of the backbone nuclei. Only a limited number of signals for rather isolated Ser residues were assigned based on the close proximity (i.e., similar chemical shifts) of the resonances for the AB-HMG construct to those assigned in the spectra of isolated AID or SB-HMG fragments (Fig. S3).
The significant spectral differences between AB-HMG and the isolated AID demonstrated that part of the AID segment binds to the other part of the AB-HMG intramolecularly (Fig. S3 A). The spectral difference between the two fragments remained in the presence of double-stranded DNA (dsDNA) (Fig. S3 B). This observation shows that the AID segment in the AB-HMG fragment still contacts other parts of the fragment without being released to behave as an isolated fragment, which would give NMR signals that match more closely to those for the isolated AID fragment.
The spectral difference between the pAB-HMG and the isolated pAID was apparent, indicating that the pAID segment in the pAB-HMG fragment has intramolecular contacts with other parts of the fragment (Fig. S3 C). The presence of the dsDNA did not completely reverse the spectral difference (Fig. S3 D), indicating that it does not block the intramolecular contacts of the pAID segment with parts of the pAB-HMG fragment.
The absence of a complete set of resonance assignments for the AB-HMG and pAB-HMG fragments allows only a limited analysis of the intramolecular interactions between the AID and BID-HMG segments. Despite the experimental limits of this study, the spectral comparison shows that intramolecular interactions between the segments happen irrespective of phosphorylation, and that these interactions remain even in the presence of dsDNA; however, the modes of binding seem to change.
The intramolecular interactions were monitored using a limited number of assigned HMG signals (Fig. S4). In addition to the limited solubility of the AB-HMG fragment, intense signals from the long unstructured part in the AB-HMG prohibited observation of the signals from the HMG.
Despite the limited residues monitored, these NMR data collectively indicate that, irrespective of the phosphorylation state, the residues in HMG and BID retain contact with the AID segment, as suggested by the spectral difference in the HMGs in the AB-HMG and pAB-HMG fragments relative to the isolated HMG (Fig. S4). In the presence of dsDNA, the intramolecular contacts of HMG with the AID segment appear to change according to the phosphorylation state of the AID segment, as evinced by the spectral comparisons for residues A606, K621, and G553 (Fig. S4). The phosphorylation-dependent spectral changes for the HMG induced by the addition of dsDNA suggest that phosphorylation of the AID segment alters its interaction with the HMG box and thus changes the HMG-mediated binding to dsDNA.
Discussion and Conclusions
The combination of HS-AFM and NMR analyses has shown that phosphorylation of multiple serine residues in the AID modulates the intramolecular interactions between the AID and the DNA-binding element, which consists of BID and HMG. Our data collectively provide mechanistic insights into dynamic interactions between AID and the DNA-binding elements in FACT, as follows.
-
1.
AID makes intramolecular contact with the DNA-binding elements. Phosphorylation of AID expands the binding epitope to the DNA-binding element by a factor of 2 when compared with the nonphosphorylated region.
-
2.
The intramolecular contact of the nonphosphorylated AID with the DNA-binding element should be relatively weak, as demonstrated by the limited spectral changes in resonances from the isolated AID (Fig. S3). This is consistent with our HS-AFM observation that the nonphosphorylated FACT retains mostly extended IDRs (Fig. 3). Thus, the intramolecular contact between the nonphosphorylated AID and the DNA-binding element remains dynamic and transient.
-
3.
Phosphorylation of AID reinforces its intramolecular interaction with the DNA-binding element, as shown by the larger spectral changes for the resonances representing the phosphorylated serines in the AID when compared with the same resonances in the isolated pAID (Fig. S3). The phosphorylated AID retained intramolecular contacts with the DNA-binding elements in the presence of DNA (Fig. S3), thus indicating that this AID element impairs FACT binding to DNA. In the presence of DNA, several resonances representing residues within the HMG showed chemical-shift changes, despite the fact that these residues retain contact with phosphorylated AID (Fig. S4). Our HS-AFM observation revealed dynamic behavior between folding and unfolding of the globular domain, which is putatively formed by pAID and BID-HMG (Fig. 3). Taken together, FACT involves dynamic and transient intramolecular interactions, even in the phosphorylated state.
The structure of the dSSRP1-HMG was similar to the yeast Nhp6a protein structure isolated from SSRP1 (30). The DNA-binding surface on the dSSRP1-HMG structure was similar to the DNA-binding surface in Nhp6A. The NMR structure of the Nhp6A-DNA complex revealed a characteristic L-shaped HMG fold, which contacts the minor groove of DNA, whereas an extended N-terminal BID region interacts with the adjacent major groove (36). The DNA-binding interface contains numerous conserved lysine and arginine residues that participate in electrostatic interactions with the phosphate backbone. Although some hydrophobic stacking/wedge interactions are formed between a number of Nhp6A residues and DNA bases, the interactions of Nhp6A with DNA are predominantly electrostatic. Our NMR data indicate that DNA and AID occupy almost the same electrostatic interaction interfaces on the HMG and BID regions regardless of the phosphorylation state of AID.
An apparently similar mechanism was proposed for the autoinhibition of DNA binding by phosphorylation of a transcription factor, Ets-1 (37,38). The affinity of Ets-1 to DNA is allosterically regulated by the flexibility of the unstructured serine-rich region and the inhibitory module, which is adjacent to the ETS domain responsible for DNA binding (38). Previous NMR experiments demonstrated that phosphorylation at several sites within the serine-rich region gradually shifts the equilibrium more to the rigid-inactive form, which is stabilized by stronger intramolecular interactions with both the inhibitory module and the DNA-binding domain (39). However, this inhibitory effect of Ets-1 is considerably different from our previous results, which showed no additive reduction of DNA-binding inhibition in response to the number of phosphorylated Ser/Thr mutations of dFACT (20). In the FACT mutant containing the SSRC motif, AID, BID, and HMG (Fig. 1), Ser/Thr-to-Ala mutations at two phosphorylation sites showed negligible inhibition of DNA binding, whereas mutations at four and six sites dramatically decreased the inhibitory effect (as shown in Fig. 3 C of Tsunaka et al. (20)). In other words, the inhibitory effect appears to be ultrasensitive, but not linear.
Our HS-AFM data provide, to our knowledge, a novel structural view, where the dynamic behavior of the FACT-IDR is drastically altered in response to phosphorylation. The NMR data showed that dense phosphorylation of the AID region increases binding sites with the DNA-binding elements containing the HMG. Presumably, the phosphorylation expands the AID region by elevating the repulsive forces, thus inducing a stronger interaction without protein folding. Moreover, the increase in binding sites caused by the phosphorylation of residues can be ascribed to a dynamic equilibrium among multiple binding states but not a simple two-state equilibrium, thereby blocking DNA binding (Fig. 8). This notion relates to the concept of encounter probability and differs to the mechanism observed for Ets-1.
Figure 8.
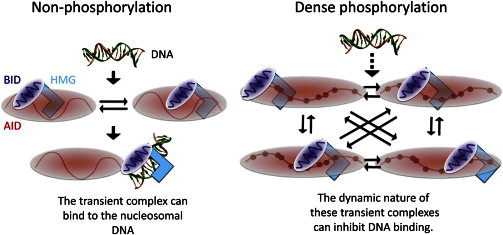
Summary of the mechanism underlying phosphorylation-dependent inhibition with respect to nucleosomal DNA. The intramolecular contact of nonphosphorylated AID with BID-HMG is nonspecific and dynamic. This situation allows BID-HMG to transiently bind with the nucleosomal DNA. In contrast, dense phosphorylation expands the AID region by elevating repulsive forces. This results in an increase of the probability of encounter between BID-HMG and pAID, thereby forming a more robust complex between BID-HMG and pAID. AID and BID are symbolically represented by the red and purple strings, respectively. Red and purple spheres indicate negative and positive net charges, respectively. The HMG is denoted by the cyan L-shaped structure. Red open circles labeled P indicate phosphorylation sites.
This dynamic binding mode is rather similar to that in the electrostatic model proposed for the interaction between Cdc4 and Sic1 (34,40); the SCF ubiquitin ligase subunit Cdc4 interacts with the cyclin-dependent kinase inhibitor Sic1 in a dense phosphorylation-dependent manner (34). Multiple phosphorylation in the N-terminal IDRs of Sic1 leads to equilibrium engagement by an interchange between phosphorylation sites (34). The degree of phosphorylation fine-tunes the complex formation of Sic1 with Cdc4 via long-range electrostatic interactions, ensuring ultrasensitivity of the Sic1-Cdc4 interaction caused by a net charge reversal (40). In agreement with this ultrasensitive change, our previous data showed that simultaneous mutations at four or six phosphorylation sites drastically enhance the binding ability of FACT to nucleosomal DNA (as shown in Fig. 3 C of Tsunaka et al. (20)). It is thus likely that the inhibitory mechanism of the phosphorylated FACT is essentially based on the electrostatic model.
Furthermore, a notable feature of this regulation mechanism is that the cooperative action of the tandemly linked AID and BID regions directs nucleosomal DNA binding through their interaction with the HMG in FACT. These tandemly linked IDRs may enhance the probability of encounter between the DNA-binding elements and the phosphorylated IDR in the inhibitory mechanism (Fig. 8). In fact, our HS-AFM analyses indicate that the lifetime of the globular domain in phosphorylated dFACT is much longer than that of the nonphosphorylated form.
Many atomic structures of protein complexes provide a general view that domain and/or subunit contacts take place on well-ordered surfaces that complement each other. However, functionally important complexes frequently involve dynamic, or transient, properties, which are derived from unstructured IDRs even upon complex formation. It is very likely that such flexibilities of protein complexes should change in response to posttranslational modifications, thereby regulating physiological functions. Thus, the combination of NMR and HS-AFM analysis used in this study potentially can be applied to many other protein complexes in which IDRs play important roles.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research on Innovative Areas (Research in a Proposed Research Area) (Ministry of Education, Culture, Sports, Science and Technology KAKENHI Grant No. 21113002, 21121006, and 23107724); a Grant-in-Aid for Japanese Society for the Promotion of Science Fellows (JSPS KAKENHI Grant No. 07J01196); the Mitsubishi Foundation; Japan Science and Technology (JST) Precursory Research for Embryonic Science and Technology; and JST Development of Systems and Technology for Advanced Measurement and Analysis.
Contributor Information
Kosuke Morikawa, Email: kmorikawa@icems.kyoto-u.ac.jp.
Shin-ichi Tate, Email: tate@hiroshima-u.ac.jp.
Supporting Material
References
- 1.Dyson H.J. Expanding the proteome: disordered and alternatively folded proteins. Q. Rev. Biophys. 2011;44:467–518. doi: 10.1017/S0033583511000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uversky V.N., Dunker A.K. Understanding protein non-folding. Biochim. Biophys. Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uversky V.N., Dunker A.K. Multiparametric analysis of intrinsically disordered proteins: looking at intrinsic disorder through compound eyes. Anal. Chem. 2012;84:2096–2104. doi: 10.1021/ac203096k. [DOI] [PubMed] [Google Scholar]
- 4.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 5.Garza A.S., Ahmad N., Kumar R. Role of intrinsically disordered protein regions/domains in transcriptional regulation. Life Sci. 2009;84:189–193. doi: 10.1016/j.lfs.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Uversky V.N., Oldfield C.J., Dunker A.K. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 7.Uversky V.N., Oldfield C.J., Dunker A.K. Unfoldomics of human diseases: linking protein intrinsic disorder with diseases. BMC Genomics. 2009;10(Suppl 1):S7. doi: 10.1186/1471-2164-10-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vuzman D., Levy Y. Intrinsically disordered regions as affinity tuners in protein-DNA interactions. Mol. Biosyst. 2012;8:47–57. doi: 10.1039/c1mb05273j. [DOI] [PubMed] [Google Scholar]
- 9.Sugase K., Dyson H.J., Wright P.E. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447:1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 10.Wright P.E., Dyson H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuxreiter M. Fuzziness: linking regulation to protein dynamics. Mol. Biosyst. 2012;8:168–177. doi: 10.1039/c1mb05234a. [DOI] [PubMed] [Google Scholar]
- 12.Mittag T., Kay L.E., Forman-Kay J.D. Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 2010;23:105–116. doi: 10.1002/jmr.961. [DOI] [PubMed] [Google Scholar]
- 13.Iakoucheva L.M., Radivojac P., Dunker A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyagi A., Tsunaka Y., Ando T. Visualization of intrinsically disordered regions of proteins by high-speed atomic force microscopy. ChemPhysChem. 2008;9:1859–1866. doi: 10.1002/cphc.200800210. [DOI] [PubMed] [Google Scholar]
- 15.Formosa T. The role of FACT in making and breaking nucleosomes. Biochim. Biophys. Acta. 2012;1819:247–255. doi: 10.1016/j.bbagrm.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakayama T., Nishioka K., Hirose S. Drosophila GAGA factor directs histone H3.3 replacement that prevents the heterochromatin spreading. Genes Dev. 2007;21:552–561. doi: 10.1101/gad.1503407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reinberg D., Sims R.J., 3rd de FACTo nucleosome dynamics. J. Biol. Chem. 2006;281:23297–23301. doi: 10.1074/jbc.R600007200. [DOI] [PubMed] [Google Scholar]
- 18.Shimojima T., Okada M., Hirose S. Drosophila FACT contributes to Hox gene expression through physical and functional interactions with GAGA factor. Genes Dev. 2003;17:1605–1616. doi: 10.1101/gad.1086803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winkler D.D., Muthurajan U.M., Luger K. Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. J. Biol. Chem. 2011;286:41883–41892. doi: 10.1074/jbc.M111.301465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsunaka Y., Toga J., Morikawa K. Phosphorylated intrinsically disordered region of FACT masks its nucleosomal DNA binding elements. J. Biol. Chem. 2009;284:24610–24621. doi: 10.1074/jbc.M109.001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brewster N.K., Johnston G.C., Singer R.A. A bipartite yeast SSRP1 analog comprised of Pob3 and Nhp6 proteins modulates transcription. Mol. Cell. Biol. 2001;21:3491–3502. doi: 10.1128/MCB.21.10.3491-3502.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xin H., Takahata S., Formosa T. yFACT induces global accessibility of nucleosomal DNA without H2A-H2B displacement. Mol. Cell. 2009;35:365–376. doi: 10.1016/j.molcel.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ando T., Kodera N., Toda A. A high-speed atomic force microscope for studying biological macromolecules. Proc. Natl. Acad. Sci. USA. 2001;98:12468–12472. doi: 10.1073/pnas.211400898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ando T., Uchihashi T., Fukuma T. High-speed atomic force microscopy for nano-visualization of dynamic biomolecular processes. Prog. Surf. Sci. 2008;83:337–437. [Google Scholar]
- 25.Uchihashi T., Kodera N., Ando T. Guide to video recording of structure dynamics and dynamic processes of proteins by high-speed atomic force microscopy. Nat. Protoc. 2012;7:1193–1206. doi: 10.1038/nprot.2012.047. [DOI] [PubMed] [Google Scholar]
- 26.Yasuno K., Yamazaki T., Kyogoku Y. Interaction of the C-terminal domain of the E. coli RNA polymerase α subunit with the UP element: recognizing the backbone structure in the minor groove surface. J. Mol. Biol. 2001;306:213–225. doi: 10.1006/jmbi.2000.4369. [DOI] [PubMed] [Google Scholar]
- 27.Lian L.-Y., Roberts G. Wiley; Chichester, United Kingdom: 2011. Protein NMR Spectroscopy: Practial Techniques and Applications. [Google Scholar]
- 28.Press W.H., Teukolsky S.A., Flannery B.P. Cambridge University Press; New York: 1992. Numerical Recipes in C. [Google Scholar]
- 29.Rivetti C., Guthold M., Bustamante C. Scanning force microscopy of DNA deposited onto mica: equilibration versus kinetic trapping studied by statistical polymer chain analysis. J. Mol. Biol. 1996;264:919–932. doi: 10.1006/jmbi.1996.0687. [DOI] [PubMed] [Google Scholar]
- 30.Kasai N., Tsunaka Y., Tate S. Solution structure of the HMG-box domain in the SSRP1 subunit of FACT. J. Biomol. NMR. 2005;32:83–88. doi: 10.1007/s10858-005-3662-3. [DOI] [PubMed] [Google Scholar]
- 31.Sarno S., Vaglio P., Pinna L.A. Protein kinase CK2 mutants defective in substrate recognition. Purification and kinetic analysis. J. Biol. Chem. 1996;271:10595–10601. doi: 10.1074/jbc.271.18.10595. [DOI] [PubMed] [Google Scholar]
- 32.Fielding L. NMR methods for the determination of protein–ligand dissociation constants. Prog. Nucl. Magn. Reson. Spectrosc. 2007;51:219–242. [Google Scholar]
- 33.Arai M., Ferreon J.C., Wright P.E. Quantitative analysis of multisite protein-ligand interactions by NMR: binding of intrinsically disordered p53 transactivation subdomains with the TAZ2 domain of CBP. J. Am. Chem. Soc. 2012;134:3792–3803. doi: 10.1021/ja209936u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mittag T., Marsh J., Forman-Kay J.D. Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase. Structure. 2010;18:494–506. doi: 10.1016/j.str.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleerekoper Q., Liu W., Putkey J.A. Identification of binding sites for bepridil and trifluoperazine on cardiac troponin C. J. Biol. Chem. 1998;273:8153–8160. doi: 10.1074/jbc.273.14.8153. [DOI] [PubMed] [Google Scholar]
- 36.Murphy E.C., Zhurkin V.B., Clore G.M. Structural basis for SRY-dependent 46-X,Y sex reversal: modulation of DNA bending by a naturally occurring point mutation. J. Mol. Biol. 2001;312:481–499. doi: 10.1006/jmbi.2001.4977. [DOI] [PubMed] [Google Scholar]
- 37.Hollenhorst P.C., McIntosh L.P., Graves B.J. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 2011;80:437–471. doi: 10.1146/annurev.biochem.79.081507.103945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pufall M.A., Lee G.M., Graves B.J. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science. 2005;309:142–145. doi: 10.1126/science.1111915. [DOI] [PubMed] [Google Scholar]
- 39.Lee G.M., Pufall M.A., McIntosh L.P. The affinity of Ets-1 for DNA is modulated by phosphorylation through transient interactions of an unstructured region. J. Mol. Biol. 2008;382:1014–1030. doi: 10.1016/j.jmb.2008.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borg M., Mittag T., Chan H.S. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc. Natl. Acad. Sci. USA. 2007;104:9650–9655. doi: 10.1073/pnas.0702580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.