

Abstract
N-methyl-D-aspartate (NMDA) receptors are obligate heterotetrameric ligand-gated ion channels that play critical roles in learning and memory. Here, using targeted molecular dynamics simulations, we developed an atomistic model for the gating of the GluN1/GluN2A NMDA receptor. Upon agonist binding, lobe closure of the ligand-binding domain produced outward pulling of the M3-D2 linkers, leading to outward movements of the C-termini of the pore-lining M3 helices and opening of the channel. The GluN2A subunits, similar to the distal (B/D) subunits in the homotetrameric GluA2 α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionate receptor, had greater M3 outward movements and thus contributed more to channel gating than the GluN1 subunits. Our gating model is validated by functional studies, including cysteine modification data indicating wider accessibility to the GluN1 M3 helices than to the GluN2A M3 helices from the lumen of the open channel, and reveals why the Lurcher mutation in GluN1 has a stronger ability in maintaining channel opening than the counterpart in GluN2A. The resulting structural model for the open state provides an explanation for the Ca2+ permeability of NMDA receptors, and the structural differences between the closed and open states form the basis for drug design.
Introduction
Inotropic glutamate receptors (iGluRs) are a family of tetrameric, cation-selective ligand-gated ion channels. Although the three main subtypes of iGluRs, α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionate (AMPA), N-methyl-D-aspartate (NMDA), and kainate receptors share sequence homology and a common structural architecture (1), they exhibit different functional characteristics. In particular, NMDA receptors are distinguished by an obligate heteromeric assembly, high Ca2+ permeability, and slow deactivation and desensitization kinetics (2), as well as by their critical roles in learning and memory (3). NMDA receptors are the target of memantine, a drug for Alzheimer’s disease. In a previous study (4) we used molecular dynamics (MD) simulations to develop an atomistic model for the activation and desensitization of the homotetrameric GluA2 AMPA receptor. Here, we extended the study to the GluN1/GluN2A NMDA receptor, to uncover the molecular details on the roles of the different subunits in channel gating and to gain insight into the functional differences between AMPA and NMDA receptors.
All iGluRs are organized into an amino-terminal domain, a ligand-binding domain (LBD), a transmembrane domain (TMD), and a carboxy-terminal domain (1). Among the three subtypes, AMPA receptors are perhaps the best characterized. Numerous structures have been determined for the amino-terminal domain and for the LBD bound with various agonists and antagonists, culminating in the antagonist-bound structure for a functional GluA2 construct encompassing the LBD and the TMD (Protein Data Bank (PDB) entry 3KG2) (5). In the LBD, the structure of each subunit resembles a clamshell, with two lobes, D1 and D2, closing up on the bound ligand. Instead of a fourfold symmetry, the four homomeric subunits (A, B, C, and D) are arranged into a twofold symmetric dimer of AD and CB dimers, such that the D2 lobes of subunits A and C are proximal, whereas those of subunits B and D are distal. This asymmetry between A/C and B/D subunits suggest that they contribute differently to channel gating. On the other hand, the TMD is fourfold symmetric; each subunit forms three transmembrane helices (M1, M3, and M4). The C-terminal half of M3, containing the signature motif SYTANLAAF, lines the channel outer pore; three of these residues (including the two in boldface in the signature motif), in the form of three rings of constriction in the TMD, constitute the activation gate. A short helix (pre-M1) lies toward the extracellular side of the membrane, whereas a reentrant helix (M2) forms the inner pore.
In our previous study of the GluA2 AMPA receptor (4), we started MD simulations from the resting-state structure of the LBD-TMD construct, and then targeted the LBD toward the activated- or desensitized-state conformation. The activated-state conformation of the LBD was provided by an agonist-bound LBD dimer (PDB entry 1FTJ) (6), whereas the desensitized-state conformation of the LBD was provided by a variant introducing a disulfide bond between the two D2 lobes in the dimer (7). The transition of the LBD from the resting-state to the activated-state conformation primed the M3-D2 linkers for concerted translational and rotational motions in the lateral plane. The translational motions resulted in an outward pull on the C-termini of the M3 helices and consequently the opening of the gate; the rotational motions made the transition from the LBD twofold symmetry to the TMD fourfold symmetry more gradual. The outward movements of the M3 helices had a greater magnitude in the B/D subunits than in the A/C subunits, indicating that the B/D subunits make a greater contribution to channel gating. This atomistic model of channel activation was validated by experimental observations, including the difference in cysteine modification rates at multiple positions between the resting and activated states (8) and a dimer of dimers configuration for the second ring of the activation gate as indicated by Cd2+ coordination (9). In our model for desensitization, the motions of the LBD-TMD linkers and the TMD largely reversed those observed upon channel activation.
NMDA receptors are assembled as dimers of GluN1/GluN2 and/or GluN1/GluN3 heterodimers (10). GluN1 and GluN3 subunits bind glycine, whereas GluN2 subunits bind glutamate. Furukawa and Gouaux (11) and Inanobe et al. (12) determined the structures of the GluN1 LBD monomer bound with glycine or antagonists including 5,7-dichlorokynurenic acid (DCKA; the latter structure in PDB entry 1PBQ). These structures reveal a clamshell that is open when bound with antagonists and closed when bound with agonists, as seen for GluA2 (6). Furukawa et al. (13) further determined the structure of the GluN1/GluN2A LBD dimer bound to glycine and glutamate (PDB entry 2A5T). The arrangements between and the overall fold of the subunits in this dimer are similar to those in the agonist-bound GluA2 LBD dimer (6), suggesting a common mechanism of channel gating. Structures have also been determined for LBD monomers of GluN2D, GluN3A, and GluN3B bound with agonists (14,15). No structural information is available for how the LBD interacts with the TMD and for the TMD itself in any NMDA receptor.
Electrophysiological studies have contributed significantly to our understanding of the gating mechanism of NMDA receptors. By introducing cysteine mutations around the M3 C termini, Salussolia et al. (16) and Riou et al. (17) observed dimer cross-linking and oxidation-induced current inhibition in GluN1 but not GluN2 mutants. This indicated that, at the level of these mutations, the GluN1 and GluN2 subunits occupy diagonal positions that are proximal and distal, respectively, thus mapping GluN1 and GluN2 to the A/C and B/D subunits of the GluA2 receptor (5). In a series of studies involving cysteine mutations around the extracellular vestibule, Wollmuth and co-workers (18–22) presented strong evidence indicating that GluN1 and GluN2 subunits are not equivalent during channel gating. In the open state, GluN1 had more positions in the M3 C-terminal halves that were accessible to methanethiosulfonate (MTS) reagents, and were accessed with higher rates. A cluster of charged residues in the M3-D2 linkers of GluN1, not GluN2, defined a Ca2+-binding site and contributed to Ca2+ influx (21). Further evidence for subunit-specific contributions is provided by the observation that the Lurcher mutation (A → T in the underlined position of SYTANLAAF) in GluN1 significantly slowed down channel deactivation and desensitization (23), implicating a much stronger ability of this mutation in GluN1 than the counterpart in GluN2A in maintaining channel opening. The studies by Wollmuth and co-workers (19,20,22,24) and by others (25–28) reported state-dependent modification rates of cysteines introduced around the extracellular vestibule, which could possibly indicate movements of the constituent segments during channel activation. However, without an atomic model for the open state, the ability to interpret these data is limited.
Here we report, to our knowledge, the first full model for the activation of an NMDA receptor. To develop this model, we built homology models for the GluN1/GluN2A receptor in the closed state and the LBD tetramer in the agonist-bound form, and then carried out targeted MD simulations. Upon the lobe closure of the LBD as it transitioned from the closed-state to the agonist-bound conformation, the C-terminal halves of the M3 helices, pulled by the M3-D2 linkers, moved outward. Reminiscent of the situation in the activation of the GluA2 AMPA receptor (4), the GluN2A subunits in the distal positions had greater outward movements and thus contributed more to the NMDA receptor gating than the GluN1 subunits. This gating model provides a molecular basis for the observed wider accessibility to the GluN1 M3 helices than to the GluN2A M3 helices from the lumen of the open channel, and reveals why the Lurcher mutation in GluN1 has a stronger ability in maintaining channel opening than the counterpart in GluN2A. The resulting structural model for the open state provides an explanation for the Ca2+ permeability of NMDA receptors. With the structural models for both the closed state and the open state at hand, we can begin to delineate the mechanism of action of memantine and to design other drugs that specifically target NMDA receptors.
Computational Methods
In this study, we first built structural models of a functional NMDA receptor (consisting of the GluN1/GluN2A LBD and TMD) in the closed state and a LBD tetramer in the agonist-bound form. In subsequent MD simulations, we forced the LBD to change from the closed-state to the agonist-bound conformation. The response of the rest of the receptor was expected to contain molecular details of the activation process. We analyzed various aspects of this gating model for validation against and rationalization of experimental observations.
Molecular modeling
The structural model for the GluN1/GluN2A receptor in the closed state was built mostly by homology using the antagonist-bound structure of the GluA2 receptor (PDB entry 3KG2) (5) as a template. The LBD monomers and the TMD helices were modeled separately and then superimposed onto the counterparts of 3KG2. Finally, the loops within the TMD and the linkers between the LBD and TMD were modeled using MODELER v9.10 (29).
For the GluN1 subunits (residues T396 to K543 and I664 to C798 in chains A and C) in the LBD, we used the structure of the DCKA-bound GluN1 LBD monomer (PDB entry 1PBQ) (11). The missing residues, i.e., T396 and P439 to T450, were built by MODELER. The GluN2A subunits (residues N404 to S538 and V662 to C800 in chains B and D) in the LBD were built by homology according to the alignment in Fig. 1 A, except that loop 1 (residues I418 to K457) was excised from PDB entry 2A5T chain B (glutamate-bound GluN2A LBD). The TMD helices were completely modeled by homology according to the alignment in Fig. 1 A. The amino-acid sequence ranges of these helices (plus short C-terminal extensions) were: pre-M1 to M1, L551 to D581 for GluN1 and P546 to E577 for GluN2A; M2, E598 to L614 for GluN1 and G596 to V612 for GluN2A; M3, S626 to E661 for GluN1 and G624 to Q655 for GluN2A; and M4, F810 to K838 for GluN1 and I814 to Y842 for GluN2A.
Figure 1.
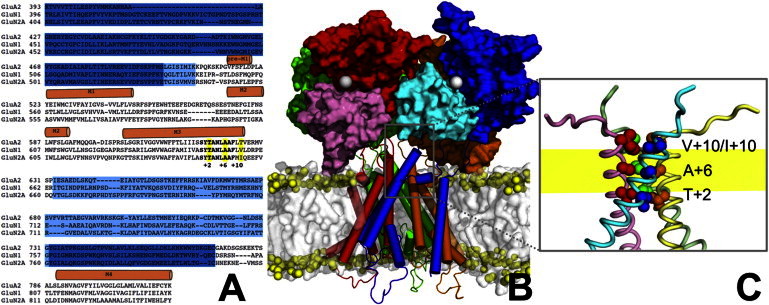
Sequence and structure of the GluN1/GluN2A receptor. (A) Sequence alignment of GluA2, GluN1, and GluN2A. Sequences for the D1 and D2 lobes of the LBD are shaded in blue and cyan, respectively. Helices in the transmembrane domains are indicated by cylinders above the sequences. The SYTANLAAF motif in M3 is in boldface. Residues forming the three rings of the activation gate are indicated by yellow shading and by the positions relative to the start of the SYTANLAAF motif (i.e., +2, +6, and +10). (B) The closed GluN1/GluN2A receptor after 20 ns simulation in the POPC bilayer. The four subunits, referred to as A, B, C, and D, with A and C being GluN1 and B and D being GluN2A, are colored in blue, red, green, and orange, respectively, except that the D2 lobes of subunits A and B are in cyan and pink, respectively. Subsequent figures follow a similar coloring scheme for the subunits. LBD dimers are formed by subunits A and D and by subunits C and B. Two gray spheres in subunits A and B indicate the agonist binding sites. The lipid bilayer is shown as a gray surface, but with phosphorus atoms shown as yellow spheres to indicate the headgroup region. (C) Enlarged view of the M3 to M3-D2 linker segments. The three rings of the activation gate are shown as spheres. The yellow band indicates the range of positions for the phosphorus atoms in the upper leaflet.
The GluN1/GluN2A LBD tetramer in the agonist-bound form was needed as the target for activation simulations. To build this, we used the structure of the agonist-bound dimer (PDB entry 2A5T) for both the AD and CB dimers and then arranged the dimers by superimposing to the model that we previously developed for the GluA2 LBD tetramer in the activated state (4).
MD simulations
A preequilibrated POPC bilayer consisting of 256 lipid molecules was obtained from CHARMM-GUI (30), and replicated and trimmed to generate a membrane with 686 lipid molecules. The GluN1/GluN2A receptor in the closed state was energy minimized while fixing the backbone atoms, and then inserted into the membrane, with clashing lipid molecules removed. The system was solvated with 91,422 water molecules, 266 Na+ ions, and 258 Cl− ions (corresponding to a salt concentration of 0.15 M), resulting in a total of ∼380,000 atoms. The receptor was positioned relative to the membrane such that charged side chains of the TMD either were surrounded by lipid headgroups or snorkeled into the aqueous phase (Fig. 1 B). The system preparation was done using VMD (31) and the Solvate and Ionize plugins.
To equilibrate the receptor with the membrane and the solvent, a 20-ns simulation was carried out while constraining the receptor backbone atoms with a force constant 20 kcal mol−1 Å−2. Simulation of the receptor in the closed state (without any constraints) was continued for another 20 ns. Thereafter two independent targeted MD simulations were carried out, each lasting 100 ns. For the first 20 ns of these simulations, the LBD tetramer was forced to change from the closed-state to the agonist-bound conformation, through constraining the root mean-square deviation (RMSD) from a target structure. This RMSD constraint was placed on the Cα atoms of GluN1 residues T396 to K543 (but excluding the modeled stretch G438-V451 in loop 1) and I664 to C798 and GluN2A residues N404 to S538 and V662 to C800. The target structure was interpolated between the closed-state and agonist-bound conformations, such that the RMSD from the latter conformation decreased linearly toward zero. The conformational transition of the LBD triggered motions of the LBD-TMD linkers and the TMD, presumably mimicking those induced by agonist binding. For the remaining 80 ns, the RMSD constraint was maintained with the agonist-bound conformation as the target, allowing the rest of the receptor to further relax. The two activation simulations followed the same protocol, except that the force constant for the RMSD constraint were 10,000 and 20,000 kcal mol−1 Å−2, respectively (4), and are referred to as sim1 and sim2.
All the simulations were conducted using NAMD 2.7 (32) under constant NPγT with pressure at 1 atm, temperature at 310 K, and surface tension at 20 dyn cm−1 (4). The RMSD constraint was realized using the colvars RMSD module of NAMD. The CHARMM27 force field was used (33). The Langevin dynamics and Nose-Hoover Langevin piston methods were used for temperature and pressure coupling, respectively.
Structural analyses
The HOLE program (34) was used to calculate the pore radii along the pore axis for each snapshot of the closed-state and activation simulations (sampled once per 50 ps). The solvent accessible surface areas (SASAs) of individual residues were calculated by the NACCESS program (35). Averages over 500 snapshots from the last 5 ns of the closed-state simulation and 5000 snapshots from the last 50 ns of sim1 were taken as values for the closed and open states, respectively. To mimic experiments for measuring state-dependent MTS modification rates, each residue of interest was mutated to cysteine in every snapshot before calculating its SASA. The electrostatic potential surfaces of the receptors were calculated using APBS (36). For these calculations, first the structure file of each receptor in PDB format was used to generate a PQR file (containing partial charges and atomic radii) by the python script pdb2pqr.py (37). This PQR file was then loaded to Pymol APBS Tools (38) to calculate the electrostatic potential with default parameters.
Results and Discussion
The membrane-bound closed GluN1/GluN2A receptor and the agonist-bound LBD tetramer
The sequence identities of GluN1 and GluN2A with GluA2 in the LBD and the TMD are all ∼30% (Fig. 1 A). This level of sequence identities ensures the reliability of our homology models of these domains (the structure of GluN1 subunits in the LBD was actually from PDB entry 1PBQ). In the closed-state structure of the GluA2 receptor (PDB entry 3KG2), the C-terminal halves of the M3 helices are formed by the sequence S615YTANLAAFLT625 in subunits B and D, and are four residues longer in subunits A and C. The boldfaced residues form three rings of constriction. In the model for the GluN1/GluN2A receptor, these positions are occupied by T648/T646, A652/A650, and V656/I654 (residues before and after “/” are those in GluN1 and GluN2A), respectively (Fig. 1, A and C). For the M3 C-terminal halves and the subsequent M3-D2 linkers, we adopt the nomenclature that the first residue of the SYTANLAAF motif is referred to as S+0 and a residue C-terminal to it as X+n, where X is the amino-acid name and n is the separation in sequence from S+0. For example, the residues forming the first and second rings of constriction are referred to as T+2 and A+6, respectively, and the residues forming the third ring are referred to as V+10 in GluN1 and I+10 in GluN2A. In the GluN2A subunits, the sequence Q+11EEFVDQ+17 constitute the M3-D2 linkers. In the GluN1 subunits, the fourth residue after V+10 is a proline, which is a helix breaker; therefore, instead of the four-residue lengthening of the M3 helices found in the A/C subunits of the GluA2 receptor, we have a three-residue lengthening, and the subsequent sequence P+14EER+17 constitutes the M3-D2 linkers. As in the GluA2 template (5), in addition to the difference in length, another important difference in the M3-D2 linkers between the A/C GluN1 subunits and the B/D GuN2A is their orientations: they are directed vertically in the former subunits but horizontally in the latter subunits (Fig. 1 C). The M3-D2 linkers are tethered to GluN1 I664 and GluN2A V662, which are referred to as the tips of the D2 lobes of the respective subunits.
In the structure of the closed GluN1/GluN2A receptor equilibrated in the lipid bilayer, A+6 are at the level of the membrane headgroup region, whereas V+10/I+10 and T+2 are just above and below it, respectively (Fig. 1, B and C). The pre-M1 helices are located in the membrane headgroup region. This location is compatible with the amphipathic nature of the pre-M1 sequences.
The GluN1/GluN2A agonist-bound LBD dimer (PDB entry 2A5T) superimposed well to the GluA2 activated LBD dimer (PDB entry 1FTJ), with a 1.0-Å RMSD over 352 Cα atoms (Fig. S1 A in the Supporting Material). However, as observed by Furukawa et al. (13), there is a notable difference at the tip of the D2 lobe of the GluN2A (but not GluN1) subunit relative to the GluA2 counterpart (largely due to different interlobe arrangements). Specifically, GluN2A V662 shifts toward the dimer interface by ∼6 Å. In the context of the closed GluN1/GluN2A receptor, this shift positions V662 to be collinear with the GluN2A M3-D2 linker (Fig. S1 B). GluN2A V662 in the agonist-bound conformation is thus optimally positioned for the outward pull of the M3-D2 linker, without having to change the direction of the tethered linker. Consequently, the GluN1/GluN2A receptor is expected to be more efficient than the GluA2 receptor in propagating the agonist-induced conformational change to the TMD, possibly resulting in some of the different functional characteristics between these two types of receptors (see below).
Gating motions of the GluN1/GluN2A receptor
We simulated the receptor in the closed state for 20 ns and subsequently, in two independent activation simulations (sim1 and sim2), we forced the LBD to change from the closed-state to the agonist-bound conformation during 20 ns and maintained the latter conformation for the remaining 80 ns. In the last 50 ns of each activation simulation, representing the open-state ensemble, the conformations of the receptor remained stable (Cα RMSDs, calculated over the LBD, M3-D2 linkers, and TMD helices, from the snapshot at 72.3 ns of sim1 averaged at 1.0 Å; the counterpart for sim2, with the snapshot at 90.0 ns as reference, was 1.1 Å). Moreover, the conformations in the two simulations were very similar (Cα RMSD between the two representative snapshots at 1.7 Å). Below we mostly present the results from sim1, noting that comparable results were obtained in sim2.
To follow the gating process, we monitored the radii of the channel pore along the pore axis, from the start of the closed-state simulation to the finish of each activation simulation (Fig. 2, A and B). In the closed-state simulation, the three rings of constriction (V+10/I+10, A+6, and T+2) were well maintained. In the first 20 ns of each activation simulation, as the LBD gradually moved toward the agonist-bound conformation, the pore radii at the V+10/I+10 level and to a lesser extent at the A+6 level, increased. In the next 80 ns, the pore became wide open at V+10/I+10 and A+6 (with radii of ∼3.5 and ∼2.5 Å), and partially open even at T+2. We characterize the channel as achieved the open state, and focus our description on the regions above T+2.
Figure 2.
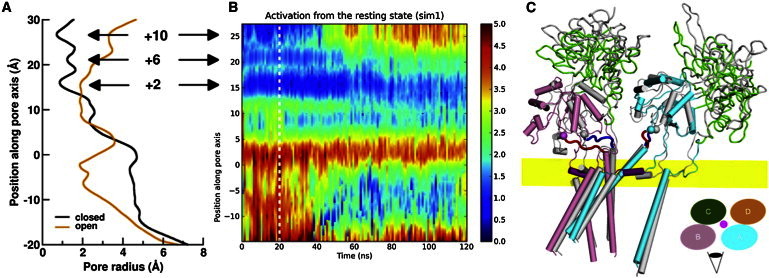
Change in pore radii and movement of the receptor during activation. (A) Pore radii along the pore axis in two snapshots. One, as black curve, is for the snapshot at the end of the 20-ns closed-state simulation; the other, as an orange curve, is for the snapshot at 72.3 ns of the activation simulation sim1. Here and in subsequent figures, these two snapshots are used as representatives of the closed and open states, respectively. The positions of the three rings of the activation gate are highlighted by arrows. (B) Time evolution of the pore radii, shown as color code in Å, in the close-state (to the left of the white dashed line at 20 ns) and the activation (to the right of that line) simulations. (C) Comparison of closed- and open-state structures. Only two subunits (A and B) are shown. The closed-state structure is shown in gray, except for the M3-D2 linkers, which are in blue. The D1 lobes (green trace) of the open-state structure are superimposed to the closed-state counterparts, and the open-state receptor is then translated downward to recover its vertical position relative to the bilayer, as indicated by the location of pre-M1 helices (purple cylinders) relative to the phosphorus band (in yellow). The rest of the open-state subunits A and B are in cyan and pink, respectively, except for the M3-D2 linkers, which are in red.
The overall gating movement of the receptor is illustrated in Fig. 2 C, by comparing the structures of two snapshots, one at the end of the 20-ns closed-state simulation and one at 72.3 ns into the activation simulation sim1. The Cα RMSDs between these two structures were 6.1 Å for the LBD and 2.7 Å for the TMD. As the D1 and D2 lobes of the LBD closed, the vertical distance between the D1 lobes and the TMD inevitably decreased. Throughout the activation simulation, the pre-M1 helices (Fig. 2 C; purple cylinders) were firmly anchored in the upper headgroup region of the lipid bilayer, indicating a steady vertical position for the TMD. Lobe closure thus resulted in a 6-Å movement of the D1 lobes toward the lipid bilayer. Recently, by atomic force microscopy imaging, Suzuki et al. (39) observed a 10-Å decrease in the height of the GluN1/GluN2A receptor upon activation. This observation is in good agreement with our activation simulations.
The movement critical for channel opening is by the M3-D2 linkers (Fig. 2 C; blue and red noodles for the closed and open states, respectively). In the open state, these linkers largely maintained their horizontal orientation in the GluN2A subunits and vertical orientation in the GluN1 subunits. Relative to the twofold symmetry axis, the movement of the GluN2A M3-D2 linkers was a radially outward translation, whereas the movement of the GluN1 M3-D2 linkers was a rotation plus modest outward translation (Fig. 2 C and Fig. S2 A). Using E+15/V+15 Cα atoms as representatives of the M3-D2 linkers, the diagonal distance between the GluN1 subunits increased by only 6.5 Å, whereas that between the GluN2A subunits increased by 17.5 Å (Fig. S2 B). On the other hand, the GluN1 diagonal rotated by ∼35° but the GluN2A diagonal rotated very little (Fig. S2 C). The pure translational motion of the GluN2A M3-D2 linkers is a result of the collinear positioning, noted above, of the agonist-bound D2 tips and the closed-state M3-D2 linkers. In comparison, the subunit B/D diagonal rotated by 7° in the GluA2 receptor upon activation (4).
Subunit-specific contributions to channel opening
The distinct motions of the M3-D2 linkers in the GluN1 and GluN2A subunits are expected to result in their differential contributions to channel opening. In particular, because an outward translational motion of the linkers would produce an outward pull of the M3 C-termini and hence likely widening of the outer pore, whereas a rotational motion may not be directly linked to pore widening, the GluN2A subunits should make greater contributions. (We suggested previously that rotational motions of the linkers were a way to relief the strain caused by the mismatch between the twofold symmetry of the LBD and the fourfold symmetry of the TMD in the closed-state GluA2 receptor (4).)
To quantify the subunit-specific contributions to pore widening, we calculated the diagonal Cα distances of the M3-helix residues forming the three rings of constriction in the outer pore. These diagonals for the second-ring A+6 residues are illustrated as dotted lines in Fig. 3 A. The time evolution of the diagonal distances is presented in Fig. 3 B, and the averages in the 20-ns closed simulation and in the last 50 ns of each activation simulation are shown in Fig. 3 C. Relative to their closed-state values, in the open state the GluN2A I+10, A+6, and T+2 distances (denoted as BD+10, BD+6, and BD+2 in Fig. 3, B and C), increased by 9.0, 4.1, and 1.3 Å, respectively, in sim1 (and similar results for sim2). In comparison, the GluN1A V+10 and A+6 distances increased by only 4.2 and 1.2 Å, and the GluN1A T+2 distance exhibited no change upon activation. These results demonstrate that the different motions of the GluN1 and GluN2A M3-D2 linkers, due to different positions and orientations, are propagated to the movement of the activation gate. The GluN2A subunits contribute more to gating than the GluN1 subunits, even though channel opening may require the cooperative movements of both types of subunits.
Figure 3.
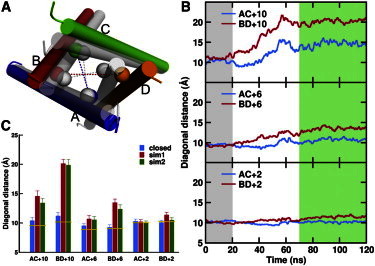
Differential contributions of the GluN1 and GluN2A subunits to gating, as indicated by diagonal distances of the Cα atoms of the residues forming the three rings of the activation gate. (A) Movement of the M3 helices, from the closed state (gray cylinders) to the open state (colored cylinders). The diagonals are shown as dotted lines. (B) Time evolution of the diagonal distances in the closed-state (0 to 20 ns, gray shaded region) and the activation (20 to 120 ns) simulations. The receptor in the last 50 ns of the trajectory is assumed to be in the open state (green shaded region). (C) Comparison of the average diagonal distances in the closed and open states. The yellow lines indicate the corresponding values in 3KG2.
The greater contribution of the GluN2A subunits is consistent with the role of the B/D subunits revealed in our previous study of the GluA2 receptor (4). However, in this study we identified a subtle but potentially significant difference between the two subtypes of iGluRs. As suggested above, the GluN2A M3-D2 linkers are better positioned for efficiently propagating the agonist-induced conformational change to the TMD. This is supported by a comparison between the two simulation studies in the magnitude of the movement of the activation gate (Fig. 3 and Fig. S3). Although the BD+10 distances were similar (20.1 vs. 20.5 Å) in the open-state GluN1/GluN2A and GluA2 receptors, both the BD+6 and BD+2 distances were larger in the GluN1/GluN2A receptor (13.4 and 11.4 Å vs. 11.0 and 10.3 Å). The more significant gating movement of the GluN1/GluN2A receptor could contribute to its larger conductance relative to the GluA2 receptor (2). Moreover, after activation, we speculate that a wider open NMDA receptor would have to traverse a longer distance and could encounter a higher energy barrier in returning to its closed state than an AMPA receptor, thus providing a possible contributing factor to the slower deactivation/desensitization of NMDA receptors (2). Recent study on a pentameric ligand-gated channel (40) raises the possibility that the isolated TMD has a higher free energy in the open state than in the closed state.
Wollmuth and co-workers found strong evidence for the differential contributions of the GluN1 and GluN2 subunits to channel gating (18–22). In the open state, deep pore-lining positions that were accessible to MTS reagents spanned 260° on the GluN1 M3 helical wheel (including V-2, T+2, A+3, N+4, and L+5) but only 60° on the GluN2A M3 helical wheel (V-5, L-2, T+2). In addition, the GluN1 accessible positions had significantly faster reaction rates than the GluN2 ones. The top view into the M3 helices in our open-state model (Fig. 3 A) shows that it indeed predicts wider accessibility to the GluN1 M3 helices than to the GluN2A M3 helices from the lumen of the open channel. The greater outward movement of the GluN2A M3 C termini leads to a parallelogram for the four positions occupied by homologous M3 residues (e.g., A+6), with the GluN1 residues at the obtuse-angle vertices and the GluN2A residues at the acute-angle vertices. It is obvious that the obtuse angle corresponds to greater access from the center of the parallelogram. Side views into the GluN1 and GluN2A M3 surfaces exposed to the lumen further illustrate the point (Fig. S4, A and B). The C-terminal half of a GluN1 subunit, sandwiched between two GluN2 M3 helices, has a wide swath of exposed surface; in comparison, the C-terminal half of a GluN2A subunit has only a narrow strip of exposed surface.
Lurcher and adjacent mutations
The Lurcher mutation A+7T in the SYTALAAF motif was originally discovered in the GluD2 receptor for resulting in a constitutively open channel (41). In the GluN1/GluN2A receptor, Lurcher mutations resulted in slow down of the desensitization and deactivation kinetics (23). Importantly, the mutation on the GluN1 subunits has a much more significant effect than the GluN2A counterpart, implicating a stronger ability of the former mutation in maintaining channel opening.
In our structural models, the A+7 residues are located in the M3-M3 helix interfaces and become exposed upon channel opening, much more so in the GluN1 subunits than the GluN2A subunits (Fig. S4, A and B). A mutation from alanine to threonine could favor the open state, because it provides additional room for threonine’s bulkier side chain. As emphasized above, the GluN2A M3 helices undergo greater gating movement than the GluN1 M3 helices. A GluN1 A+7T residue can be accommodated in the interface with the adjacent GluN2A M3 helix (Fig. S4 A), thereby locking it into the open-state position. A GluN2A A+7T residue would be less effective in slowing down deactivation and desensitization, because such a residue is less well accommodated in the interface with the adjacent GluN1 M3 helix (Fig. S4 B) and the latter helix contributes less to channel gating.
Our rationalization of the subunit-specific effects of Lurcher mutations is bolstered by the observation that the GluN1 A+3C mutation, but not the homologous GluN2 mutation, resulted in a constitutively open channel (22). The A+3 residues are one helix turn down from and hence right next to the A+7 residues, occupying the same M3-M3 helix interfaces (Fig. S4, A and B). Again, we expect the GluN1 A+3C residues, upon being accommodated in the interfaces with the adjacent GluN2A M3 helices, to lock them in the open-state positions.
Position-specific inhibition and potentiation effects
Using cysteine mutations around the extracellular vestibule, Beck et al. (18), Watanabe et al. (21), and Sobolevsky et al. (19,20,22) identified positions where MTS modifications resulted in either inhibition or potentiation of agonist-induced currents. Our structural model for the open state can now provide rationalization of these results. To that end, in Fig. 4, A and B, we display these inhibitory and potentiating positions in the open-state model. Inhibition effects may be observed either when MTS modifications shift the equilibrium between the open and closed states toward the latter, or when the covalently attached MTS reagents act as open-pore blockers. Potentiation effects may be observed when the attached MTS reagents shift the equilibrium toward the open state and yet do not block the open pore.
Figure 4.
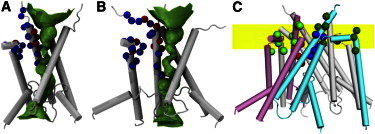
Positions around the extracellular vestibule exhibiting different behaviors upon MTS modifications. (A) GluN1 positions exhibiting inhibitory (blue spheres) or potentiating (red spheres) effects. Data are from Beck et al. (18) and Watanabe et al. (21). (B) GluN2A positions exhibiting inhibitory (blue spheres) and potentiating (red spheres) effects. Data are from Sobolevsky et al. (20,22). In (A) and (B), a HOLE image (34) is shown to indicate the channel pore. (C) Positions showing either voltage-independent (dark and light green for GluN1 and GluN2, respectively) or voltage-dependent (blue and red for GluN1 and GluN2, respectively) MTS modification rates. Data are from Sobolevsky et al. (19,20,22). The yellow band indicates the range of positions for the phosphorus atoms in the upper leaflet.
Fig. 4, A and B, shows that the GluN1 and GluN2A subunits have similar patterns of inhibition and potentiation. Both GluN1 and GluN2A harbor two groups of inhibitory positions. One is in the interface between the M3 to M3-D2 linker segment and the pre-M1 to M1 segment (GluN1 N+4, L+5, F+8, L+9, L+11, D+12, R+13, E+15, E+16 and D552, F554, M555, P557, F558, and L562; GluN2A F+8, M+9, Q+11, E+13, F+14, V+15 and S547, A548, F549, L550, P552, F553, A555, S556, V557, and W558). We suggest that the attachment of bulky MTS reagents in this interface hinders the outward movement of the M3 helix during channel activation, thus shifting the equilibrium toward the closed state. The second group of inhibitory positions is deep into the outer pore (GluN1 V-2, T+2, and A+3 and GluN2A V-5, L-2, T+2). We suggest that MTS reagents attached to these positions act as open-pore blockers.
Both GluN1 and GluN2A harbor potentiating positions at the lumen of the channel (GluN1 A+6, A+7, and V+10; GluN2A A+3, N+4, A+6, A+7, I+10, and E+12). We suggest that like-charge repulsion between MTS reagents attached at diagonal positions on GluN1 or GluN2A can force the C-termini of the M3 helices to move apart, thereby shifting the equilibrium toward the open state. It is likely that the lumen of the open channel is too wide for the attached MTS reagents to block it. This explanation is supported by the observation that the A+6R mutation on either GluN1 or GluN2 resulted in a constitutively open channel (28). Moreover, when GluN1 A+6R was paired with GluN2 A+6E or GluN2 A+6R was paired with GluN1 A+6E, the constitutive activity reduced. The latter result can be understood because the attraction between opposite charges in neighboring GluN1 and GluN2 subunits would mitigate the repulsion of like charges in diagonal GluN1 and GluN2 subunits. The fact that the V+10/I+10 and A+6 positions produced potentiating effects but the T+2 positions produced inhibitory effects fits well with our gating model, which, due to the outward pull of the M3-D2 linkers, resulted in a wider opening at V+10/I+10 and A+6 than at T+2 (Fig. 2, A and B and Fig. 4, A and B).
We found a simple set of criteria that is able to identify most of these inhibitory and potentiating residues: 1), all residues in the interface between the M3 to M3-D2 linker segment and the pre-M1 to M1 segment are putatively inhibitory; and 2), a pore-facing M3 residue, depending on the pore size there, is either inhibitory or potentiating. With these criteria, 13 of the 18 inhibitory GluN1 residues and 11 of the 19 inhibitory GluN2A residues are correctly identified; for potentiating residues on the pore-facing side of M3, 3 out of 3 such residues in GluN1 and 3 out of 6 such residues in GluN2A are identified (Fig. S5).
The L+5 position in GluN1 produced inhibitory effects but in GluN2 was inaccessible (22). Chen and Lipton (42) identified GluN1 L+5 as the shallow, low-affinity site for memantine binding, in addition to strong binding deep in the pore (near the so-called Q/R/N site at the tip of the M2 helix). A number of studies have suggested that binding to the shallow site contributes to memantine’s unique therapeutic use (42–46). We suggest that memantine binding, similar to MTS modification, at the GluN1 L+5 position produces an inhibitory effect. A memantine molecule bound at this position, in the interface of the M3 and pre-M1 helices (Fig. S6) could hinder the outward movement of the M3 helix and thereby maintain channel closing.
Staggering of GluN1 and GluN2 M3 helices?
In 2002 Sobolevsky et al. (20) proposed that the GluN1 and GluN2 M3 helices were staggered, with the latter shifted upward by approximately one helix turn (or four residues). Given that the M3 helices in the four subunits of the GluA2 receptor are positioned at the same height (5) and the strong sequence homology between the TMDs of the AMPA and NMDA receptors, including the conserved SYTANLAAF motif in the M3 helices, subunit staggering seems an unlikely scenario. In this regard, we note that the hydrophobic sequence and the bordering charged residues in a transmembrane helix are important features in determining the positioning of the helix relative to the membrane (47). These features are conserved between the M3 helices of the AMPA and NMDA receptors (Fig. 1 A).
The staggering proposal was based on three observations. These observations now serve as important tests of our structural model for the GluN1/GluN2A receptor, which has the four M3 helices positioned at the same height. The first observation is the putatively distinct patterns of voltage dependence for the MTS modification rates (in the presence of agonists) at homologous positions on the GluN1 and GluN2 M3 helices (19,20). A voltage-dependent position is likely embedded in the membrane, whereas a voltage-independent position is likely outside the membrane. In Fig. 4 C we display the voltage-dependent GluN1 and GluN2 M3 positions in blue and red, respectively, and the voltage-independent GluN1 and GluN2 M3 positions in dark and light green, respectively, on our structural model for the open state. The blue and red positions indeed show disparity in height, which could be relieved by staggering. However, when the results from the pre-M1, M1, and M4 helices (19,22) are also displayed (Fig. 4 C), the border between voltage-dependent and independent positions become fuzzier. Overall, the pattern of voltage dependence for the MTS modification rates in the various positions is consistent with a widened border that is coincident with the membrane headgroup region.
The second observation implying a one-turn upward shift for the GluN2 M3 helix is that 2-aminoethyl MTS was accessible down to GluN2 V-5 but only to GluN1 V-2 (19,20). In our structural model for the open-state GluN1/GluN2A receptor, GluN2A V-5 projects into the pore but the homologous GluN1 M-5 is buried in the interface with the adjacent GluN2A M3 helix (Fig. S7 A). The latter position explains why GluN1 M-5 is not accessible to the MTS reagent.
The third observation that apparently favored staggering is that the T+2C/L-2C mutant coordinated Cu2+ (while in the presence of agonists). Upon introducing the GluN1 T+2C and GluN2A L-2C mutations into our open-state GluN1/GluN2A receptor, we find that Cu2+ can indeed be coordinated between adjacent GluN1 T+2C and GluN2A L-2C residues, with Cu2+-sulfur distances of 2.8–2.9 Å (Fig. S7 B). In short, all the observations that led Sobolevsky et al. (20) to propose staggering can be explained by our structural model for the GluN1/GluN2A receptor.
State-dependent MTS modification rates
Wollmuth and co-workers (19,20,22,24) and others (25–28) used the substituted-cysteine accessibility method (SCAM) (48) to obtain state-dependent MTS modification rates at positions around the extracellular vestibule. In previous work (4,49,50), we found that such SCAM results, low-resolution in themselves, can be very valuable in validating gating models developed from computation. In this validation we examine whether the differences in modification rates ( and ) of substituted cysteines in the presence and absence of agonists can be correlated with the differences in the solvent accessible surface areas of the corresponding residues between the open- and closed-state structural models.
Fig. S8 displays the comparison of and for positions in the pre-M1 to M1 segment and the M3 to M3-D2 linker segment of the GluN1/GluN2 receptor. As noted by Sobolevsky et al. (22), overall, the M3 to M3-D2 linker segment showed more extensive state-dependent changes in MTS modification rates than the pre-M1 to M1 segment. For most positions in the M3 to M3-D2 linker segment, was significantly higher than , whereas many positions in the pre-M1 to M1 segment had comparable and . This general trend is well captured by the values.
Ca2+ permeability of NMDA receptors
A main difference of NMDA receptors from other iGluRs is the former’s high Ca2+ permeability (2). This Ca2+ influx plays an important role in learning and memory, possibly via triggering a CaMKII-related signaling cascade (3,51). Ca2+ permeability is mainly controlled by the selectivity filter at the Q/R/N site (52). However, an additional extracellular Ca2+-binding site was suggested, based on the observation that Ca2+ can block the channel in a voltage-independent manner (53,54). The existence of an additional site was further supported by the difference in Ca2+ transport between NMDA receptors and Ca2+-permeable AMPA receptors (55). Watanabe et al. (21) investigated the role of a highly charged motif D+12RPEER+17 that is unique to the GluN1 subunits and concluded that this motif acts as the external Ca2+-binding site.
With the structural model for the GluN1/GluN2A receptor at hand, we can now analyze this putative external Ca2+-binding site in detail. In Fig. 5 A we display the electrostatic surface of the open-state receptor. In our structural model, the D+12RPEER+17 motif borders the large fenestration between the D2 lobes (e.g., from subunits A and B) of two different LBD dimers, which likely serves as the entrance to the channel pore. The electrostatic potential in this region is strongly negative. In addition to the three anionic residues (D+12, E+15, and E+16) in the DRPEER motif, this region includes a number of other anionic residues, including D552 from the pre-M1 helix and D669 from the D2 lobe of the same GluN1 subunit, as well as D742 and E743 from the D2 lobe and E803 and E806 of the GluN2A subunit across the fenestration (Fig. 5 B). The strong negative electrostatic potential is expected to attract cations to the mouth of the channel pore and contribute to Ca2+ influx through NMDA receptor channels.
Figure 5.
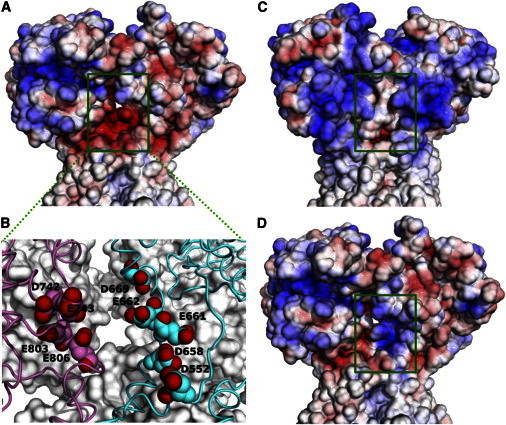
Electrostatic potential surfaces of the GluN1/GluN2A and GluA2 receptors in the activated state. The view is into the dimer-dimer interface (between subunits A on the right and B on the left), where a fenestration is likely to serve as the entrance to the channel pore. The color scale for the electrostatic potentials is from –5 kBT/e for red to +5 kBT/e for blue. (A) Electrostatic surface of the wild-type GluN1/GluN2A receptor. (B) A cluster of anionic residues around the interdimer fenestration. (C) Electrostatic surface of the GluA2 receptor. (D) Electrostatic surface of a mutant GluN1/GluN2A receptor, in which the GluN1 DRPEER motif is changed to RRPRRR.
That expectation was borne out by our activation simulations. In the last 50 ns of the simulations, on average 6.2 Na+ ions (but only 0.2 Cl− ions) were within 5 Å of the aforementioned anionic residues. Ca2+ has an ionic radius very similar to Na+ but has twice the charge, and is therefore expected to bind even more strongly to this site. We also traced the movement of a Na+ ion that was eventually positioned in the pore (blue sphere in Fig. S9). Starting from the bulk solution, this ion came to the putative binding site at the dimer-dimer interface, bound there for a few ns, and diffused into the interior of the LBD. After some excursion there, this ion then entered the channel pore and was solvated there. Although we did not observe the passage of any ion between the extracellular and intracellular vestibules due to the partially open pore and the relatively short duration of our simulations, a continuous chain of water molecules connecting the two vestibules indicates the potential pathway for ion transport (Fig. S9).
In contrast to the strong negative electrostatic potential in the GluN1/GluN2A receptor, the electrostatic potential calculated on the open-state GluA2 receptor from our previous study (4) is neutral to mildly positive around the interdimer fenestration (Fig. 5 C). This difference provides further support for the contributions of anionic residues at the mouth of the channel pore in NMDA receptors. Watanabe et al. (21) found that mutation of the anionic residues in the DRPEER motif to arginine resulted in a decrease in the fractional Ca2+ currents. Consistent with this observation, this mutation changes the electrostatic potential around the interdimer fenestration from strongly negative to moderately negative (Fig. 5 D).
Concluding Remarks
We have used homology modeling and targeted MD simulations to develop structural models for the GluN1/GluN2A receptor in the closed state and open state as well as a gating model. Our simulations showed that, upon lobe closure induced by agonist binding, the M3-D2 linkers moved outward. The resulting pull on the C-termini of the M3 helices led to the outward tilt of the C-terminal halves of these helices and the opening of the channel pore. The GluN2A subunits had greater M3 outward movements and thus contributed more to channel gating than the GluN1 subunits.
Our gating model is validated by a range of experimental observations, including a decrease in the height of the receptor upon activation and state-dependent MTS modification rates. It now provides resolution to paradoxical observations, such as wider exposure of the GluN1 M3 helices to the lumen of the open channel and Cu2+ coordination between GluN1 T+2 and GluN2 L-2 positions. Our gating model also reveals a possible molecular basis for the action of the Lurcher mutations, especially in the GluN1 subunits, in slowing down the deactivation and desensitization kinetics of the GluN1/GluN2A receptor.
We have provided explanations for position-specific inhibition and potentiation effects of MTS modifications and in doing so, identified a region of potential pharmacological interest in NMDA receptors. This is the interface of the M3 and pre-M1 interface. Here, MTS modifications resulted in inhibition of agonist-induced currents, presumably by hindering the outward movement of the M3 helices. It is also here that the anti-Alzheimer’s disease drug memantine putatively binds. With a structural model for the GluN1/GluN2A developed, it will be interesting to further explore the binding mode of memantine at this external site and screen for other compounds that target this site. More broadly, differences between the closed- and open-state structural models can be the basis for design drugs that specifically act on NMDA receptors.
This study has uncovered important clues to the unique functional characteristics of NMDA receptors. A shift of the tip of the GluN2A D2 lobe toward the dimer interface, relative to an AMPA receptor, appears to allow the NMDA receptor to more efficiently propagate the agonist-induced conformational changes from the LBD to the TMD. A cluster of anionic residues, including in the GluN1 DRPEER motif, are positioned for attracting cations into the likely mouth to the channel pore, thereby contributing to the Ca2+ permeability of NMDA receptors.
The accuracy of our structural models is subject to the limitations of homology modeling and targeted MD simulations. In our opinion, the models are more reliable in regions above the second ring (i.e., A+6) of the activation gate, where we have focused analyses. Future experiments will provide information for refinement of these models, leading to a better understanding of this important subtype of inotropic glutamate receptors.
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) grant GM88187.
Supporting Material
References
- 1.Kumar J., Mayer M.L. Functional insights from glutamate receptor ion channel structures. Annu. Rev. Physiol. 2013;75:313–337. doi: 10.1146/annurev-physiol-030212-183711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Traynelis S.F., Wollmuth L.P., Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lynch M.A. Long-term potentiation and memory. Physiol. Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- 4.Dong H., Zhou H.X. Atomistic mechanism for the activation and desensitization of an AMPA-subtype glutamate receptor. Nat Commun. 2011;2:354. doi: 10.1038/ncomms1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sobolevsky A.I., Rosconi M.P., Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armstrong N., Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- 7.Armstrong N., Jasti J., Gouaux E. Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell. 2006;127:85–97. doi: 10.1016/j.cell.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 8.Sobolevsky A.I., Yelshansky M.V., Wollmuth L.P. Different gating mechanisms in glutamate receptor and K+ channels. J. Neurosci. 2003;23:7559–7568. doi: 10.1523/JNEUROSCI.23-20-07559.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sobolevsky A.I., Yelshansky M.V., Wollmuth L.P. The outer pore of the glutamate receptor channel has 2-fold rotational symmetry. Neuron. 2004;41:367–378. doi: 10.1016/s0896-6273(04)00008-x. [DOI] [PubMed] [Google Scholar]
- 10.Schüler T., Mesic I., Laube B. Formation of NR1/NR2 and NR1/NR3 heterodimers constitutes the initial step in N-methyl-D-aspartate receptor assembly. J. Biol. Chem. 2008;283:37–46. doi: 10.1074/jbc.M703539200. [DOI] [PubMed] [Google Scholar]
- 11.Furukawa H., Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO J. 2003;22:2873–2885. doi: 10.1093/emboj/cdg303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inanobe A., Furukawa H., Gouaux E. Mechanism of partial agonist action at the NR1 subunit of NMDA receptors. Neuron. 2005;47:71–84. doi: 10.1016/j.neuron.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 13.Furukawa H., Singh S.K., Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438:185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- 14.Vance K.M., Simorowski N., Furukawa H. Ligand-specific deactivation time course of GluN1/GluN2D NMDA receptors. Nat. Commun. 2011;2:294. doi: 10.1038/ncomms1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao Y., Harrison C.B., Mayer M.L. Molecular mechanism of ligand recognition by NR3 subtype glutamate receptors. EMBO J. 2008;27:2158–2170. doi: 10.1038/emboj.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salussolia C.L., Prodromou M.L., Wollmuth L.P. Arrangement of subunits in functional NMDA receptors. J. Neurosci. 2011;31:11295–11304. doi: 10.1523/JNEUROSCI.5612-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riou M., Stroebel D., Paoletti P. An alternating GluN1-2-1-2 subunit arrangement in mature NMDA receptors. PLoS ONE. 2012;7:e35134. doi: 10.1371/journal.pone.0035134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beck C., Wollmuth L.P., Kuner T. NMDAR channel segments forming the extracellular vestibule inferred from the accessibility of substituted cysteines. Neuron. 1999;22:559–570. doi: 10.1016/s0896-6273(00)80710-2. [DOI] [PubMed] [Google Scholar]
- 19.Sobolevsky A.I., Beck C., Wollmuth L.P. Molecular rearrangements of the extracellular vestibule in NMDAR channels during gating. Neuron. 2002;33:75–85. doi: 10.1016/s0896-6273(01)00560-8. [DOI] [PubMed] [Google Scholar]
- 20.Sobolevsky A.I., Rooney L., Wollmuth L.P. Staggering of subunits in NMDAR channels. Biophys. J. 2002;83:3304–3314. doi: 10.1016/S0006-3495(02)75331-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe J., Beck C., Wollmuth L.P. DRPEER: a motif in the extracellular vestibule conferring high Ca2+ flux rates in NMDA receptor channels. J. Neurosci. 2002;22:10209–10216. doi: 10.1523/JNEUROSCI.22-23-10209.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sobolevsky A.I., Prodromou M.L., Wollmuth L.P. Subunit-specific contribution of pore-forming domains to NMDA receptor channel structure and gating. J. Gen. Physiol. 2007;129:509–525. doi: 10.1085/jgp.200609718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohda K., Wang Y., Yuzaki M. Mutation of a glutamate receptor motif reveals its role in gating and delta2 receptor channel properties. Nat. Neurosci. 2000;3:315–322. doi: 10.1038/73877. [DOI] [PubMed] [Google Scholar]
- 24.Talukder I., Borker P., Wollmuth L.P. Specific sites within the ligand-binding domain and ion channel linkers modulate NMDA receptor gating. J. Neurosci. 2010;30:11792–11804. doi: 10.1523/JNEUROSCI.5382-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones K.S., VanDongen H.M.A., VanDongen A.M.J. The NMDA receptor M3 segment is a conserved transduction element coupling ligand binding to channel opening. J. Neurosci. 2002;22:2044–2053. doi: 10.1523/JNEUROSCI.22-06-02044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan H., Erreger K., Traynelis S.F. Conserved structural and functional control of N-methyl-D-aspartate receptor gating by transmembrane domain M3. J. Biol. Chem. 2005;280:29708–29716. doi: 10.1074/jbc.M414215200. [DOI] [PubMed] [Google Scholar]
- 27.Wada A., Takahashi H., Chen H.S. NR3A modulates the outer vestibule of the “NMDA” receptor channel. J. Neurosci. 2006;26:13156–13166. doi: 10.1523/JNEUROSCI.2552-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang H.R., Kuo C.C. The activation gate and gating mechanism of the NMDA receptor. J. Neurosci. 2008;28:1546–1556. doi: 10.1523/JNEUROSCI.3485-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 30.Jo S., Kim T., Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 31.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 32.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 34.Smart O.S., Neduvelil J.G., Sansom M.S. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996;14:354–360. doi: 10.1016/s0263-7855(97)00009-x. 376. [DOI] [PubMed] [Google Scholar]
- 35.Hubbard S.J., Thornton J.M. Department of Biochemistry and Molecular Biology, University College; London: 1993. NACCESS. [Google Scholar]
- 36.Baker N.A., Sept D., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dolinsky T.J., Nielsen J.E., Baker N.A. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32(Web Server issue):W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lerner M.G., Carlson H.A. University of Michigan; Ann Arbor: 2006. APBS Plugin for PyMOL. [Google Scholar]
- 39.Suzuki Y., Goetze T.A., Edwardson J.M. Visualization of structural changes accompanying activation of N-methyl-D-aspartate (NMDA) receptors using fast-scan atomic force microscopy imaging. J. Biol. Chem. 2013;288:778–784. doi: 10.1074/jbc.M112.422311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu F.Q., Hummer G. Pore opening and closing of a pentameric ligand-gated ion channel. Proc. Natl. Acad. Sci. USA. 2010;107:19814–19819. doi: 10.1073/pnas.1009313107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zuo J., De Jager P.L., Heintz N. Neurodegeneration in Lurcher mice caused by mutation in delta2 glutamate receptor gene. Nature. 1997;388:769–773. doi: 10.1038/42009. [DOI] [PubMed] [Google Scholar]
- 42.Chen H.S., Lipton S.A. Pharmacological implications of two distinct mechanisms of interaction of memantine with N-methyl-D-aspartate-gated channels. J. Pharmacol. Exp. Ther. 2005;314:961–971. doi: 10.1124/jpet.105.085142. [DOI] [PubMed] [Google Scholar]
- 43.Blanpied T.A., Boeckman F.A., Johnson J.W. Trapping channel block of NMDA-activated responses by amantadine and memantine. J. Neurophysiol. 1997;77:309–323. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- 44.Sobolevsky A.I., Koshelev S.G., Khodorov B.I. Interaction of memantine and amantadine with agonist-unbound NMDA-receptor channels in acutely isolated rat hippocampal neurons. J. Physiol. 1998;512:47–60. doi: 10.1111/j.1469-7793.1998.047bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sobolevsky A., Koshelev S. Two blocking sites of amino-adamantane derivatives in open N-methyl-D-aspartate channels. Biophys. J. 1998;74:1305–1319. doi: 10.1016/S0006-3495(98)77844-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kotermanski S.E., Wood J.T., Johnson J.W. Memantine binding to a superficial site on NMDA receptors contributes to partial trapping. J. Physiol. 2009;587:4589–4604. doi: 10.1113/jphysiol.2009.176297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou H.X., Cross T.A. Influences of membrane mimetic environments on membrane protein structures. Annu. Rev. Biophys. 2013 doi: 10.1146/annurev-biophys-083012-130326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karlin A., Akabas M.H. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 49.Yi M.G., Tjong H.T., Zhou H.X. Spontaneous conformational change and toxin binding in alpha7 acetylcholine receptor: insight into channel activation and inhibition. Proc. Natl. Acad. Sci. USA. 2008;105:8280–8285. doi: 10.1073/pnas.0710530105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du J., Dong H., Zhou H.X. Gating mechanism of a P2X4 receptor developed from normal mode analysis and molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 2012;109:4140–4145. doi: 10.1073/pnas.1119546109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bayer K.U., De Koninck P., Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- 52.Kuner T., Wollmuth L.P., Sakmann B. Structure of the NMDA receptor channel M2 segment inferred from the accessibility of substituted cysteines. Neuron. 1996;17:343–352. doi: 10.1016/s0896-6273(00)80165-8. [DOI] [PubMed] [Google Scholar]
- 53.Premkumar L.S., Auerbach A. Identification of a high affinity divalent cation binding site near the entrance of the NMDA receptor channel. Neuron. 1996;16:869–880. doi: 10.1016/s0896-6273(00)80107-5. [DOI] [PubMed] [Google Scholar]
- 54.Sharma G., Stevens C.F. Interactions between two divalent ion binding sites in N-methyl-D-aspartate receptor channels. Proc. Natl. Acad. Sci. USA. 1996;93:14170–14175. doi: 10.1073/pnas.93.24.14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wollmuth L.P., Sakmann B. Different mechanisms of Ca2+ transport in NMDA and Ca2+-permeable AMPA glutamate receptor channels. J. Gen. Physiol. 1998;112:623–636. doi: 10.1085/jgp.112.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.