

Abstract
The endoplasmic reticulum (ER) provides a specialized environment for the folding and modification of trans-membrane proteins, including receptor tyrosine kinases (RTKs), which are vital for the growth and survival of malignancies. To identify compounds which disrupt the function of the ER and thus could potentially impair cancer cell survival signaling, we adapted a set of glycosylation-sensitive luciferase reporters for the development and optimization of a cell-based high-throughput screen (HTS). Secondary screens for false-positive luciferase activation and tertiary lectin-based and biochemical analyses were also devised for compound triage. Through a pilot screen of 2802 compounds from the National Cancer Institute (NCI) chemical libraries, we identified aclacinomycin (Acm) as a compound that preferentially affects ER function. We report that Acm reduces plasma membrane expression of glycoproteins including epidermal growth factor receptor (EGFR) and Met but does not inhibit N-linked glycosylation or generalized protein translation. Fluorescence microscopy co-localization experiments were also performed and demonstrated Acm accumulation in the ER in further support of the overall HTS design. The consequences of Acm treatment on cell survival were analyzed through clonogenic survival analysis. Consistent with the reduction of EGFR levels, pretreatment with Acm sensitizes the EGFR-mutant non-small cell lung cancer (NSCLC) cell lines HCC827 and HCC2935 to ionizing radiation and did not affect the sensitivity of the RTK-independent and KRAS-mutant A549 NSCLC cell line. Thus, Acm and similar compounds targeting the ER may represent a novel approach for radiosensitizing tumor cells dependent on RTK function.
Introduction
The endoplasmic reticulum (ER) is a multifunctional eukaryotic organelle. The ER's main functions are to guide the proper folding and sorting of transmembrane and secretory proteins through a set of specialized ER-resident chaperones and biosynthetic pathways. To accomplish this, the ER provides a highly oxidative environment to allow for formation of disulfide bonds, is the subcellular site of N-glycosylation to aid in tertiary structure adoption and protein sorting, and contains internal quality control mechanisms to insure proper protein folding [1]. Compounds that disrupt the function of the ER, such as the N-glycosylation inhibitor tunicamycin (Tn) or the Ca++ ATPase inhibitor thapsigargin, are known to interfere with protein function through interruption of these processes and induction of the unfolded protein response, an ER-dependent stress response intended to restore protein folding and normalize cell function [2,3].
One key family of proteins that is exceptionally dependent on the function of the ER is the receptor tyrosine kinase (RTK) family. RTKs are conserved, multifunctional transmembrane proteins that are proto-oncogenes and both cause transformation and promote growth of malignant tumors [4]. RTKs adapt their tertiary structures with the assistance of ER-resident chaperones, are N-glycosylated by the ER-resident oligosaccharyltransferase complex, and are stabilized by disulfide bonds catalyzed in the ER. RTKs that adapt the proper structure are allowed to continue on to the Golgi apparatus for further modification [5,6] and eventually to the plasma membrane where they can respond to their ligands and elicit the appropriate cellular responses.
Because of their importance to cell signaling and well-established role in malignancies, RTKs are attractive therapeutic targets. The epidermal growth factor receptor (EGFR), a member of the ErbB RTK family, is a salient example and is overexpressed in head and neck squamous cell carcinomas [7], amplified with an oncogenic extracellular domain deletion in malignant gliomas [8], and amplified with oncogenic kinase domain mutations in lung adenocarcinomas [9]. For head and neck squamous cell carcinomas and EGFR kinase domain mutant lung adenocarcinoma, EGFR-targeted therapies either with or without radiation therapy have been demonstrated to improve patient outcomes in randomized trials [10,11].
However, a limitation to therapeutic agents that inhibit individual RTKs is that tumors activate parallel signaling pathways, such as Met or ErbB3 following EGFR inhibition [12,13], or alternatively acquire resistance to these inhibitors through mutation (such as the resistant EGFR T790M), and thus circumvent inhibition of the targeted receptor [14]. Due to these compensatory mechanisms and cooperative signaling between RTK pathways [15,16], the inhibition of multiple targets may be necessary to fully disable receptor-mediated survival signaling. We have previously demonstrated that inhibition of N-linked glycosylation is one ER-directed approach to reduce signaling from multiple coexpressed RTKs and we have used this strategy to sensitize tumor cells to ionizing radiation [17,18]. To further explore this therapeutic concept, we sought to identify small molecules that interfere with ER homeostasis and consequently impair the survival signaling initiated by RTKs. We now report on the development of a bioluminescent cell-based high-throughput screen (HTS) designed to identify compounds that preferentially disrupt ER function. Our results demonstrate the proficiency of this screening approach and identify aclacinomycin (Acm), a compound with previously unrecognized effects on the ER, which can be used to radiosensitize EGFR-addicted cell models.
Materials and Methods
Reagents
Cell culture reagents and ER-Tracker Blue-White DPX dye were obtained from Gibco/Life Technologies (Carlsbad, CA). The anti-EGFR antibody was a gift from J. Schlessinger (Yale University, New Haven, CT). The c-Met antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies recognizing Grp78 and HSP70 were obtained from Cell Signaling Technologies (Danvers, MA). Acm was obtained from the National Cancer Institute (NCI) Developmental Therapeutics Program (NCI No. 208734). Tn, concanavalin A (Con A), and swainsonine (Sw) were purchased from Calbiochem/Merck (Rockland, MA). Protease inhibitor tablets were purchased from Roche (Basel, Switzerland), and phosphatase inhibitor cocktails were purchased from Sigma (St Louis, MO). All Western blot analysis supplies were obtained from Bio-Rad (Hercules, CA). The ONE-Glo Luciferase Assay (No. E6120) and MTS Assay kits (No. G4000) were purchased from Promega (Madison, WI).
Cell Lines
D54-ERLucT and D54-LucT cells were generated by Lipofectamine (Life Technologies) transfection of vectors containing either the control luciferase gene or a luciferase gene preceded by an ER translation sequence. Clones with luciferase expression were isolated by serial dilution and selection with 0.5 µg/ml G418 [18]. CHO and Lec-15 cells were a gift from Mark Lehrman (University of Texas Southwestern, Dallas, TX) and, unless otherwise indicated, grown and maintained in Ham's F-12 media supplemented with 10% FBS and penicillin and streptomycin. HCC2935, HCC827, and A549 cell lines were obtained from the American Type Culture Collection (ATCC, Rockville, MD). Unless otherwise indicated, human cells were kept in a 37°C incubator with 5% CO2 and grown as monolayers in RPMI 1640 + 10% FBS supplemented with penicillin and streptomycin.
High-throughput Bioluminescent Screens
Screen optimization. For the DMSO dose-response experiment, five serial 1:2 dilutions of DMSO in growth medium starting at a top concentration of 0.3% DMSO were added in eight replicates to D54-ERLucT cells in an opaque white 96-well plate by a multichannel pipette. For HTS cell titer optimization, a range of D54-ERLucT cells (5 x 102 to 5 x 103 per well) were plated in 20 µl of growth medium in opaque white 384-well plates and allowed to attach overnight. Each 384-well plate reserved 16 wells each for positive (1 µM Tn) and negative controls (0.1% DMSO). The following day, Tn and DMSO were added by a 20-nl pin tool and plates were returned to the incubator for 48 hours. For the Tn dose-response experiment, 15 serial 1:2 dilutions of Tn starting at a top concentration of 30 µM were added in eight replicates to growing D54-ERLucT cells by a multichannel pipette and the 10 µM positive control was omitted. To quantify luminescence for the optimization experiments, one plating volume (100 µl for 96-well plates or 20 µl for 384-well plates) of ONE-Glo luciferase assay reagent (Promega) was added to each well, then plates were mixed and incubated for 5 minutes at room temperature, and counts per second were read in a luminescence-equipped multiplate reader (BioTek Synergy HT). Data were represented as relative light unit (RLU) or as fold increase RLU over DMSO-only controls for each experiment.
Bioluminescent screening. D54-ERLucT cells (3 x 103) were plated in 100 µl of RPMI 1640 + 10% FBS (HyClone) supplemented with penicillin and streptomycin in opaque 384-well plates and allowed to attach overnight. The following day, 2802 compounds from the NCI Diversity Set II, Mechanistic Set, and Natural Products libraries were added using pin tool and incubated for 24 hours. Each plate contained Tn and DMSO as positive and negative controls, respectively. Luminescence values were read using a FloStar Optima multiplate reader (BMG Labtech, Ortenberg, Germany) and converted from RLU to “percent effect” by the formula %E = [(observed RLU - negative control RLU)/(positive control RLU - negative control RLU)]. For the secondary screen, 3 x 103 D54-LucT cells were plated in 100 µl of RPMI 1640 + 10% FBS supplemented with penicillin and streptomycin in opaque 96-well plates and allowed to attach overnight. The following day, hit compounds from the primary screen were added using multichannel pipette and incubated for 24 hours. Luminescence values were read using a multiplate reader (BioTek Synergy HT). A ratio of the fold increase RLU in the primary screen to the increase in RLU in the secondary screen was generated for each hit compound.
Western Blot Analysis
Cells were washed with 1x phosphate-buffered saline (PBS) and harvested on ice. Lysates were prepared in Western lysis buffer as previously described [17] (25 mM Tris, 10 mM EDTA, 15% glycerol, 0.1% Triton X-100, 1x protease inhibitor cocktail, and 1x phosphatase inhibitor cocktails 2 and 3) by repeatedly passing cells through a 22-gauge syringe needle. Lysates were quantified using a Bradford protein assay (Bio-Rad) and equal protein amounts were subjected to polyacrylamide gel electrophoresis.
In Vitro Luciferase Assays
CHO cells with the ERLucT reporter were grown to confluence in 15-cm dishes and lysed in 2 ml of freshly prepared lysis buffer [20 mM Tris-Cl (pH 7.5), 10 mM NaCl, 1 mM DTT, 15 mM MgSO4, and 0.1% NP-40] by vortexing. Lysates were centrifuged at 15,000 rpm for 5 minutes in a desktop centrifuge at 4°C and placed on ice. Lysates were then incubated with drug or control compounds for 5 minutes at room temperature, and then added to 12 x 75 mM tubes containing 100 µl of pre-aliquoted 1x Luc assay buffer [20 mM Tris-Cl (pH 7.5), 10 mM NaCl, 1 mM DTT, 15 mM MgSO4, 2 mM ATP, and 500 µM luciferin] and read immediately in a single-read luminometer (EG&G Berthold Lumat 9507B) with a 10-second flash read. Linear range for the luminometer was determined by diluting a control luciferase-containing lysate.
Lectin Selection Assays
Positive growth selection after Con A treatment was performed in 96-well plates seeded with 1 x 103 D54 cells per well. The day after plating, Acm (25 or 10 nM), Sw (2 µg/ml), or Con A (12.5 µg/ml) was added as indicated. Cells were then grown for 72 hours and assayed by tetrazolium reduction (MTS; Promega) and quantified using a multi-plate reader set to read absorbance at 490 nM, and values were normalized to vehicle-only controls.
Lipid-linked Oligosaccharide Extraction and Fluorophore-Assisted Carbohydrate Electrophoresis
Lipid-linked oligosaccharides (LLOs) were extracted essentially as described [19,20]. Briefly, adherent cells were pretreated with 10 µM Acm for 6 hours, washed with cold 1x PBS, and scraped into cold 1x PBS and pelleted at 500g for 5 minutes. The cell pellet was washed twice with 4°C PBS and then resuspended in 1 ml of 4°C methanol. The methanol suspension was evaporated to near dryness under a vacuum using a SpeedVac (Thermo Fisher Savant SC100) modified to accept 15-ml centrifuge tubes. The near-dry pellet was resuspended in 0.9 ml of water and sonicated for 2 minutes with 1-second on-off pulses. Methanol (1.5 ml) was added to the aqueous cell suspension and this mixture was vortexed for 30 seconds and sonicated again. Chloroform (3 ml) was added to this mixture, vortexed for 1 minute, and centrifuged at 3000g for 10 minutes to resolve three phases (organic, aqueous, and a middle LLO-containing phase). LLOs were collected and resuspended in 3 ml of a freshly prepared mixture of chloroform, methanol, and water (10:10:3), centrifuged at 3000g for 10 minutes, and the entire solute was dried under a vacuum. LLOs were extracted from this pellet by mild acid hydrolysis; the pellet was resuspended in 80% tetrahydrofuran, 37% hydrochloric acid was added to a final concentration of 0.1 M, and the tubes were placed in a 50°C water bath for 90 minutes. After hydrolysis, the tetrahydrofuran (THF)/HCl mixture was dried completely under vacuum, and the pellet was resuspended in 400 µl of water and 1 ml of a freshly prepared mixture of chloroform and methanol (2:1) was added. This suspension was vortexed and spun at 3000g for 10 minutes to resolve two phases; the aqueous phase was dried under vacuum for labeling and gel analysis. LLOs were labeled with 8-aminonaphthalene1,3,6-trisulfonic acid using a commercially available labeling kit (Prozyme No. GK50004), resolved by electrophoresis through oligosaccharide profiling gels (Prozyme No. GK60020), and observed using a UV gel doc system (Syngene G:BOX) fitted with a digital camera and 302/365 nm UV converter plate (VWR No. 95025-406).
Microscopy
Localization of Acm and the ER was accomplished in live HCC2935 or HCC827 cells grown in 6-cm dishes and treated with 10 µM Acm for 24 hours before visualization. Plates were washed once with 1x Hank's balanced salt solution (HBSS) and then incubated with 1 µM ER-Tracker Blue-White DPX dye in 1x HBSS for 15 minutes. Cells were rinsed once more with 1x HBSS to remove unbound dye and kept in HBSS without dye for observation. Cells were observed without fixation using equivalent exposure settings and a 470/525 nM filter (Acm) or a wide-pass DAPI filter (ER Tracker) using an EVOS-FL digital inverted microscope equipped with monochrome camera and a 40x objective (AMG, Bothell, WA). Images for two-channel merges were created by the built-in microscope software.
Clonogenic Survival Assays
Clonogenic survival assays were performed by standard methods in triplicate wells using six-well plates. Growing cells were pretreated in 10-cm tissue culture plates with 500 nM Acm A for 2 hours, then washed, trypsinized, and replated in triplicate wells using six-well plates. Six-well plates were treated with a single dose of either 0 (control), 2, 4, or 6 Gy at approximately 2 Gy/45 seconds in a Precision X-ray 320-kV orthovoltage unit. Cells were grown for 14 (A549) or 21 days (HCC2935 and HCC827) to produce colonies of >50 cells/colony, washed once with 1x PBS, then stained with 0.25% crystal violet in 80% methanol.
Data Analysis
Data are plotted as experimental mean ± SEM. Statistical significance was determined as P < .05 using a two-tailed Student's t test. The Z factor for high-throughput experiments was calculated as described [21]: Z′ = [1 - (3(σρ + ση)/(µρ - µη))], where ρ and η represent the positive and negative controls.
Results
HTS Optimization
To directly monitor posttranslational processes in the ER of living cells, we have developed a functional bioluminescent reporter vector, ERLucT (Figure 1A), which targets the Photinus pyralis luciferase gene for ER translation through in-frame addition of the EGFR N-terminal amino acid leader sequence. Previously, we have detailed incorporation of Asn-X-Ser/Thr glycosylation consensus sites into the luciferase coding sequence to produce a highly sensitive molecular imaging strategy for dynamically measuring aspargine (N)-linked glycosylation in vivo [17,18]. Under normal cell growth conditions, the reporter is translated into the ER and glycosylated, inactivating the enzymatic activity of luciferase. However, under conditions where the reporter does not undergo glycosylation or glycosylation is insufficient to block enzymatic activity, functional changes in the ER compartment can be measured through an increase in bioluminescence. A second identical luciferase gene lacking the ER translation signal (LucT) is used as a control vector for nonspecific luciferase activation because it cannot be modified by ER-resident glycosylation machinery.
Figure 1.
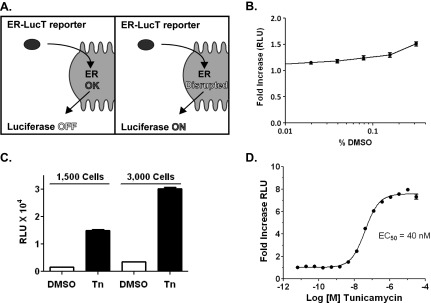
Optimization of a bioluminescent HTS to detect changes in N-linked glycosylation. (A) Schematic demonstrating the principle for signal detection using the ERLucT reporter vector: Luciferase is translated into the ER, glycosylated, and inactivated. However, when ER function is disrupted, the luciferase retains bioluminescent activity. (B) DMSO concentration-activity relationship in the D54-ERLucT cell line; representative experiment showing average of eight wells per DMSO concentration. (C) Cell density optimization for 96-well plate format using 1 µM of the GlcNac-phosphotransferase inhibitor Tn; representative experiment showing average of eight wells per treatment. (D) Dose response of Tn under optimized conditions. Data represent average of eight wells per Tn dose. Tn activates luciferase activity with a half-maximal effective concentration of 40 nM.
We hypothesized that the ERLucT/LucT reporter system could be optimized as an HTS to identify compounds that preferentially disrupt biologic processes of the ER such as N-linked glycosylation (NLG). Using D54 glioma cells with stable expression of ERLucT, we experimentally established conditions for HTS in 96-well and 384-well plate formats and used Tn, a GlcNAc-phosphotransferase inhibitor, as a positive control. First, the effects of DMSO on reporter function were determined to quantitate potential effects on assay performance, as this vehicle is used to solubilize compounds in chemical libraries. We found that DMSO concentrations do not cause significant increases in luminescence in the D54-ERLucT line at assay relevant concentrations, although a 1.5 ± 0.1-fold increase was observed at 0.3% DMSO (Figure 1B). To avoid this minimal effect, DMSO was kept at or below a final concentration of 0.1% for all experiments. Next, we evaluated the effects of cell plating density and found similar levels of Tn-induced reporter activity. To increase signal differences between positive and negative controls, a cell titer of 3000 cells per well was used for all subsequent experiments (Figure 1C). Dose-response experiments with the positive control, Tn, were then performed (Figure 1D) and demonstrated an 8.0 ± 0.1-fold peak induction of luciferase activity over control cells. The half-maximal effective concentration of Tn using this reporter is 40.3 ± 1.0 nM. The mean Z factor for positive and negative controls was calculated to be 0.71 (SD ± 0.04).
HTS Identifies Acm as a Preferential ERLucT Activator
Using this live cell bioluminescent assay, we performed an HTS to find compounds that preferentially activate the ERLucT reporter and to identify small molecules that interfere with biologic functions of the ER. Two thousand eight hundred and two compounds from the NCI Diversity Set, Mechanistic Set, and Natural Products libraries were screened and analyzed for effects on luciferase activity using the HTS. Compounds were scored for percent effect relative to the positive control Tn (Figure 2A). In total, the primary screen identified 117 compounds with at least a 40% enhancement of luciferase activity.
Figure 2.
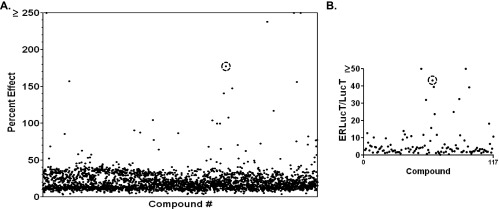
HTS of NCI compound libraries. (A) Primary screens of 2802 compounds from the NCI Diversity, Mechanistic, and Natural Compound libraries were screened at 10 µM using the D54-ERLucT cell line. The activity of each compound relative to Tn is plotted. (B) Secondary screen of 117 compounds in the D54-LucT cell line, a false-positive screen for nonspecific luciferase activators. Y-axis represents the ratio of primary/secondary screen activity (ERLucT/LucT ratio). Acm is circled in A and B.
These compounds were further analyzed in D54-LucT cells, a control cell line expressing the LucT (luciferase without the ER translation sequence), which serves as a secondary screen to identify false-positive luciferase activators. The ratio of luciferase activity in each screen (ERLucT/LucT) for the 117 compounds is presented in Figure 2B and shows that 11 of the compounds demonstrated an activation ratio greater than 20-fold. This activation profile suggested preferential activity in the ER compartment and was thus identified as HTS “hits.” One of these compounds, Acm, a glycosylated antibiotic and antitumor drug, showed among the highest increases in luminescence in the primary screen as well as the primary screen/counterscreen ratio (Figure 2, A and B, circles), and a literature search suggested that Acm may disrupt NLG [22]. On the basis of the ER-specific effect in the tiered screen, Acm was selected for further investigation in both cell-based and biochemical analyses.
Acm Reduces Cell Surface Glycoprotein Expression
Acm preferentially activated the ERLucT reporter, similar to Tn, and we therefore investigated whether this compound is an NLG inhibitor using tertiary analyses. Inhibition of NLG with Tn produces molecular weight changes and increased protein migration on acrylamide gels, and we therefore performed Western blots to examine the effect of Acm on luciferase Western blot migration (Figure 3A, top panel). Unlike Tn, Acm did not increase the gel mobility, suggesting that it did not prevent addition of N-linked glycans to the protein. Precipitation of protein lysates with the lectin Con A, a maneuver that removes glycosylated proteins from the lysate, was also performed to confirm that luciferase remained glycosylated after Acm treatment.
Figure 3.

Acm does not prevent N-linked glycosylation. (A) Western blot analysis of luciferase size and quantity following incubation with 10 µM of compounds identified in the HTS (top panel). Parallel lysates were also depleted of glycosylated proteins through precipitation with the lectin Con A agarose. Tn was used as a positive control. (B) Activity of Acm (10 µM) in cell-free luciferase assays compared to resveratrol, a known luciferase inhibitor and false positive in cell-based luciferase assays. Data represent average of three independent measurements for each treatment. (C) Lectin-positive growth selection assays. MTT assays were performed in 96-well plates for wild-type CHO and the glycosylation-defective Lec15 CHO cell line with combinations of 2 µg/ml of the mannosidase inhibitor Sw and 12.5 µg/ml Con A (left panel). Data represent an average of three wells per treatment. D54 cells were grown under identical conditions with or without 25 nM Acm and treated with Sw, Con A, or a combination of the two (middle panel). Data represent three wells per treatment. Using the same methods, D54 cells were incubated with increasing concentrations of Con A either with or without Acm pretreatment (right panel). (D) FACE was performed to determine the levels and composition of LLOs extracted from D54 cultures pretreated with 10 µM of each compound for 6 hours. Tn (1 µM) was used as a positive control. The mature 14-carbohydrate LLO (Glc3Man9GlcNAc2) is identified with an arrowhead. An oligosaccharide ladder (left) and a glucose-4 standard (right) provide molecular weight standards.
One paradoxical class of compounds that affect luciferase-based HTSs is luciferase inhibitors [23,24]. In cell-based luciferase assays, luciferase inhibitors sequester and stabilize luciferase, increase the amount of protein, and generate a false-positive signal. Acm-treated cells appeared to have subtle increases in luciferase protein expression (Figure 3A), and thus to test whether Acm is a luciferase inhibitor, we determined whether this compound could block luciferase activity in a cell-free assay using luciferase from crude cell lysates. The results show that preincubation with resveratrol, a known luciferase inhibitor [25], significantly reduced the luminescence of our CHO ERLucT crude lysates (P < .0001) with a mean reduction of 90.4%. However, preincubation with Acm did not reduce luminescence, eliminating luciferase inhibition as the mechanism for activity (Figure 3B).
Because Acm has been previously linked with protein NLG [21], we next investigated its effects on LLO synthesis, as deficiencies in these enzymatic steps could yield either hypoglycosylation or abnormal transfer of truncated glycan precursors. The effects of Acm on extracellular glycan expression were first evaluated through adaptation of a lectin selection assay to a 96-well plate format [26]. The premise of this positive growth selection assay is that defects late in the ER LLO synthesis pathway provide a survival advantage when cells are treated with a combination of the mannosidase inhibitor Sw and the lectin Con A. This effect is demonstrated in Figure 3C, where dolichyl-phosphate mannosyltransferase polypeptide 2-defective CHO cells that do not synthesize the mature 14-carbohydrate LLO demonstrate significantly increased survival after Sw + Con A treatment (P < .01) relative to equivalently treated parental CHO cells. We therefore used this strategy to test whether Acm could protect cells from Con A-dependent toxicity. The results show that Acm does not protect D54 cells from Sw + Con A toxicity, suggesting that Acm does not alter biosynthesis of mature LLO species. However, this assay did show a significant reduction of D54 cell killing by Con A after Acm treatment (Figure 3C, middle and right panels; P < .05), a test sensitive to the general state of surface glycosylation, suggesting that Acm does reduce cell surface expression of glycosylated proteins.
To confirm that glycan precursor biosynthesis was not reduced by Acm, we performed direct analysis of LLOs with fluorophore-assisted carbohydrate electrophoresis (FACE; Figure 3D). Our results show that pretreatment with Tn reduced synthesis of the mature 14-sugar LLO in D54-ERLucT cells; however, no change in the abundance of this LLO species was observed on pretreatment with Acm (or with three additional screen hits), providing direct biochemical evidence that Acm does not inhibit synthesis of LLOs in these cells. Taken together, these experiments provide clear evidence that Acm does not affect protein glycosylation through glycan transfer to ER proteins or by causing aberrant glycosylation through inhibition of mature LLO biosynthesis.
Acm Reduces RTK Protein Levels
Though our data show that Acm does not block NLG, the HTS did demonstrate an ER pathway-specific effect and the lectin selection assay did suggest that Acm reduces cell surface expression of glycosylated proteins. We therefore tested whether Acm could alter ER proteins important for tumor cell survival and analyzed protein levels for RTKs, a class of ER-dependent proteins that require folding and processing in the ER to function. In the D54 glioma cell line, Western blot analysis demonstrated that, like Tn [17], Acm reduced EGFR and Met protein levels (Figure 4A). However, Acm did not induce the Grp78/Bip expression characteristic of NLG inhibition and ER stress, further demonstrating that Tn and Acm have distinct ER/secretory pathway targets. The effects of Acm on RTK protein levels were also tested in the EGFR-amplified and exon 19 kinase domain mutant-expressing HCC827 lung adenocarcinoma cell line. Treating HCC827 cells with Acm reduced total EGFR and Met protein levels in this cell line as well without an observed increase in Grp78/Bip expression (Figure 4B).
Figure 4.

Acm reduces RTK protein levels. (A) D54 cells were treated with 10 µM Acm (A) or 1 µM Tn (T) for 24 hours, and protein levels of EGFR, Met, and Grp78 were evaluated with Western blots. (B) The effects of Acm A on RTK protein levels were analyzed in the HCC827 cell line using identical conditions. Blots are representative of three independent experiments. HSP70 was used as a loading control.
Acm Accumulates in the ER
To more clearly identify the relationship between the cellular effects of Acm and the ER, we directly observed Acm distribution in live cell cultures. Acm is a fluorescent molecule with two major emission peaks in the green wavelength range [27], and we therefore examined Acm cellular localization in two lung adenocarcinoma cell lines using fluorescence microscopy (Figure 5). Acm accumulates in a perinuclear cellular compartment and is excluded from the nucleus in both the HCC827 and HCC2935 cell lines. To determine if Acm was accumulating in the ER, we co-treated cell cultures with an ER-Tracker dye and observed co-localization, suggesting that Acm at least partially accumulates in the ER and post-ER cellular compartments.
Figure 5.

Localization of Acm in NSCLC. HCC2935 and HCC827 were treated with or without 10 µM Acm for 24 hours followed by incubation with ER-Tracker Blue-White DPX dye to observe co-localization. Cells were observed using an EVOS-FL digital inverted microscope, and merged images were created using the EVOS-FL on-board software. Images were captured using a x40 objective. Scale bar represents 100 µM.
Acm Radiosensitizes EGFR-Mutant Cell Lines
Because Acm treatment reduced RTK protein levels, we hypothesized that Acm would radiosensitize tumor cells that are dependent on RTK signaling pathways for survival. To test this hypothesis, we investigated the consequences of Acm treatment in EGFR-dependent lung adenocarcinoma cell lines (HCC827 and HCC2935). We found that pretreatment with 500 nM Acm for 2 hours before radiation significantly reduced clonogenic survival after radiation at 4 and 6 Gy for both cell lines (P < .05 and P < .001, respectively). The calculated dose enhancement ratio at 10% survival was 1.4 for HCC827 and 1.3 for HCC2935 (Figure 6, A and B). To confirm that RTK signaling was essential to Acm's radiosensitizing effect in these cells, we tested A549 lung carcinoma cells. A549 cells carry a G12S activating mutation in KRAS and are resistant to EGFR-targeted therapeutics [28]. In contrast to the RTK-addicted lung lines, we observed no difference in survival between Acm-treated and untreated A549 cells (Figure 6C). Together, these data suggest that Acm disrupts ER function, reduces RTK protein levels, and enhances radiosensitivity.
Figure 6.

Acm radiosensitizes EGFR-mutant lung cancer cell lines. Clonogenic survival analysis was performed on NSCLC cell lines harboring EGFR kinase domain mutations [(A) HCC827 and (B) HCC2935] or a KRAS mutation [(C) A549]. Cells were pretreated with (triangles) or without (circles) 500 nM Acm for 24 hours. Points represent the average of three experiments performed in triplicate (HCC-2935 and HCC-827) or two experiments performed in triplicate (A549), and error bars represent the SEM.
Discussion
In this work, we have devised an optimized cell-based assay for screening small molecule compounds in high-throughput format. The HTS employs a modified luciferase that is designed to lose activity under normal cell growth conditions and to gain activity when ER function is compromised. Combined with methods for sensitive detection of luciferase activity, the ERLucT HTS demonstrated a robust signal-to-noise ratio in microplate formats with statistically significant predictive power for identifying “hits” and thus provides a novel method for identifying compounds that preferentially disrupt biologic functions of the ER in tumor cell lines. To test performance of the HTS, we used NCI chemical libraries formulated to represent chemical diversity from both synthetic and natural compound sources. These libraries have previously been shown to be vital research tools, having been screened to identify modulators of posttranslational protein modification [29], proteasome function [30], and major developmental signal transduction pathways [31]. We found that our ERLucT assay successfully triaged the vast majority of compounds in the chemical libraries that were tested, validating its use as a primary HTS and the initial step in a tiered strategy for selecting compounds with ER-specific functions.
To advance the findings from the ERLucT primary screen, we also established a secondary cell-based screen in D54 cells that expresses luciferase without the N-terminal ER translation leader sequence. This assay served as a screen for false positives and was a practical method for eliminating compounds that nonspecifically enhanced luciferase activity in the D54 cell line. Although this secondary screen was effective for selecting and limiting compounds for further biochemical analyses, we found that it was not sufficient for excluding all false-positive luciferase activators. The addition of a cell-free luciferase assay to identify compounds that directly interact with and block luciferase activity showed that several HTS hits were in fact luciferase inhibitors (data not shown), consistent with other published work [22,23]. These results suggest that small molecule/ reporter vector interactions vary according to cellular compartment, and we therefore recommend both cell-based and cell-free assays for the identification of false-positive reporter activation in HTS designs.
Our HTS design was complemented by a tiered approach for evaluating hits in tertiary experimental assays. Because one goal of this screening program is to identify small molecule inhibitors of N-linked glycosylation, a site-specific posttranslational modification that is carried out in the ER, we determined the effects of these compounds on NLG. One of the hits in the screen, Acm, was previously suggested to be a small molecule that could disrupt NLG [21] and validating this biologic role for Acm was therefore an emphasis of our work. However, our results demonstrate that Acm does not directly inhibit either synthesis of glycan precursors or protein glycosylation in vitro. This disparity is most likely due to differences in the experimental approach; however, our data are consistent with the previously observed reduction in cell surface glycoprotein activity attributed to Acm. The assays presented here focused on the end results of proper NLG and NLG precursor biosynthesis: molecular weight increases of glycoproteins, cell surface expression of glycoproteins detectable by lectin binding assays, and interrogation of NLG precursor synthesis by FACE. Our results show that implementation of this novel primary HTS, secondary counterscreen, and tertiary glycosylation assays can both identify inhibitors of N-linked glycosylation and also be used to rule out compounds that do not affect NLG per se.
Although Acm was excluded as an inhibitor of NLG, it did produce ER-specific cellular effects in the primary and secondary screens. Additionally, and somewhat unexpectedly, treatment with Acm demonstrated a protective effect on Con A-induced cell death, suggesting that it reduced cell surface expression of glycoproteins. Furthermore, direct observation of Acm subcellular localization in living cells demonstrated that this compound accumulates in the ER and secretory compartments. We have previously hypothesized that blockade of the ER biosynthetic machinery would reduce protein levels of RTKs and provide a strategy for therapeutic radio-sensitization [17,18], and we therefore examined the effects of Acm on the RTK glycoproteins, EGFR and Met, in both the D54 and HCC8327 cell lines. Our results showed that Acm significantly reduced protein levels of these transmembrane receptors without a generalized effect on protein translation. Together, these experimental results provided evidence for a preferential effect of Acm on the ER and suggest that Acm accumulates in the ER and disrupts protein levels and cell surface expression of glycoproteins important for cell survival signaling such as EGFR and Met.
Acm was a tantalizing compound for study because of its status as an existing cancer drug. Acm is used primarily in the treatment of refractory acute myelogenous leukemia (AML) [32], and Acm is known to cause DNA damage by inhibiting topoisomerases I and II [33] and can cause DNA damage indirectly by generating reactive oxygen species (ROS) [34]. In this work, we have demonstrated that in addition to its role in causing DNA damage, Acm has at least one additional mechanism of action and at least part of Acm's toxicity is mediated by disrupting the function of the ER. However, exactly how Acm disrupts the ER is as of yet unclear. Acm does not cause a general repression of translation as non-ER proteins are not affected, including our non-ER luciferase reporter. There are several remaining possible explanations that are not mutually exclusive. One explanation is that Acm generates ROS that have a deleterious effect on the ER or ER proteins and causes ER stress [35]. Our data suggest that Acm is not simply activating ER stress pathways, but an ROS-mediated mechanism cannot be excluded. A second possible explanation is that Acm affects RTK transport through the ER or indirectly affects transport by disrupting proper folding and association with chaperone proteins. ER chaperones including calreticulin and calnexin are vital for RTK folding and stability and promote RTK transport through the ER [36]. Consistent with this, chaperone inhibitors, such as geldanamycin, an inhibitor of HSP90, have been shown to inhibit folding of ErbB3 within the ER [37] and destabilize cytoplasmic EGFR [38] and FGFR3 [39]. Thus, Acm may disrupt RTK-chaperone interactions that are vital for stability and folding of RTKs at the early stages in their life cycle.
Acm is a glycosylated natural compound antibiotic, and typical for the NCI libraries, as it has undergone extensive preclinical and clinical testing in multiple tumor sites, though it has not been advanced past phase II trials. This compound has also been evaluated as a potential radiosensitizer in vitro with conflicting results. Acm pretreatment was shown to radiosensitize a colon cancer cell line [40,41], although pretreatment did not radiosensitize HeLa cells [42]. Because we have demonstrated an ER-specific effect of Acm and a corresponding reduction of RTK proteins, we tested whether an enhancement in Acm radiosensitivity could be explained by cell dependence on ER-translated proteins such as RTKs. To perform these experiments, we used the HCC827 and H2935 EGFR kinase domain mutant non-small cell lung cancer (NSCLC) cell lines that are EGFR-dependent and compared them to the KRAS-mutant (and EGFR-independent) A549 cell line. The results of clonogenic survival experiments demonstrate that both EGFR-mutant cell lines were radiosensitized by Acm, but the RAS-mutant cell line was not. While the effect of Acm on DNA damage is likely a major contributor to the enhancement of radiosensitivity, our findings suggest that Acm's effect on ER function can also modify cellular radiation responses in susceptible tumor cells. Together, these findings support the hypothesis that disrupting ER function is a feasible strategy to enhance tumor cell radiosensitivity.
In summary, we report a set of assays using modified luciferase reporters that can be used successfully in HTSs to identify compounds that preferentially target the ER. The unique design of our screening strategy allows the identification of a wide range of ER-targeting compounds, including cancer therapeutics that can be difficult to assay secondary to cell toxicity. Using this strategy, we demonstrated that Acm targets the ER but does not affect the N-linked glycosylation pathway and that Acm radiosensitizes EGFR-mutant NSCLC cell lines. This implies that drugs targeting the ER may represent a novel approach to inducing radiosensitivity in cancer cells and demonstrates one plausible mechanism to identify those drugs through a succession of tiered screens.
Footnotes
D.C.B. was supported by the National Institutes of Health (NIH) and the National Research Service Award postdoctoral training grant T32CA009259 and J.C. was supported by the ASTRO Junior Faculty Award and the Yale Clinical Center for Investigation Scholar Award (UL1RR024139/KL2RR024138). This work was also supported by NIH grants P01-CA085878 (A.R.) and NIH-GM038545 (M.L.). The authors declare no conflicts of interest.
References
- 1.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 2.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, Kaufman RJ. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci USA. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shang J, Gao N, Kaufman RJ, Ron D, Harding HP, Lehrman MA. Translation attenuation by PERK balances ER glycoprotein synthesis with lipid-linked oligosaccharide flux. J Cell Biol. 2007;176:605–616. doi: 10.1083/jcb.200607007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laisney JA, Mueller TD, Schartl M, Meierjohann S. Hyper-activation of constitutively dimerized oncogenic EGF receptors by autocrine loops. Oncogene. 2012 doi: 10.1038/onc.2012.267. E-pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 5.Wu YM, Liu CH, Hu RH, Huang MJ, Lee JJ, Chen CH, Huang J, Lai HS, Lee PH, Hsu WM, et al. Mucin glycosylating enzyme GALNT2 regulates the malignant character of hepatocellular carcinoma by modifying the EGF receptor. Cancer Res. 2011;71:7270–7279. doi: 10.1158/0008-5472.CAN-11-1161. [DOI] [PubMed] [Google Scholar]
- 6.Liu YC, Yen HY, Chen CY, Chen CH, Cheng PF, Juan YH, Chen CH, Khoo KH, Yu CJ, Yang PC, et al. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc Natl Acad Sci USA. 2011;108:11332–11337. doi: 10.1073/pnas.1107385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pectasides E, Rampias T, Kountourakis P, Sasaki C, Kowalski D, Fountzilas G, Zaramboukas T, Rimm D, Burtness B, Psyrri A. Comparative prognostic value of epidermal growth factor quantitative protein expression compared with FISH for head and neck squamous cell carcinoma. Clin Cancer Res. 2011;17:2947–2954. doi: 10.1158/1078-0432.CCR-10-2040. [DOI] [PubMed] [Google Scholar]
- 8.Pelloski CE, Ballman KV, Furth AF, Zhang L, Lin E, Sulman EP, Bhat K, McDonald JM, Yung WK, Colman H, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol. 2007;25:2288–2294. doi: 10.1200/JCO.2006.08.0705. [DOI] [PubMed] [Google Scholar]
- 9.Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Jänne PA, Joshi VA, McCollum D, Evans TL, Muzikansky A, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26:2442–2449. doi: 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- 10.Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, Raben D, Baselga J, Spencer SA, Zhu J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11:21–28. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 11.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 12.Engelman JA, Jänne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14:2895–2899. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 13.Contessa JN, Abell A, Mikkelsen RB, Valerie K, Schmidt-Ullrich RK. Compensatory ErbB3/c-Src signaling enhances carcinoma cell survival to ionizing radiation. Breast Cancer Res Treat. 2006;95:17–27. doi: 10.1007/s10549-005-9023-9. [DOI] [PubMed] [Google Scholar]
- 14.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 15.Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA. 2005;102:7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang S, Li C, Armstrong EA, Peet CR, Saker J, Amler LC, Sliwkowski MX, Harari PM. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2013;73:824–833. doi: 10.1158/0008-5472.CAN-12-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, Lawrence TS. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res. 2008;68:3803–3809. doi: 10.1158/0008-5472.CAN-07-6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Contessa JN, Bhojani MS, Freeze HH, Ross BD, Rehemtulla A, Lawrence TS. Molecular imaging of N-linked glycosylation suggests glycan biosynthesis is a novel target for cancer therapy. Clin Cancer Res. 2010;16:3205–3214. doi: 10.1158/1078-0432.CCR-09-3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao N, Lehrman MA. Analyses of dolichol pyrophosphate-linked oligosaccharides in cell cultures and tissues by fluorophore-assisted carbohydrate electrophoresis. Glycobiology. 2002;12:353–360. doi: 10.1093/glycob/12.5.353. [DOI] [PubMed] [Google Scholar]
- 20.Hori H, Elbein AD. Characterization of the oligosaccharides from lipid-linked oligosaccharides of mung bean seedlings. Plant Physiol. 1982;70:12–20. doi: 10.1104/pp.70.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 22.Bakhtiarova A, Taslimi P, Elliman SJ, Kosinski PA, Hubbard B, Kavana M, Kemp DM. Resveratrol inhibits firefly luciferase. Biochem Biophys Res Commun. 2006;351:481–484. doi: 10.1016/j.bbrc.2006.10.057. [DOI] [PubMed] [Google Scholar]
- 23.Morin MJ, Sartorelli A. Inhibition of glycoprotein biosynthesis by the inducers of HL-60 cell differentiation, aclacinomycin A and marcellomycin. Cancer Res. 1984;44:2807–2812. [PubMed] [Google Scholar]
- 24.Auld DS, Southall NT, Jadhav A, Johnson RL, Diller DJ, Simeonov A, Austin CP, Inglese J. Characterization of chemical libraries for luciferase inhibitory activity. J Med Chem. 2008;51:2372–2386. doi: 10.1021/jm701302v. [DOI] [PubMed] [Google Scholar]
- 25.Auld DS, Thorne N, Nguyen D-T, Inglese J. A specific mechanism for nonspecific activation in reporter-gene assays. ACS Chem Biol. 2008;3:463–470. doi: 10.1021/cb8000793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ware FE, Lehrman MA. Expression cloning of a novel suppressor of the Lec15 and Lec35 glycosylation mutations of Chinese hamster ovary cells. J Biol Chem. 1996;271:13935–13938. doi: 10.1074/jbc.271.24.13935. [DOI] [PubMed] [Google Scholar]
- 27.Bachur NR, Moore AL, Bernstein JG, Liu A. Tissue distribution and disposition of daunomycin (NCS-82151) in mice: fluorometric and isotopic methods. Cancer Chemother Rep. 1970;54:89–94. [PubMed] [Google Scholar]
- 28.Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, Halmos B, Pearlberg J, Tsuchihashi Z, Cantley LC, et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005;97:1185–1194. doi: 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- 29.Francis NJ, Rowlands M, Workman P, Jones K, Aherne W. Small-molecule inhibitors of the protein methyltransferase SET7/9 identified in a high-throughput screen. J Biomol Screen. 2012;17:1102–1109. doi: 10.1177/1087057112452137. [DOI] [PubMed] [Google Scholar]
- 30.Lavelin I, Beer A, Kam Z, Rotter V, Oren M, Navon A, Geiger B. Discovery of novel proteasome inhibitors using a high-content cell-based screening system. PLoS One. 2009;4:e8503. doi: 10.1371/journal.pone.0008503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pinchot SN, Jaskula-Sztul R, Ning L, Peters NR, Cook MR, Kunnimalaiyaan M, Chen H. Identification and validation of Notch pathway activating compounds through a novel high-throughput screening method. Cancer. 2011;117:1386–1398. doi: 10.1002/cncr.25652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei G, Ni W, Chiao JW, Cai Z, Huang H, Liu D. A meta-analysis of CAG (cytarabine, aclarubicin, G-CSF) regimen for the treatment of 1029 patients with acute myeloid leukemia and myelodysplastic syndrome. J Hematol Oncol. 2011;4:46. doi: 10.1186/1756-8722-4-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stoyanova T, Roy N, Kopanja D, Bagchi S, Raychaudhuri P. DDB2 decides cell fate following DNA damage. Proc Natl Acad Sci USA. 2009;106:10690–10695. doi: 10.1073/pnas.0812254106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurz EU, Douglas P, Lees-Miller SP. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J Biol Chem. 2004;279:53272–53281. doi: 10.1074/jbc.M406879200. [DOI] [PubMed] [Google Scholar]
- 35.Chinta SJ, Rane A, Poksay KS, Bredesen DE, Andersen JK, Rao RV. Coupling endoplasmic reticulum stress to the cell death program in dopaminergic cells: effect of paraquat. Neuromolecular Med. 2008;10:333–342. doi: 10.1007/s12017-008-8047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramos RR, Swanson AJ, Bass J. Calreticulin and Hsp90 stabilize the human insulin receptor and promote its mobility in the endoplasmic reticulum. Proc Natl Acad Sci USA. 2007;104:10470–10475. doi: 10.1073/pnas.0701114104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerbin CS, Landgraf R. Geldanamycin selectively targets the nascent form of ERBB3 for degradation. Cell Stress Chaperones. 2010;15:529–544. doi: 10.1007/s12192-009-0166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahsan A, Ramanand SG, Whitehead C, Hiniker SM, Rehemtulla A, Pratt WB, Jolly S, Gouveia C, Truong K, Van Waes C, et al. Wild-type EGFR is stabilized by direct interaction with HSP90 in cancer cells and tumors. Neoplasia. 2012;14:670–677. doi: 10.1593/neo.12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laederich MB, Degnin CR, Lunstrum GP, Holden P, Horton WA. Fibroblast growth factor receptor 3 (FGFR3) is a strong heat shock protein 90 (Hsp90) client: implications for therapeutic manipulation. J Biol Chem. 2011;286:19597–19604. doi: 10.1074/jbc.M110.206151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bill CA, Garrett KC, Harrell R, Tofilon PJ. Enhancement of radiation-induced cell killing and DNA double-strand breaks in a human tumor cell line using nanomolar concentrations of aclacinomycin A. Radiat Res. 1992;129:315–321. [PubMed] [Google Scholar]
- 41.Bill CA, Mendoza EA, Vrdoljak E, Tofilon PJ. Enhancement of tumor cell killing in vitro by pre- and post-irradiation exposure to aclacinomycin A. Radiother Oncol. 1993;28:63–68. doi: 10.1016/0167-8140(93)90187-d. [DOI] [PubMed] [Google Scholar]
- 42.Miyamoto T, Wakabayashi M, Terasima T. Aclarubicin (aclacinomycin A) and irradiation: evaluation using HeLa cells. Radiology. 1983;149:835–839. doi: 10.1148/radiology.149.3.6580675. [DOI] [PubMed] [Google Scholar]