

Abstract
Increased exploration and exploitation of resources in the Arctic is leading to a higher risk of petroleum contamination. A number of Arctic microorganisms can use petroleum for growth-supporting carbon and energy, but traditional approaches for stimulating these microorganisms (for example, nutrient addition) have varied in effectiveness between sites. Consistent environmental controls on microbial community response to disturbance from petroleum contaminants and nutrient amendments across Arctic soils have not been identified, nor is it known whether specific taxa are universally associated with efficient bioremediation. In this study, we contaminated 18 Arctic soils with diesel and treated subsamples of each with monoammonium phosphate (MAP), which has successfully stimulated degradation in some contaminated Arctic soils. Bacterial community composition of uncontaminated, diesel-contaminated and diesel+MAP soils was assessed through multiplexed 16S (ribosomal RNA) rRNA gene sequencing on an Ion Torrent Personal Genome Machine, while hydrocarbon degradation was measured by gas chromatography analysis. Diversity of 16S rRNA gene sequences was reduced by diesel, and more so by the combination of diesel and MAP. Actinobacteria dominated uncontaminated soils with <10% organic matter, while Proteobacteria dominated higher-organic matter soils, and this pattern was exaggerated following disturbance. Degradation with and without MAP was predictable by initial bacterial diversity and the abundance of specific assemblages of Betaproteobacteria, respectively. High Betaproteobacteria abundance was positively correlated with high diesel degradation in MAP-treated soils, suggesting this may be an important group to stimulate. The predictability with which bacterial communities respond to these disturbances suggests that costly and time-consuming contaminated site assessments may not be necessary in the future.
Keywords: 16S rRNA, Arctic, betaproteobacteria, bioremediation, disturbance, nutrient amendment
Introduction
The thawing of Arctic sea ice is facilitating the exploration and exploitation of Arctic resources by northern countries. Petroleum is a targeted resource, and is also used to power many aspects of Arctic work, so there is an increasing risk of contamination in a biome that has had limited exposure to human industrial activity. In many environments, a variety of native microorganisms are capable of petroleum degradation (Greer et al., 2010), but petroleum-degrading activity is primarily limited to the few months in which soil temperatures exceed 0 °C (Walworth et al., 2001). In the interest of decreasing the lifespan of these pollutants in the environment, nutrients are often added to contaminated Arctic soils to stimulate microbial growth and activity, although studies have shown that treatment effectiveness can vary substantially between geographically similar soil environments (Greer, 2009; Yergeau et al., 2009).
Several recent studies have determined that pH and nitrogen concentration are major determinants of microbial diversity across a variety of soils in uncontaminated ecosystems (Lauber et al., 2009; Chu et al., 2010; Ramirez et al., 2012), but the factors governing diversity in hydrocarbon-contaminated soils have not been firmly established. Comparisons of microbial community composition across sites have mostly been conducted in soils with existing petroleum contamination (Greer et al., 2010; Powell et al., 2010), but the discovery of consistent trends has often been confounded by variations in pollutant composition and age. In addition, despite the identification of many hydrocarbon-degrading taxa across ecosystems, it is generally not known which indigenous microorganisms should be promoted to optimize the rate and extent of bioremediation in contaminated environments. Added nutrients are not incorporated equally by bacterial taxa in hydrocarbon-contaminated soils (Bell et al., 2011), and because of the fact that many taxa are able to remain active following hydrocarbon contamination, it is not clear that the most effective hydrocarbon-degrading taxa are favoured by bioremediation treatments or the natural environment.
Ecological disturbance is generally defined as a distinct event that alters the taxonomic composition, resource availability or physical properties of an environment (Horner-Devine et al., 2004), which classifies both the petroleum contamination of soils and the subsequent application of nutrients as disturbance events. Although petroleum contaminants can provide growth-supporting energy and carbon to many bacteria, they are toxic in high concentrations, primarily due to negative interactions with cell walls (Ramos et al., 2002; Kang and Park, 2010), and thus require adaptations and resource trade-offs. Microbial communities can also be disrupted by nitrogen inputs, and reductions in respiration, biomass and diversity have been observed following nitrogen addition (Allison et al., 2007; Ramirez et al., 2012). Experiments with protists have shown that competition can intensify at high levels of disturbance, when multiple surviving organisms are at the limit of their tolerance (Violle et al., 2010), so a competitive advantage in a specific environment may have more of a role in taxonomic dominance than the ability to quickly metabolize the petroleum carbon source.
In this study, we collected soil samples from across the Arctic and sub-Arctic, and treated them with equal amounts of diesel, both with and without a nutrient amendment. The amendment that we selected is comparable in both type and concentration to those that have been used previously at hydrocarbon-contaminated Arctic sites (Thomassin-Lacroix et al., 2002; McCarthy et al., 2004; Greer, 2009; Bell et al., 2011). We expected that the success of hydrocarbon-degrading bacteria in disturbed soils would be governed by adaptations to the soil environment, and that we would observe clear differences in the relative abundance of hydrocarbon-degrading groups across soil gradients. In addition, we expected to see a shift in the extent of hydrocarbon degradation depending on which taxa dominated the bacterial community. A clear understanding of the factors that drive microbial community shifts following disturbance with petroleum and nutrient amendments will help in predicting optimal bioremediation treatments without site-specific assessments that are time-consuming and expensive (Dorn and Salanitro, 2000).
Materials and methods
Soil collection and analyses
The top 15 cm of soil was collected from 18 Arctic locations between June and August 2011. We aimed to find patterns in microbial community response to petroleum contamination and nutrient amendment regardless of where a contamination event occurred, so we collected soils that varied widely in pH and organic matter content. Soil sampling locations and properties are presented in Table 1 and Supplementary Figure 1. Hydrocarbon analyses were adapted from the Canada-Wide Standard for Petroleum Hydrocarbons in Soil, as used previously for the determination of hydrocarbon concentrations in sub-Arctic soils (Chang et al., 2011). Detailed procedures for soil and hydrocarbon analyses are presented in the Supplementary Material.
Table 1. Soil characteristics and percent diesel degradation.
| Sample code | Coordinates | pH |
Soil characteristics |
Diesel degradation (%) |
||||
|---|---|---|---|---|---|---|---|---|
| Water content (%) | Organic matter (%) | Nitrate+nitrite (mg kg−1) | Total nitrogen (mg kg−1) | DSL | DSL–MAP | |||
| AH1 | 79°N, 90°W | 7.20 | 70.70 | 27.15 | 0.64 | 1230.78 | 28.32 | 52.97 |
| AH2 | 79°N, 90°W | 7.30 | 12.33 | 5.20 | <0.05 | 876.73 | 22.67 | 41.54 |
| AK1 | 69°N, 150°W | 5.95 | 80.97 | 96.25 | 0.20 | 1560.34 | 51.84 | 55.75 |
| AK2 | 69°N, 150°W | 6.74 | 66.19 | 50.07 | 1.18 | 4868.21 | 38.67 | 41.66 |
| AK3 | 69°N, 150°W | 6.97 | 88.53 | 79.45 | 2.80 | 1994.91 | 27.76 | 34.86 |
| AL1 | 82°N, 62°W | 8.10 | 21.44 | 9.35 | 5.99 | 2356.67 | 4.85 | 41.01 |
| AL2 | 82°N, 62°W | 8.58 | 16.21 | 4.13 | 1.82 | 586.52 | 12.91 | 20.72 |
| AL3 | 82°N, 62°W | 8.49 | 15.88 | 3.44 | 1.88 | 672.94 | 8.57 | 28.04 |
| AVK | 61°N, 78°W | 6.63 | 76.66 | 80.52 | 1.87 | 3734.74 | 0.00 | 50.22 |
| BDE | 61°N, 73°W | 6.25 | 23.30 | 11.07 | <0.05 | 1840.87 | 15.95 | 30.09 |
| BY1 | 73°N, 78°W | 6.61 | 9.35 | 6.73 | <0.05 | 1722.31 | 22.11 | 26.68 |
| BY2 | 73°N, 78°W | 6.72 | 10.93 | 0.67 | 0.75 | 178.13 | 24.33 | 4.29 |
| EBA | 64°N, 82°W | 7.67 | 1.88 | 3.73 | 44.16 | 11587.60 | 17.39 | 44.06 |
| IQA | 63°N, 68°W | 6.29 | 76.27 | 76.42 | 0.35 | 5765.36 | 8.75 | 53.67 |
| NOR | 70°N, 19°E | 6.00 | 65.61 | 79.81 | 0.33 | 1684.98 | 17.91 | 43.48 |
| RAN | 62°N, 92°W | 6.93 | 45.51 | 22.47 | <0.05 | 7682.68 | 33.22 | 50.22 |
| RUS | 69°N, 70°E | 7.18 | 22.40 | 4.20 | 1.45 | 1552.06 | 23.97 | 48.25 |
| THU | 76°N, 68°W | 7.83 | 24.28 | 8.14 | 0.90 | 984.30 | 0.77 | 8.73 |
Abbreviations: DSL, diesel-treated soils; DSL–MAP, diesel+monoammonium phosphate-treated soils.
Microcosm setup and incubation
For each sample, 250 g of soil was contaminated to a final concentration of 5500 mg kg−1 of ultra-low sulfur diesel fuel. We selected diesel fuel as it is a common contaminant of polar soils, as is jet fuel (Aislabie et al., 2006; Bell et al., 2011; Yergeau et al., 2012c), which is similar in composition to diesel. Soils were incubated for 1 week at 1 °C to allow abiotic loss of the volatile components of diesel, as most short-term evaporative weathering can occur within 1 week of contamination (Neff et al., 2000). Following weathering, a 50-g sample was removed to determine baseline hydrocarbon concentrations. Each soil was then split into 2 × 100 g portions, and one portion was treated with 250 mg kg−1 monoammonium phosphate (MAP), a nutrient amendment that has previously been shown to effectively stimulate bioremediation in hydrocarbon-contaminated Arctic soils (Greer, 2009). This led to three separate treatments per soil: (1) initial soil, (2) DSL (diesel added) and (3) DSL–MAP (both diesel and MAP added). Both DSL and DSL–MAP treatments were split into five replicates of 20 g, and were incubated at 10 °C in loose-top 50-ml Falcon tubes for 4 weeks to simulate the high point of the Arctic summer. At the end of the incubation, soil was used for both hydrocarbon analysis and total DNA extraction.
Soil DNA extraction, 16S rRNA amplification and Ion Torrent sequencing
Total soil DNA was extracted from three replicates of each soil under each treatment condition using the protocol described in Yergeau et al. (2007). A detailed protocol is available in the SupplementaryMaterial. Partial 16S ribosomal RNA (rRNA) gene amplicons were produced using the universal primers E786 (5′-GATTAGATACCCTGGTAG-3′) and U926 (5′-CCGTCAATTCCTTTRAGTTT-3′), which target the V5 variable region (Baker et al., 2003). These primers were initially compared with other 16S rRNA sets that produced amplicons below the Ion Torrent size threshold (at the time, 220 bp including adapters). The primers were compared for inclusivity in the Ribosomal Database Project database, and matched between 95.5 and 98.3% of all bacteria with one mismatch or less, with no clear bias against any specific groups. We conducted a sequencing run with the candidate primers, and selected the set that yielded the highest bacterial diversity.
Primers for each sample contained unique multiplex identifier codes and adapter sequences for Ion Torrent sequencing. Full primer sequences can be found in Supplementary Table 1. Reactions were carried out using 12 μl of KAPA2G Robust DNA polymerase mix (KapaBiosystems Inc., Boston, MA, USA), 12 μl of nuclease-free deionized water, and 40 pmol of each of the appropriate forward and reverse primers. PCR cycling conditions involved an initial 5 min denaturing step at 95 °C, followed by 30 cycles of 30 s at 95 °C, 30 s at 55 °C and 30 s at 72 °C, and a final elongation step of 7 min at 72 °C. Amplicons were gel purified using the illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare, Piscataway, NJ, USA), and then quantified using the Quant-iT PicoGreen dsDNA assay kit (Invitrogen, Burlington, Ontario, Canada), pooled in equimolar ratios, and diluted to a concentration of 5 × 107 molecules for sequencing. Sequencing was conducted on an Ion Torrent Personal Genome Machine using the Ion Xpress Template Kit (Life Technologies, Carlsbad, CA, USA) and the Ion 314 chip (Life Technologies) following the manufacturer's protocols. Ion Torrent sequencing has been shown to produce microbial community profiles that are highly comparable to those produced by 454 sequencing (Yergeau et al., 2012b).
Sequence classification and operational taxonomic unitanalysis
Taxonomic identities were assigned to by classifying sequences in parallel using the MultiClassifier tool available on the Ribosomal Database Project website (Release 9; Wang et al., 2007). A bootstrap of 50% was used as is recommended for sequences of <250 bp (Claesson et al., 2009). At each taxonomic level, the proportion of sequence identities was calculated as a percent of all sequences classified to that level. Operational taxonomic unit analysis was performed in Mothur (Schloss et al., 2009), and was used to determine Shannon diversity values and UniFrac distance between samples. To standardize between samples, the number of sequences representing each sample was reduced to the lowest number among all sample replicates (156 sequences) using random numbers as in Fortunato et al. (2012). Analysis was performed following mainly the workflows outlined on the Mothur website with the Sogin et al. (2006) and Costello et al., (2009) data sets. Detailed sequence processing is described in the Supplementary Material. The sequence data generated in this study were deposited in the NCBI Sequence Read Archive and are available under the project number SRA058896. All statistical analyses are described in the Supplementary Material.
Results
Soil characteristics
Soil parameters are presented in Table 1. Differences in degradation between DSL and DSL–MAP soils are also depicted in Supplementary Figure 2. Organic matter content covaried with both water content (R=0.921, P<0.001) and pH (R=−0.640, P=0.004), and pH was also correlated with water content (R=−0.549, P=0.018). Total nitrogen was correlated with nitrates/nitrites (R=0.721, P<0.001). Diesel degradation in DSL–MAP soils correlated with water content (R=0.565, P=0.015), log total nitrogen (R=0.670, P=0.002), and log organic matter content (R=0.657, P=0.003). Degradation in DSL soils was not linearly related to any of the environmental variables measured, but peaked at∼pH 7.
Diversity and community composition across soils
After sequence filtering, we obtained a total of 154 418 usable reads, with an average of 903 reads per sample replicate. Shannon Diversity declined with increasing disturbance (P<0.001) using a 3% dissimilarity cutoff to define operational taxonomic units (Figure 1), and all treatments were significantly different from each other according to paired Student's t-tests (P<0.05). Principle coordinate analysis ordination showed that microbial community composition spread farther across the first axis in the DSL–MAP soils than in the initial or DSL soils (Figure 2a,b). Average weighted UniFrac distances between soils within the same treatment were higher in the DSL–MAP soils than in the initial or DSL soils (Figure 2c; P<0.05). Bacterial community composition grouped almost exclusively based on whether the soil of origin contained more or less than 10% organic matter (Figure 2d). This clustering was also linked to differences in pH, although slightly less closely, as there was overlap in the pH range of these two groups. The pH of soils with <10% organic matter ranged from 6.61–8.58, while the pH of soils with >10% organic matter ranged from 5.95–7.20.
Figure 1.

Decline in Shannon diversity following soil disturbance with DSL and DSL–MAP, as measured by differences in 16S rRNA gene composition using a 3% dissimilarity cutoff. Error bars represent s.e. Different letters above columns indicate significant differences between means (P<0.05), as determined by paired Student's t-tests.
Figure 2.

Representation of differences between samples using distance matrices. Principle coordinate analysis (PCoA) ordination plots show relatedness of samples separated by treatment using (a) weighted UniFrac distances, and (b) Bray–Curtis distances of classified 16S rRNA gene sequences at the phylum level (including classes of Proteobacteria). The average weighted UniFrac distance between samples is displayed in (c) as a column graph. Error bars represent s.e. while different letters above columns indicate significant differences between means (P<0.05), as determined by paired Student's t-tests. Weighted UniFrac distances were also used to create an ordination plot of (d) samples separated by % organic matter.
The abundance of Actinobacteria and Proteobacteria correlated strongly and significantly with organic matter (Figure 3). Despite the strong covariance between organic matter and water content, organic matter correlated better than water content with the relative abundance of the two major phyla, Actinobacteria and Proteobacteria, with the exception of the Proteobacteria in the DSL soils (Supplementary Figure 3). This was further confirmed using forward selection on all environmental variables in canonical redundancy analysis (P=0.003, Radj=0.471 in reduced model). Before treatment, Actinobacteria dominated soils with low organic matter, but abundance dropped off quickly with increasing organic matter content (R=−0.790, P<0.001), giving way to the Proteobacteria (R=0.792, P<0.001) (Figure 4a). Following disturbance, Actinobacteria represented an even larger proportion of the microbial communities in low-organic matter soils (RDSL=−0.849, PDSL<0.001; RDSL–MAP=−0.807, PDSL–MAP<0.001), while Proteobacteria represented a greater proportion of the communities in high-organic matter soils (RDSL=0.763, PDSL<0.001; RDSL–MAP=0.759, PDSL–MAP<0.001). This was especially true in DSL–MAP soils, with Actinobacteria representing 11.10–83.71% and Proteobacteria representing 11.70–75.16% of classified sequences, as compared with 15.20–59.02% and 28.76–54.66%, respectively, in the initial soils.
Figure 3.
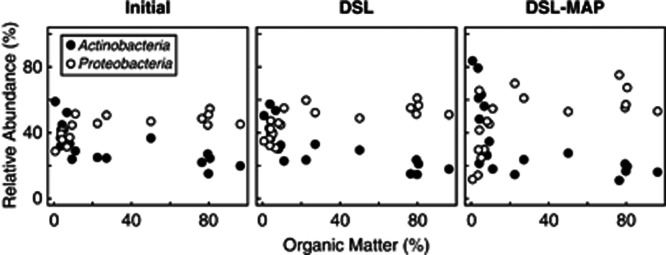
Correlations between Actinobacteria, Proteobacteria and organic matter content in initial, DSL, and DSL–MAP soils.
Figure 4.

Percent shifts in the DSL and DSL–MAP soils in the relative abundance of classified sequences of (a) the major phyla (and classes of Proteobacteria), and (b) families of Actinobacteria and Proteobacteria that were among the ten most abundant in at least one of the treatments. Significant differences in sequence abundance shifts between soils with low- and high-organic matter content are denoted by stars (P<0.05) or circles (P<0.10) as determined by the Student's t-test.
No major taxonomic group shifted in the same direction following disturbance in all samples (Table 2). When samples were grouped by percent organic matter, The Betaproteobacteria and Alphaproteobacteria were both significantly more successful in soils with over 10% organic matter in response to DSL and DSL–MAP treatments (P<0.05), although on average, the Alphaproteobacteria still declined in abundance in both soil types (Figure 4a). There were also significant differences in the relative abundance of Actinobacteria between low- and high-organic matter soils (P<0.05), as they increased dramatically in abundance in low-organic matter soils, and declined in high-organic matter soils in response to disturbance, while the Gammaproteobacteria increased marginally in both low- and high-organic matter soils. Most other phyla tended to decline following disturbance (Figure 4a). When the abundance of Ribosomal Database Project-classified families was compared, shifts in the Nocardioidaceae (Actinobacteria) varied significantly between low- and high-organic matter soils (P<0.05), as they increased the most of any group in low-organic matter soils in the presence of disturbance, but declined in high-organic matter soils, while the reverse was true for Burkholderiales inc. sed. (Betaproteobacteria) (P<0.05; Figure 4b). Hyphomicrobiaceae (Actinobacteria) declined in both low- and high-organic matter soils, but the decline was significantly greater in low-organic matter soils (P<0.05; Figure 4b).
Table 2. Number of samples in which major taxonomic groups increased or decreased between treatments.
| Taxonomic group | Δ Initial to DSL | Δ Initial to DSL–MAP | Δ DSL to DSL–MAP | |||
|---|---|---|---|---|---|---|
| + | − | + | − | + | − | |
| Alphaproteobacteria | 10 | 8 | 2 | 16 | 1 | 17 |
| Betaproteobacteria | 12 | 6 | 11 | 7 | 13 | 5 |
| Gammaproteobacteria | 12 | 6 | 11 | 7 | 7 | 11 |
| Deltaproteobacteria | 2 | 16 | 1 | 17 | 7 | 11 |
| Actinobacteria | 9 | 9 | 8 | 10 | 8 | 10 |
| Acidobacteria | 6 | 12 | 4 | 14 | 3 | 15 |
| Firmicutes | 7 | 11 | 2 | 16 | 4 | 14 |
| Bacteroidetes | 6 | 12 | 5 | 13 | 7 | 11 |
Abbreviations: DSL, diesel-treated soils; DSL–MAP, diesel+monoammonium phosphate-treated soils.
Predictability of diesel degradation
As the Actinobacteria and Betaproteobacteria responded most strongly to disturbance from diesel and nutrients, we decided to further investigate these groups for correlations with degradation across soils. For correlations with these two phyla and degradation, we used a Bonferroni-corrected P-value cutoff of 0.025 (α=0.05/2 pairwise comparisons). There was a negative correlation between the abundance of Actinobacteria in the initial soils and degradation in DSL–MAP soils (R=−0.564, P=0.0149), whereas the abundance of Betaproteobacteria in the initial soils was not predictive of degradation in either DSL or DSL–MAP soils. We also investigated correlations between degradation and each bacterial family that made up ⩾5% of sequences identified from either the Actinobacteria or Betaproteobacteria in at least one sample. As different families are likely promoted depending on environmental factors, we also examined assemblages of all families meeting the above criteria (⩾5% of sequences in at least one sample) from each phylum, grouping by those that correlated either positively or negatively with degradation. For these comparisons, we used a Bonferroni-corrected P-value cutoff of 0.0026 (α=0.05/19 pairwise comparisons). No single family in the initial soils was significantly predictive of degradation in either DSL or DSL–MAP soils, but when the six most commonly identified Betaproteobacteria families were normalized to the total abundance of Betaproteobacteria in each sample, the relative abundance of Burkholderiaceae+Oxalobacteraceae+Nitrosomonadaceae in the initial soils was significantly and positively correlated with degradation in DSL soils (R=0.775, P<0.001), while the relative abundance of Burkholderiales inc. sed.+Comamonadaceae+Rhodocyclaceae in the initial soils was significantly and negatively correlated with degradation in DSL soils (R=−0.771, P<0.001). The resulting abundance of these assemblages following disturbance in DSL soils was not significantly correlated with degradation (Figure 5a), nor were these assemblages predictive of degradation in DSL–MAP soils (data not shown). Degradation in DSL–MAP soils could be predicted by bacterial diversity in the initial soils (R=0.660, P<0.003), but diversity in DSL–MAP soils did not correlate with degradation (Figure 5b). Although the relative abundance of Betaproteobacteria in the initial soils was not predictive of degradation in DSL–MAP soils (Figure 5b), the abundance of Betaproteobacteria in DSL–MAP soils was significantly correlated with DSL–MAP degradation (R=0.597, P=0.009). The relative abundance of Rhodocyclaceae (Betaproteobacteria) in DSL–MAP soils was negatively correlated with DSL–MAP degradation (R=−0.740, P<0.001).
Figure 5.

Correlations between (a) degradation in DSL soils and the relative abundance of major families of Betaproteobacteria (Burk, Burkholderiaceae; Oxal, Oxalobacteraceae; Nitr, Nitrosomonadaceae; Burk i.s., Burkholderiales incertae sedis; Com, Comamonadaceae; Rho, Rhodocyclaceae) as a proportion of all Betaproteobacteria, (b) degradation in DSL–MAP soils and Shannon Diversity (3% dissimilarity), and degradation in DSL–MAP soils and the relative abundance of Betaproteobacteria.
Discussion
Environmental controls on disturbed soil communities
From both an applied and ecological standpoint, it was interesting to observe that disturbance and environment combined to reliably shape bacterial communities across soils collected from geographically disparate Arctic sites. Other recent studies have also shown that disturbance from shifts in climate (Yergeau et al., 2012a), differences in local environmental factors (Pommier et al., 2012), and distance from the litter layer (Schulz et al., 2012) have a greater effect than geography in determining microbial community composition. Metagenomic studies have shown that discrete taxonomic units exist within bacteria (Caro-Quintero and Konstantinidis, 2012), and our data demonstrate that taxonomic affiliation does affect which organisms will dominate in particular contaminated soils. Hydrocarbons are found almost ubiquitously across natural environments, so the disturbance of soil communities by petroleum contaminants may in fact be an exaggerated analogy to natural fluxes in the Arctic environment, such as biomass decay, plant inputs and the disruption of organic matter by freeze-thaw. Similarly, the addition of nutrients may resemble fluxes such as nutrient pulses at snowmelt and the periodic input of animal waste products. In many biological systems, community composition shifts temporally in response to changing environmental conditions, helping to support a greater range of taxa in a single environment through time (Fuhrman et al., 2006; Magurran and Henderson, 2010). Thus, the bacteria that responded positively to diesel and nutrient inputs may have a history of increasing in abundance with such fluxes. Bacteria that live in variable-resource environments have been shown to quickly process pulsed carbon sources (Lennon and Cottingham, 2008), so resource history could be important in predicting bioremediation capacity.
Although some bacteria may be naturally adapted to high carbon and nutrient sources, diesel contamination and subsequent nutrient amendments were shown to be significant disturbances to bacterial communities in Arctic soils. Shannon diversity of operational taxonomic units declined in both DSL and DSL–MAP soils, with the greatest declines observed in DSL–MAP soils. The addition of both diesel and MAP appeared to have a disruptive effect on soil microbial communities, as the average weighted UniFrac distance between samples increased relative to both initial and DSL soils, and community composition was polarized depending on the soil environment. Only Actinobacteria and the major classes of Proteobacteria represented large proportions of the microbial community in DSL and DSL–MAP soils, with subsequent declines in other major phyla relative to the uncontaminated soils, such as the Acidobacteria, Bacteroidetes and Firmicutes. Although pH has previously been shown to shape phylum-level microbial community structure in uncontaminated Arctic soils (Fierer and Jackson, 2006; Chu et al., 2010), the effect was less prominent in our study. This may be due to the fact that most pH effects on community composition have been observed at pH values of <6 (Fierer and Jackson, 2006; Lauber et al., 2009; Chu et al., 2010), while the lowest pH in our soils was 5.95.
Bioaugmentation, the inoculation of contaminated soils with additional hydrocarbon-degrading microorganisms, has done little to increase rates of bioremediation (for example, Thomassin-Lacroix et al., 2002; van Herwijnen et al., 2006). This lack of success has been blamed on a failure to consider other environmental controls on hydrocarbon degraders (Thompson et al., 2005), and is not surprising in light of our results. Although hydrocarbon contamination can reduce the number of bacterial taxa in Arctic soils, there are still many that remain active (Bell et al., 2011), and competitive interactions between these remaining groups are likely to be governed by other environmental parameters. Our experimental setup controlled for contaminant composition, age and concentration, and found that differences in phylum-level community composition was best explained by soil organic matter content, especially in DSL–MAP soils. Total carbon was previously identified as one of several factors that linked with community composition in hydrocarbon-contaminated Antarctic soils, but this effect was confounded by variability in soil contaminants (Powell et al., 2010). In our study, organic matter primarily affected the relative abundance of Actinobacteria and Proteobacteria, with Actinobacteria dominating in soils with <10% organic matter and Proteobacteria dominating higher-organic matter soils. Actinobacteria have previously been linked with low carbon concentrations by phospholipid fatty acid analysis in temperate soils, and are reduced when higher concentrations of carbon are present or added (Griffiths et al., 1999; Fierer et al., 2003). Similarly, Actinobacteria dominated at snowmelt in an alpine tundra soil, while Gram-negative bacteria, likely including Proteobacteria, dominated during the growing season when plant carbon inputs were higher (Björk et al., 2008).
Soil resource history
Ramirez et al. (2012) recently showed that Actinobacteria reliably increase in abundance across nitrogen gradients and environments in response to nitrogen inputs, and suggest that the Actinobacteria may represent a copiotrophic group. However, our observation of dominant Actinobacteria populations in low-organic matter soils suggests that this may not be the case. It has been suggested that fast-growing copiotrophic microorganisms that primarily use labile carbon sources are likely to most rapidly and positively respond to nutrient inputs (Ramirez et al., 2012), and that intrinsic growth rate is the main factor leading to success following disturbance (Haddad et al., 2008). However, we show that positive response to nutrient addition in the presence of hydrocarbon contaminants is not limited to specific groups, but that different bacterial taxa are favoured depending on environmental factors, in this case, organic matter content. Copiotrophs are expected to outcompete oligotrophs in the presence of abundant resources (Fierer et al., 2007), so the fact that Proteobacteria and Actinobacteria respond positively to carbon and nutrient inputs in different soil environments suggests that they have either separately adopted the copiotrophic life strategy, or that each has capitalized on reduced competition following soil disturbance. Fierer et al. (2007) suggest that copiotrophism may be a fixed characteristic of bacterial phyla, in which case the latter hypothesis appears more probable.
Degradation in DSL–MAP soils correlated linearly and positively with organic matter, water content and total nitrogen, while degradation in DSL soils was not linearly related to any of the environmental variables that we examined. Despite the fact that low water content has previously been correlated with ineffective nutrient stimulation of degradation (Walworth et al., 1997), we observed successful stimulation in soils with water contents as low as 1.88%. It was interesting to note that higher total nitrogen content in soils indicated higher degradation in the presence of added nutrients, which suggests that microorganisms that are adapted to using nitrogen naturally may more effectively use nitrogen additives. Historical resource regimes in soils have been shown to affect future microbial function, suggesting that evolved community adaptations are important even in the face of a changing soil environment (Keiser et al., 2011). Similarly, microbial communities that have evolved in high-organic matter environments may be better adapted to quickly process added diesel.
When microbial communities were inoculated into new litter environments, the resource regimes under which they had historically evolved affected decomposition rates, but these communities became more effective at decomposing particular substrates following successive 100-day exposures (Keiser et al., 2011). As hydrocarbon contaminants can persist in Arctic soils (Atlas, 1986), exposure to the contaminant may be so prolonged that the resource history of the microbial communities before contamination may no longer be relevant. Metagenomic analysis of a site that had experienced chronic heavy-metal contamination over several decades showed that the microbial community had simplified significantly, both in terms of functional potential and taxonomic diversity (Hemme et al., 2010). Further studies of extended hydrocarbon contamination will help in determining how important it is to consider microbial resource history when designing bioremediation treatments.
Predictability of hydrocarbon degradation in Arctic soils
We found bacterial diversity in the initial soils to be predictive of degradation in DSL–MAP soils, but not in DSL soils. Traditionally, the debate of how diversity influences productivity centers around the theories of sampling effect and niche complementarity; essentially whether the selection of a single efficient organism or a group of complementary organisms are responsible for a given function (Bell et al., 2005; Fargione et al., 2007). Interestingly, diversity in DSL–MAP soils was not related to degradation, suggesting that diversity may be important in the initial selection of organisms, but less important for actual hydrocarbon degradation. Other recent studies have also shown that the diversity-function relationship depends on the function being considered, and that in some cases, the presence of specific taxa is more important than actual diversity (Salles et al., 2009; Peter et al., 2011). Most of the major taxonomic groups were present in all soils, so it may be that evenness increased the number of groups that could potentially respond to nutrient addition, as the presence of dominant bacterial taxa can sometimes limit the establishment of other bacteria (Heczko et al., 2000).
We were also able to link specific taxonomic assemblages with diesel degradation. Although a number of taxa correlated significantly with degradation in both DSL and DSL–MAP soils, groupings of Betaproteobacteria were the best predictor of degradation across all soils. When the proportion of Betaproteobacteria consisting of Burkholderiaceae, Oxalobacteraceae and Nitrosomonadaceae in the initial undisturbed Arctic soils was high, hydrocarbon degradation could be expected to proceed well without treatment, while the opposite was true when mostly Burkholderiales inc. sed., Comamonadaceae and Rhodocyclaceae were present. Members of most of these families are known to be capable of degrading hydrocarbons (Pepi et al., 2003; Castorena et al., 2006; Owsianiak et al., 2009; Mbadinga et al., 2011), so it was interesting that the relative abundance of these assemblages in contaminated soils did not correlate with degradation. Instead, they likely identify an initial soil parameter that is important for hydrocarbon degradation, such as moderate pH.
In contrast to this was the relationship between Betaproteobacteria and hydrocarbon degradation in DSL–MAP soils. The abundance of specific Betaproteobacteria families in initial soils was predictive of degradation in DSL soils, but not degradation in DSL–MAP soils. However, the relative abundance of Betaproteobacteria after incubation in DSL–MAP soils was strongly and positively related to degradation, suggesting a potentially important active role for this group. In a study that aimed to determine optimal bioremediation cultures for diesel degradation using microautoradiography, Betaproteobacteria represented over half of radioactively labeled cells (Hesselsoe et al., 2008), and Betaproteobacteria have previously been shown to be positively correlated with carbon mineralization across a range of soils (Fierer et al., 2007), suggesting that this correlation could represent a functional relationship. Despite the fact that key genes involved in hydrocarbon degradation are located on mobile elements and can potentially be exchanged between taxa (Whyte et al., 1997, Ma et al., 2006), it appears that certain taxa may be more efficient at petroleum degradation than others. The apparent discrepancy between the active role for Betaproteobacteria in nutrient-amended but not unamended contaminated soils suggests that its effectiveness as a degrader may be dependent on sufficiently available nutrients. If Betaproteobacteria can be confirmed as the most efficient petroleum degraders in nutrient-amended soils, future approaches to bioremediation should attempt to favor this group, or limit the growth of competing taxa that may hinder its success.
Conclusions
As petroleum contamination is increasingly likely throughout the Arctic, it is important to understand the factors that will shape petroleum hydrocarbon-degrading microbial communities, as well as rates of contaminant degradation in soils with different physicochemical characteristics. Despite the apparent geographic isolation between the soils used in our study, we observed consistent microbial community shifts when both diesel and nutrients were added, towards Actinobacteria dominance in low-organic matter soils and Proteobacteria dominance in high-organic matter soils. Bioremediation potential was also predictable across soils, based on the initial 16S rRNA gene diversity or Betaproteobacteria composition of uncontaminated soils. In addition, total Betaproteobacteria abundance was correlated with effective degradation in the presence of nutrient amendments, suggesting that this may be an important taxonomic group to stimulate. Site-specific assessments of bioremediation potential are expensive and time-consuming (Dorn and Salanitro, 2000), so predictability will be invaluable in responding to future contamination events. As the concentration, age and composition of petroleum contaminants at affected sites is highly variable, future work should aim to identify how consistently these patterns apply, especially in soils with aged contaminants in which the original resource history of soils may no longer be relevant or available for study.
Acknowledgments
We thank Matt Wallenstein, Claudia Boot, Nicolas Lecomte, Emilie Champagne, Jen Allan, Isabelle Laurion, Chantal Lemieux, Karita Neghandi, Philippe Galipeau, Marie-Jeanne Rioux, Katryne Larrivée, JF Lamarre, Elise Bolduc, and the Center d'Études Nordiques for soil collection, Julie Champagne for sequencing assistance, and Chantale Beaulieu and Stéphane Deschamps for assistance with GC analysis. This work was supported by the NRCan PERD Program, and an NSERC postgraduate scholarship to TH Bell.
Footnotes
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Aislabie J, Saul DJ, Foght JM. Bioremediation of hydrocarbon-contaminated polar soils. Extremophiles. 2006;10:171–179. doi: 10.1007/s00792-005-0498-4. [DOI] [PubMed] [Google Scholar]
- Allison SD, Hanson CA, Treseder KK. Nitrogen fertilization reduces diversity and alters community structure of active fungi in boreal ecosystems. Soil Biol Biochem. 2007;39:1878–1887. [Google Scholar]
- Atlas RM. Fate of petroleum pollutants in Arctic ecosystems. Water Sci Technol. 1986;18:59–67. [Google Scholar]
- Baker GC, Smith JJ, Cowan DA. Review and re-analysis of domain-specific 16S primers. J Microbiol Meth. 2003;55:541–555. doi: 10.1016/j.mimet.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Bell T, Newman JA, Silverman BW, Turner SL, Lilley AK. The contribution of species richness and composition to bacterial services. Nature. 2005;436:1157–1160. doi: 10.1038/nature03891. [DOI] [PubMed] [Google Scholar]
- Bell TH, Yergeau E, Martineau C, Juck D, Whyte LG, Greer CW. Identification of nitrogen-incorporating bacteria in petroleum-contaminated Arctic soils by using [15N]DNA-based stable isotope probing and pyrosequencing. Appl Environ Microbiol. 2011;77:4163–4171. doi: 10.1128/AEM.00172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk RG, Björkman MP, Andersson MX, Klemedtsson L. Temporal variation in soil microbial communities in Alpine tundra. Soil Biol Biochem. 2008;40:266–268. [Google Scholar]
- Caro-Quintero A, Konstantinidis KT. Bacterial species may exist, metagenomics reveal. Environ Microbiol. 2012;14:347–355. doi: 10.1111/j.1462-2920.2011.02668.x. [DOI] [PubMed] [Google Scholar]
- Castorena G, Mugica V, Le Borgne S, Acuña ME, Bustos-Jaimes I, Aburto J. Carbazole biodegradation in gas oil/water biphasic media by a new isolated bacterium Burkholderia sp strain IMP5GC. J Appl Microbiol. 2006;100:739–745. doi: 10.1111/j.1365-2672.2005.02799.x. [DOI] [PubMed] [Google Scholar]
- Chang WJ, Klemm S, Beaulieu C, Hawari J, Whyte L, Ghoshal S. Petroleum hydrocarbon biodegradation under seasonal freeze-thaw soil temperature regimes in contaminated soils from a sub-Arctic site. Environ Sci Technol. 2011;45:1061–1066. doi: 10.1021/es1022653. [DOI] [PubMed] [Google Scholar]
- Chu HY, Fierer N, Lauber CL, Caporaso JG, Knight R, Grogan P. Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ Microbiol. 2010;12:2998–3006. doi: 10.1111/j.1462-2920.2010.02277.x. [DOI] [PubMed] [Google Scholar]
- Claesson MJ, O'Sullivan O, Wang Q, Nikkila J, Marchesi JR, Smidt H, et al. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One. 2009;4:e6669. doi: 10.1371/journal.pone.0006669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn PB, Salanitro JP. Temporal ecological assessment of oil contaminated soils before and after bioremediation. Chemosphere. 2000;40:419–426. doi: 10.1016/s0045-6535(99)00304-5. [DOI] [PubMed] [Google Scholar]
- Fargione J, Tilman D, Dybzinski R, Lambers JH, Clark C, Harpole WS, et al. From selection to complementarity: shifts in the causes of biodiversity-productivity relationships in a long-term biodiversity experiment. Proc Roy Soc B-Biol Sci. 2007;274:871–876. doi: 10.1098/rspb.2006.0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, Schimel JP, Holden PA. Variations in microbial community composition through two soil depth profiles. Soil Biol Biochem. 2003;35:167–176. [Google Scholar]
- Fierer N, Jackson RB. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA. 2006;103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, Bradford MA, Jackson RB. Toward an ecological classification of soil bacteria. Ecology. 2007;88:1354–1364. doi: 10.1890/05-1839. [DOI] [PubMed] [Google Scholar]
- Fortunato CS, Herfort L, Zuber P, Baptista AM, Crump BC. Spatial variability overwhelms seasonal patterns in bacterioplankton communities across a river to ocean gradient. ISME J. 2012;6:554–563. doi: 10.1038/ismej.2011.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrman JA, Hewson I, Schwalbach MS, Steele JA, Brown MV, Naeem S. Annually reoccurring bacterial communities are predictable from ocean conditions. Proc Natl Acad Sci USA. 2006;103:13104–13109. doi: 10.1073/pnas.0602399103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer CW.2009Bioremediation of contaminated sites in the Canadian Arctic: monitoring performance and the effects of biostimulation using molecular methodsIn: Bej AK, Aislabie J, Atlas RM, (eds)Polar Microbiology: The Ecology, Diversity, And Bioremediation Potential of Microorganisms in Extremely Cold Environments CRC Press, Inc.: Boca Raton, FL; 319–338. [Google Scholar]
- Greer CW, Whyte LG, Niederberger TD.2010Microbial communities in hydrocarbon-contaminated temperate, tropical, alpine, and polar soilsIn: Timmis KN, (eds)Handbook of Hydrocarbon and Lipid Microbiology Springer: Berlin Heidelberg; 2313–2328. [Google Scholar]
- Griffiths BS, Ritz K, Ebblewhite N, Dobson G. Soil microbial community structure: effects of substrate loading rates. Soil Biol Biochem. 1999;31:145–153. [Google Scholar]
- Haddad NM, Holyoak M, Mata TM, Davies KF, Melbourne BA, Preston K. Species' traits predict the effects of disturbance and productivity on diversity. Ecol Lett. 2008;11:348–356. doi: 10.1111/j.1461-0248.2007.01149.x. [DOI] [PubMed] [Google Scholar]
- Heczko U, Abe A, Finlay BB. Segmented filamentous bacteria prevent colonization of enteropathogenic Escherichia coli O103 in rabbits. J Infect Dis. 2000;181:1027–1033. doi: 10.1086/315348. [DOI] [PubMed] [Google Scholar]
- Hemme CL, Deng Y, Gentry TJ, Fields MW, Wu LY, Barua S, et al. Metagenomic insights into evolution of a heavy metal-contaminated groundwater microbial community. ISME J. 2010;4:660–672. doi: 10.1038/ismej.2009.154. [DOI] [PubMed] [Google Scholar]
- Hesselsoe M, Bjerring ML, Henriksen K, Loll P, Nielsen JL. Method for measuring substrate preferences by individual members of microbial consortia proposed for bioaugmentation. Biodegradation. 2008;19:621–633. doi: 10.1007/s10532-007-9167-x. [DOI] [PubMed] [Google Scholar]
- Horner-Devine MC, Carney KM, BJM Bohannan. An ecological perspective on bacterial biodiversity. Proc Roy Soc Lond B Bio. 2004;271:113–122. doi: 10.1098/rspb.2003.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YS, Park W. Protection against diesel oil toxicity by sodium chloride-induced exopolysaccharides in Acinetobacter sp strain DR1. J Biosci Bioeng. 2010;109:118–123. doi: 10.1016/j.jbiosc.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Keiser AD, Strickland MS, Fierer N, Bradford MA. The effect of resource history on the functioning of soil microbial communities is maintained across time. Biogeosciences. 2011;8:1477–1486. [Google Scholar]
- Lauber CL, Hamady M, Knight R, Fierer N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol. 2009;75:5111–5120. doi: 10.1128/AEM.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon JT, Cottingham KL. Microbial productivity in variable resource environments. Ecology. 2008;89:1001–1014. doi: 10.1890/07-1380.1. [DOI] [PubMed] [Google Scholar]
- Ma YF, Wang L, Shao ZZ. Pseudomonas, the dominant polycyclic aromatic hydrocarbon-degrading bacteria isolated from Antarctic soils and the role of large plasmids in horizontal gene transfer. Environ Microbiol. 2006;8:455–465. doi: 10.1111/j.1462-2920.2005.00911.x. [DOI] [PubMed] [Google Scholar]
- Magurran AE, Henderson PA. Temporal turnover and the maintenance of diversity in ecological assemblages. Philos T R Soc B. 2010;365:3611–3620. doi: 10.1098/rstb.2010.0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbadinga SM, Wang LY, Zhou L, Liu JF, Gu JD, Mu BZ. Microbial communities involved in anaerobic degradation of alkanes. Int Biodeter Biodegr. 2011;65:1–13. [Google Scholar]
- McCarthy K, Walker L, Vigoren L, Bartel J. Remediation of spilled petroleum hydrocarbons by in situ landfarming at an arctic site. Cold Reg Sci Technol. 2004;40:31–39. [Google Scholar]
- Neff JM, Ostazeski S, Gardiner W, Stejskal I. Effects of weathering on the toxicity of three offshore Australian crude oils and a diesel fuel to marine animals. Environ Toxicol Chem. 2000;19:1809–1821. [Google Scholar]
- Owsianiak M, Szulc A, Chrzanowski L, Cyplik P, Bogacki M, Olejnik-Schmidt AK, et al. Biodegradation and surfactant-mediated biodegradation of diesel fuel by 218 microbial consortia are not correlated to cell surface hydrophobicity. Appl Microbiol Biot. 2009;84:545–553. doi: 10.1007/s00253-009-2040-6. [DOI] [PubMed] [Google Scholar]
- Pepi M, Minacci A, Di Cello F, Baldi F, Fani R. Long-term analysis of diesel fuel consumption in a co-culture of Acinetobacter venetianus, Pseudomonas putida and Alcaligenes faecalis. Anton Leeuw Int J G. 2003;83:3–9. doi: 10.1023/a:1022930421705. [DOI] [PubMed] [Google Scholar]
- Peter H, Beier S, Bertilsson S, Lindström ES, Langenheder S, Tranvik LJ. Function-specific response to depletion of microbial diversity. ISME J. 2011;5:351–361. doi: 10.1038/ismej.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier T, Douzery EJP, Mouillot D. Environment drives high phylogenetic turnover among oceanic bacterial communities. Biol Lett. 2012;8:562–566. doi: 10.1098/rsbl.2011.0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SM, Bowman JP, Ferguson SH, Snape I. The importance of soil characteristics to the structure of alkane-degrading bacterial communities on sub-Antarctic Macquarie Island. Soil Biol Biochem. 2010;42:2012–2021. [Google Scholar]
- Ramirez KS, Craine JM, Fierer N. Consistent effects of nitrogen amendments on soil microbial communities and processes across biomes. Glob Change Biol. 2012;18:1918–1927. [Google Scholar]
- Ramos JL, Duque E, Gallegos MT, Godoy P, Ramos-González MI, Rojas A, et al. Mechanisms of solvent tolerance in Gram-negative bacteria. Annu Rev Microbiol. 2002;56:743–768. doi: 10.1146/annurev.micro.56.012302.161038. [DOI] [PubMed] [Google Scholar]
- Salles JF, Poly F, Schmid B, Le Roux X. Community niche predicts the functioning of denitrifying bacterial assemblages. Ecology. 2009;90:3324–3332. doi: 10.1890/09-0188.1. [DOI] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Giebler J, Chatzinotas A, Wick LY, Fetzer I, Welzl G, et al. Plant litter and soil type drive abundance, activity and community structure of alkB harbouring microbes in different soil compartments. ISME J. 2012;6:1763–1774. doi: 10.1038/ismej.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, et al. Microbial diversity in the deep sea and the underexplored ‘rare biosphere'. Proc Natl Acad Sci USA. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomassin-Lacroix EJM, Eriksson M, Reimer KJ, Mohn WW. Biostimulation and bioaugmentation for on-site treatment of weathered diesel fuel in Arctic soil. Appl Microbiol Biot. 2002;59:551–556. doi: 10.1007/s00253-002-1038-0. [DOI] [PubMed] [Google Scholar]
- Thompson IP, van der Gast CJ, Ciric L, Singer AC. Bioaugmentation for bioremediation: the challenge of strain selection. Environ Microbiol. 2005;7:909–915. doi: 10.1111/j.1462-2920.2005.00804.x. [DOI] [PubMed] [Google Scholar]
- van Herwijnen R, Joffe B, Ryngaert A, Hausner M, Springael D, Govers HAJ, et al. Effect of bioaugmentation and supplementary carbon sources on degradation of polycyclic aromatic hydrocarbons by a soil-derived culture. FEMS Microbiol Ecol. 2006;55:122–135. doi: 10.1111/j.1574-6941.2005.00001.x. [DOI] [PubMed] [Google Scholar]
- Violle C, Pu ZC, Jiang L. Experimental demonstration of the importance of competition under disturbance. Proc Natl Acad Sci USA. 2010;107:12925–12929. doi: 10.1073/pnas.1000699107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth J, Braddock J, Woolard C. Nutrient and temperature interactions in bioremediation of cryic soils. Cold Reg Sci Technol. 2001;32:85–91. [Google Scholar]
- Walworth JL, Woolard CR, Braddock JF, Reynolds CM. Enhancement and inhibition of soil petroleum biodegradation through the use of fertilizer nitrogen: an approach to determining optimum levels. J Soil Contam. 1997;6:465–480. [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte LG, Bourbonnière L, Greer CW. Biodegradation of petroleum hydrocarbons by psychrotrophic Pseudomonas strains possessing both alkane (alk) and naphthalene (nah) catabolic pathways. Appl Environ Microbiol. 1997;63:3719–3723. doi: 10.1128/aem.63.9.3719-3723.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yergeau E, Bokhorst S, Huiskes AHL, Boschker HTS, Aerts R, Kowalchuk GA. Size and structure of bacterial, fungal and nematode communities along an Antarctic environmental gradient. FEMS Microbiol Ecol. 2007;59:436–451. doi: 10.1111/j.1574-6941.2006.00200.x. [DOI] [PubMed] [Google Scholar]
- Yergeau E, Arbour M, Brousseau R, Juck D, Lawrence JR, Masson L, et al. Microarray and real-time PCR analyses of the responses of high-Arctic soil bacteria to hydrocarbon pollution and bioremediation treatments. Appl Environ Microbiol. 2009;75:6258–6267. doi: 10.1128/AEM.01029-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yergeau E, Bokhorst S, Kang S, Zhou JZ, Greer CW, Aerts R, et al. Shifts in soil microorganisms in response to warming are consistent across a range of Antarctic environments. ISME J. 2012a;6:692–702. doi: 10.1038/ismej.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yergeau E, Lawrence JR, Sanschagrin S, Waiser MJ, Korber DR, Greer CW. Next-generation sequencing of microbial communities in the Athabasca river and its tributaries in relation to oil sands mining activities. Appl Environ Microbiol. 2012b;78:7626–7637. doi: 10.1128/AEM.02036-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yergeau E, Sanschagrin S, Beaumier D, Greer CW. Metagenomic analysis of the bioremediation of diesel-contaminated Canadian high Arctic soils. PLoS One. 2012c;7:e30058. doi: 10.1371/journal.pone.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.