

Abstract
AIM: To investigate the effects of long term pretreatment with low-, medium- and high-dose aspirin (acetylsalicylic acid, ASA) on a model of acute pancreatitis (AP) induced in rats.
METHODS: Forty male Wistar rats were used. Three experimental groups, each consisting of eight animals, received low- (5 mg/kg per day), medium- (150 mg/kg per day) and high-dose (350 mg/kg per day) ASA in supplemented pellet chow for 100 d. Eight animals, serving as the AP-control group, and another eight, serving as reference value (RV) group, were fed with standard pellet chow for the same period. After pretreatment, AP was induced in the experimental animals by intraperitoneal administration of cerulein (2 × 50 μg/kg), while the RV group received saline in the same way. Twelve hours after the second injection, the animals were sacrificed. Pancreatic tissue and plasma samples were collected. One part of the collected pancreatic tissues was used for histopathological evaluation, and the remaining portion was homogenized. Cytokine levels [tumor necrosis factor, interleukin (IL)-1β, IL-6], hemogram parameters, biochemical parameters (amylase and lipase), nuclear factor-κB, aspirin triggered lipoxins and parameters related to the antioxidant system (malondialdehyde, nitric oxide, hemeoxygenase-1, catalase and superoxide dismutase) were measured.
RESULTS: Cerulein administration induced mild pancreatitis, characterized by interstitial edema (total histopathological score of 5.88 ± 0.44 vs 0.25 ± 0.16, P < 0.001). Subsequent pancreatic tissue damage resulted in an increase in amylase (2829.71 ± 772.48 vs 984.57 ± 49.22 U/L, P = 0.001) and lipase (110.14 ± 75.84 U/L vs 4.71 ± 0.78 U/L, P < 0.001) in plasma, and leucocytes (6.89 ± 0.48 vs 4.36 ± 0.23, P = 0.001) in peripheral blood. Cytokines, IL-1β (18.81 ± 2.55 pg/μg vs 6.65 ± 0.24 pg/μg, P = 0.002) and IL-6 (14.62 ± 1.98 pg/μg vs 9.09 ± 1.36 pg/μg, P = 0.04) in pancreatic tissue also increased. Aspirin pretreatment reduced the increase in the aforementioned parameters to a certain degree and partially improved the histopathological alterations caused by cerulein. No evidence of side effects related to chronic ASA administration (e.g., inflammation or bleeding) was observed in the gastrointestinal tract in macroscopic and histopathological examination.
CONCLUSION: Long term ASA pretreatment could prevent and/or ameliorate certain hematological, serological and histological alterations caused by cerulein-induced AP.
Keywords: Aspirin, Acute pancreatitis, Cerulein, Antioxidant, Cytokines
Core tip: Acute pancreatitis (AP) is an inflammatory and potentially life-threatening disease. An estimated 80000 cases of AP occur each year in the United States. There is no specific cure for AP; therefore, research interest has focused on prevention strategies. In the present study, the effects of a long-term pretreatment with different doses of aspirin, the oldest and most widely used non-steroidal anti-inflammatory drug, were investigated on a AP model in rats. Our results indicated that aspirin pretreatment dose-dependently prevents or ameliorates some hematological, serological and histological alterations caused by cerulein-induced AP.
INTRODUCTION
Acute pancreatitis (AP) is an inflammatory disease with broad clinical variation, ranging from a mild and self-limiting condition to a severe, life-threatening necrotizing inflammation[1,2]. Furthermore, it can lead to the development of systemic inflammatory response syndrome (SIRS) and multisystem organ failure[3,4].
AP may have numerous causes, such as bile duct obstructions, alcohol abuse, metabolic abnormalities, various toxins and infections[5]. One of the aforementioned incidents may trigger pancreatic acinar cell injury and premature activation of pancreatic zymogens[6]. The initial acinar cell damage is followed by local activation of the immune system and induction of transcription factors, such as nuclear factor-κB (NF-κB)[4,7]. Activation of inflammatory cells and transcription factors leads to elaboration of various proinflammatory mediators, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1β and IL-6[8]. If this proinflammatory response to acinar cell damage is balanced by an anti-inflammatory response, the pancreatitis and local inflammation resolve at this stage. However, in some cases, an overwhelming proinflammatory response upsets this balance and the proinflammatory mediators migrate into systemic circulation leading to a generalized inflammation and SIRS[4,8]. In this context, proinflammatory mediators and NF-κB, which play a key role in expression of these mediators, emerge as potential therapeutic targets[6,8].
Acetylsalicylic acid (ASA) exerts analgesic, antipyretic and antiplatelet effects, and is the oldest and most widely used nonsteroidal anti-inflammatory drug[9]. In addition to its conventional effects, ASA has a preventative effect on a wide range of diseases, including gastrointestinal cancer[10], ischemic stroke[11], myocardial infarction[12] and Alzheimer’s disease[13]. The anti-inflammatory, analgesic and antipyretic efficacy of ASA is attributed mainly to its inhibitory impact on the enzymatic activity of cyclooxygenases (COX), which convert arachidonic acid to prostaglandins (PGs)[14]. On the other hand, it has been speculated that simple inhibition of PG production cannot fully account for the wide spectrum of effects of ASA[9,15]. Indeed, substantial data have been gathered, indicating that COX-independent mechanisms play a significant role in ASA’s efficacy[15]. Kopp et al[16] discovered that ASA inhibits NF-κB activation, which is a pivotal transcription factor in cytokine network. NF-κB regulates the expression of proinflammatory enzymes, cytokines, chemokines, immunoreceptors, acute phase proteins and cell adhesion molecules; therefore, it has often been termed a “central mediator of the immune system”[15,17,18]. In this regard, it has been stated that even partial inhibition of NF-κB by ASA could have a substantial effect on inflammation[16]. Another major finding was the discovery that acetylation of COX-2 by ASA can lead to transcellular biosynthesis of a new class of eicosanoids, the 15-epi-lipoxins or so-called aspirin-triggered lipoxins (ATL), which promote the resolution of inflammation[19,20]. Lipoxins have potent counter-regulatory effects in vivo and in vitro on proinflammatory mediators such as TNF-α, IL-1β, IL-6 and IL-4[19,21,22]. Furthermore it has been speculated that ASA’s unique ability to trigger the synthesis of ATLs causes an increase in nitric oxide (NO) synthesis and this aspirin-elicited NO exerts anti-inflammatory effects[23]. Grosser et al[24] found that ASA stimulates the expression and enzymatic activity of hemeoxygenase-1 (HO-1) protein in a COX-independent manner. HO-1 is a crucial mediator of the cellular antioxidant defense system and has anti-inflammatory, antiapoptotic, and antiproliferative effects[25,26]. Recent data[27] elucidated the underlying mechanism of HO-1 expression stimulated by ASA: ATL is mainly responsible for the aforementioned stimulation.
Taken together, this wide spectrum of therapeutic effects of ASA is a consequence of its efficacy in regulating a network of biochemical and cellular events in a more complex manner than was initially thought[9,28].
The significant role of proinflammatory mediators (e.g., TNF-α, IL-1β, IL-6 and platelet activating factor) and transcription factors (e.g., NF-κB and AP-1) in the pathogenesis and complications of AP are well documented in the literature[6,8]. Considering the inhibitory efficacy of ASA on these agents, it would be reasonable to suggest that ASA may be efficient in preventing or attenuating AP and its subsequent complications. Furthermore, ASA’s antioxidant efficacy exerted via the stimulation of HO-1 expression and the anti-inflammatory efficacy of ATL supports and strengthens the aforementioned hypothesis that ASA may be a therapeutic agent for the prevention and/or treatment of AP. However, to the best of our knowledge, there are no studies investigating the preventive and/or therapeutic effects of ASA on AP. Therefore, this study aimed to investigate the effects of ASA pretreatment on experimental AP in rats. By designing an experimental study with a long-term pretreatment, we focused on the preventive effects of ASA, rather than the curative ones, because the multiple and diverse mechanisms of action of ASA seem to be most effective on the initial proinflammatory progress in the pathogenesis of AP.
MATERIALS AND METHODS
Animals and grouping
Studies were performed on 40 male Wistar rats weighing 350-400 g. Animals were housed in polycarbonate cages (four rats/cage) with wood chip bedding and fed standard laboratory chow (supplemented with ASA for treatment groups) and tap water ad libitum. They were maintained in a climate-controlled animal room (temperature: 22 ± 3 °C; relative humidity: 60% ± 5%) with a 12 h/12 h light/dark cycle.
The Istanbul University’s Local Ethics Committee approved all the experimental procedures. The animals were randomly allocated to five groups as shown in Table 1.
Table 1.
Grouping and experimental design
| Group No. | n | Group name | ASA pretreatment(mg/kg) | AP induction |
| 1 | 8 | Reference value | No | No |
| 2 | 8 | Acute pancreatitis control | No | Yes |
| 3 | 8 | Low-dose ASA | 5 | Yes |
| 4 | 8 | Medium-dose ASA | 150 | Yes |
| 5 | 8 | High-dose ASA | 350 | Yes |
ASA: Acetylsalicylic acid; AP: Acute pancreatitis.
ASA pretreatment and dosing
Low, medium and high doses of ASA pretreatment were performed as diet supplements for 100 d[29]. Doses of 80 mg/d, 2-4 and 6-8 g/d ASA have been regarded as low, medium and high doses for humans, respectively[30]. Based on average human body weight of 70 kg, these doses correspond to 1.1, 28-56 and 86-114 mg/kg per day, respectively[31]. These human doses were scaled to rats according to Kleiber’s rule[32] using the following equation: dose (rat)/dose (human) = BW-0.25 (rat)/BW-0.25 (human) (BW= body weight)[33]. Based on the dose intervals derived from the above equation, the following doses were chosen for ASA pretreatment: 5 mg/kg (low-dose), 150 mg/kg (medium-dose) and 350 mg/kg (high-dose). Considering the daily food consumption of rats, standard rat chow material was supplemented with the corresponding amounts of ASA before pelleting to achieve the aforementioned low, medium and high doses. Groups 3-5 were fed these ASA supplemented pellets, while the other groups (Groups 1 and 2) received standard chow during the 100-d pretreatment.
Induction of AP
After pretreatment, all the animals, except those in the reference value (RV) group (Group 1), received two intraperitoneal injections of cerulein in 0.9% NaCl at an hourly interval at a dose of 50 μg/kg to induce AP[34]. Animals in the RV group received injections of the same volume of sterile saline solution (0.9% NaCl) in the same way.
Sample collection and preparation
Twelve hours after the induction of AP, all animals were anesthetized (xylazine/ketamine, 10/75 mg/kg) and exsanguinated via cardiac puncture. Blood samples were collected into ethylene diamine tetraacetic acid-coated tubes and plasma samples were separated via centrifugation after performing a complete blood count. The plasma samples were aliquoted and frozen at -80 °C. After sacrificing the animals, necropsies were performed and pancreatic tissues were removed. One part of the pancreas of each animal was used for homogenization, while the remaining portion was fixed in formol-saline (10%) for histopathological examination.
Pancreas samples were homogenized in a 20 mmol/L Tris-HCl buffer (pH 7.4) containing 0.5 mol/L sucrose, 25 mmol/L KCl and 5 mmol/L MgCl2 using a rotor-stator homogenizer. The homogenates were centrifuged at 1000 g for 10 min at 4 °C, and the supernatants containing the cytosolic fraction were removed, aliquoted and frozen at -80 °C until assayed. Sedimented pellets containing the nuclear fraction were used to obtain nuclear protein extracts using a commercial protein extraction kit (Intron Biotechnology Inc., Sungnam, South Korea).
Statistical analysis
TNF-α, IL-1β and IL-6 levels were determined in tissue homogenates containing the cytosolic fraction and in plasma samples using commercial enzyme-linked immunosorbent assay (ELISA) kits (Invitrogen, Camarillo, CA, United States). NF-κB levels were measured in nuclear protein extractions, using a commercial ELISA kit (USCN Life Science Inc., Wuhan, Hubei Province, China).
Catalase (CAT) activities (Cell Biolabs, San Diego, CA, United States), superoxide dismutase (SOD) activities (Assay Designs, Ann Arbor, MI, United States), and malondialdehyde (MDA) levels (Cell Biolabs) were measured in pancreas homogenates and in plasma using commercial test kits. HO-1 levels were determined in pancreas homogenates and plasma using commercial ELISA kits (Assay Designs).
Amylase and Lipase levels in plasma were measured using an automated analyzer (Architect 16200, Abbott, IL, United States). Total NO levels were determined in plasma and pancreas homogenates using commercial test kits (Assay Designs). Plasma ATL levels were measured using commercial ELISA kits (Neogen, Lexington, KY, United States). Total protein contents of homogenates were determined using the method described by Lowry et al[35] and all parameters measured in homogenates were proportioned to the total protein content of the homogenate in mg.
Tissue samples fixed in formol-saline were embedded in paraffin blocks, sectioned using a microtome and stained with hematoxylin-eosin. Histopathological scoring was performed as described by Gülçubuk et al[36], graded on a score of 0 to 3.
Statistical analysis of the obtained data was performed using the SPSS-software package (Version 11.5.2.1, SPSS Inc., Chicago, IL, United States). Results are expressed as mean ± SEM. Data for all groups were first tested for normality using the Shapiro-Wilk test. Data of groups found to be normally distributed were than compared using one-way analysis of variance. If the normality assumption was found to be violated, data were analyzed using the non-parametric Kruskal-Wallis test. Planned (a priori) contrasts and Mann Whitney U tests were used for pairwise comparisons following parametric and nonparametric tests, respectively. The ordinal data of histopathological scoring were analyzed using aforementioned non-parametric tests.
RESULTS
Intraperitoneal administration of cerulein (2 × 50 μg/kg) caused AP in all tested rats, as indicated by the marked increase in serum amylase and lipase levels (Figure 1) and histopathological changes (Figure 2).
Figure 1.
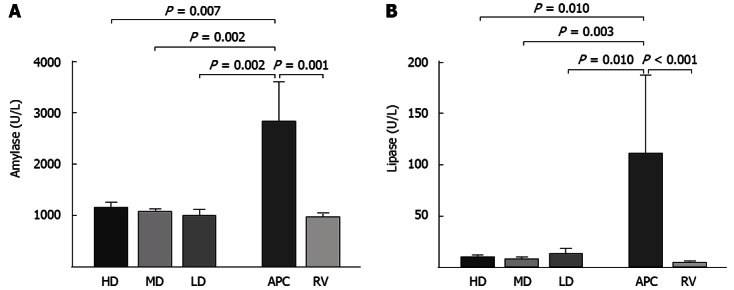
Plasma amylase (A) and lipase (B) activities. Columns show the mean and the error bars represent SEM. All groups are compared with the acute pancreatitis control (APC) group and the statistical significance is expressed as a vertical P value over the column. LD: Low-dose; MD: Medium-dose; HD: High-dose; RV: Reference values.
Figure 2.
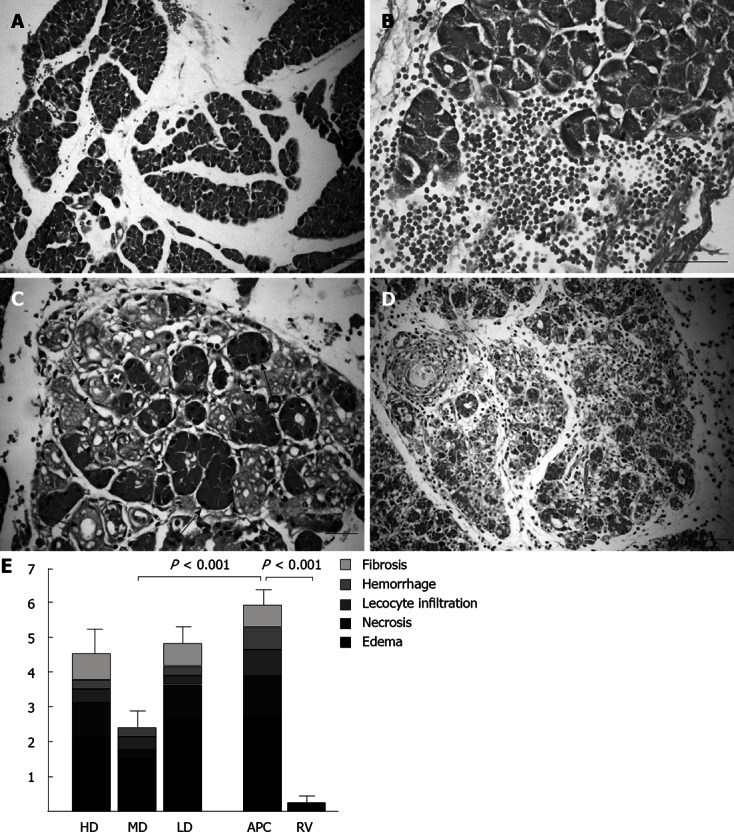
Histopathological alterations in rat pancreas caused by cerulein-induced acute pancreatitis and histopathological scores. A: Pancreatic acini were separated because of interlobular edema in cerulein treated animals (bar = 100 μm); B: Occasionally mild hemorrhages were observed (bar = 50 μm); C: Several acinar cells lost their zymogen granules (arrows) and ductus-like structures (arrowheads) occurred (bar = 50 μm); D: In some animals, leucocyte, fibrocyte and fibroblast infiltrations and collagen bands were detected (bar = 200 μm); E: Histopathological scores of each group are shown as stacked columns representing means. The whole column corresponds to the mean of the total score and the error bars represent the SEM of the total score. All groups are compared with the APC group and the statistical significance is expressed as a vertical P value over the column. HD: High-dose; MD: Medium-dose; LD: Low-dose; APC: Acute pancreatitis control; RV: Reference values.
Cerulein induced AP caused almost 3- and 23-fold increases in plasma amylase and lipase levels, respectively. ASA pretreatment significantly decreased these levels to close to those of the RV group (Figure 1). Cerulein-induced AP increased the peripheral white blood cell (WBC) count significantly compared to the RV group (6.89 ± 0.48 and 4.36 ± 0.23, respectively, P = 0.001). This increase was abolished by medium- and low-dose ASA.
Cytokine levels, lipid peroxidation and WBCs
Columns show the mean and error bars represent SEM in all figures. All groups were compared with the APC group and statistical significance was expressed as a vertical P value over the column.
The histopathological scores are shown in Table 2. Marked interstitial edema was observed in the APC group, with a score of 2.75 ± 0.16. In contrast, the edema score of the RV group was 0.25 ± 0.16 and the difference was significant (P < 0.001). Concerning the total score (Figure 2), which indicates the overall level of pathological changes, a marked difference was found between the APC and RV group scores (5.88 ± 0.44 and 0.25 ± 0.160, respectively) with a high level of significance (P < 0.001). Considering the histopathological scores, ASA pretreatment generally improved the histopathological changes. However, only the effect of the medium-dose was statistically significant (P < 0.001 for the total score). No evidence of side effects related to chronic ASA administration (e.g., inflammation or bleeding) for any of the three doses was observed in the gastrointestinal tract by macroscopic and histopathological examinations.
Table 2.
Histopathological scores
| HD-ASA (n = 8) | MD-ASA (n = 8) | P value | LD-ASA (n = 8) | APC (n = 8) | RV (n = 8) | P value | |
| Edema | 2.13 ± 0.30 | 1.50 ± 0.19 | 0.002 | 2.63 ± 0.18 | 2.75 ± 0.16 | 0.25 ± 0.16 | 0.001 |
| Hemorrhage | 0.25 ± 0.16 | 0.25 ± 0.16 | 0.25 ± 0.16 | 0.63 ± 0.18 | 0.00 ± 0.00 | 0.01 | |
| Leukocyte infiltration | 0.38 ± 0.18 | 0.38 ± 0.18 | 0.25 ± 0.16 | 0.75 ± 0.16 | 0.00 ± 0.00 | ||
| Necrosis | 1.00 ± 0.19 | 0.25 ± 0.16 | 0.007 | 1.00 ± 0.19 | 1.13 ± 0.13 | 0.00 ± 0.00 | 0.001 |
| Fibrosis | 0.75 ± 0.25 | 0.00 ± 0.00 | 0.63 ± 0.26 | 0.63 ± 0.18 | 0.00 ± 0.00 | ||
| Total score | 4.50 ± 0.68 | 2.38 ± 0.50 | 0.001 | 4.75 ± 0.49 | 5.88 ± 0.44 | 0.25 ± 0.16 | 0.001 |
ASA: Acetylsalicylic acid; RV: Reference values; APC: Acute pancreatitis control; LD: Low-dose; MD: Medium-dose; HD: High-dose.
Histopathological scores
All groups were compared with the APC group and statistical significance was expressed as P values under the corresponding mean ± SEM values.
As shown in Figure 3A, cerulein-induced AP caused a marked elevation of the IL-1β level in pancreatic tissue compared to that of the RV group (18.81 ± 2.55 pg/μg and 6.65 ± 0.24 pg/μg, respectively, P = 0.002). This elevation was suppressed significantly by ASA in all treatment groups. A similar increase was observed in the pancreatic IL-6 level (14.62 ± 1.98 pg/μg vs 9.09 ± 1.36 pg/μg, P = 0.04, Figure 3B); however, the low-dose could not prevent this increase, whereas the medium- and high-dose ASA pretreatments could suppress the IL-6 elevation. There were no statistical differences between the groups regarding the TNF-α and NF-κB levels.
Figure 3.
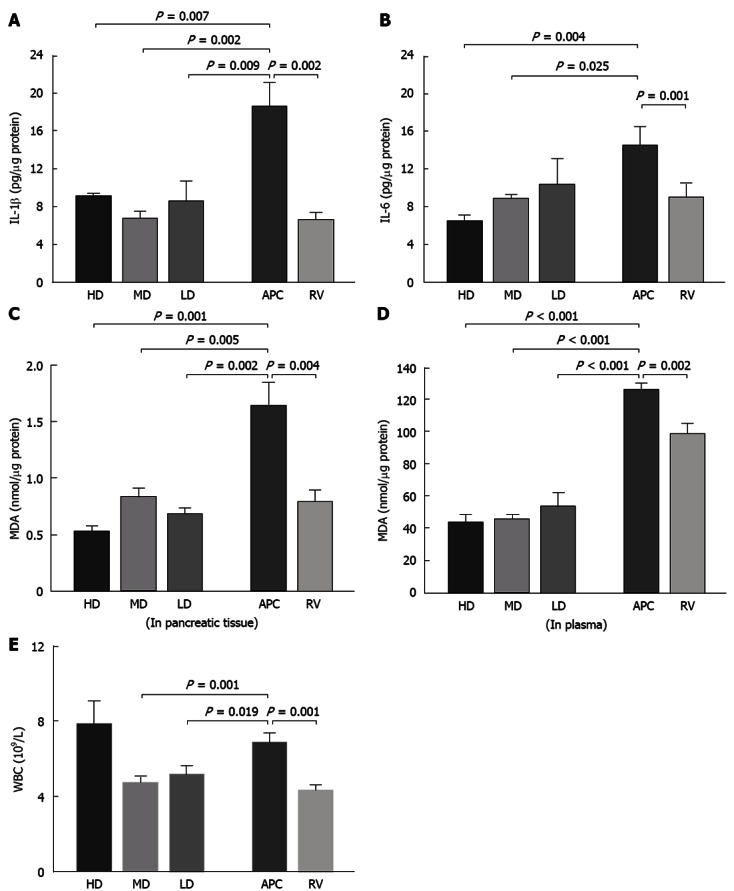
Cytokine levels, lipid peroxidation and white blood cells. A: Cerulein induced acute pancreatitis (AP) caused a marked elevation of interleukin (IL)-1β level in pancreatic tissue compared to that of the reference values (RV) group (18.81 ± 2.55 and 6.65 ± 0.24 pg/μg, respectively, P = 0.002); B: This elevation was suppressed significantly by acetylsalicylic acid (ASA) in all treatment groups. A similar increase was observed in the pancreatic IL-6 level. However, this time, the low-dose seemed to be ineffective against it, while medium- and high-dose ASA pretreatments suppressed the IL-6 elevation. There was no statistical difference between the groups regarding the tumor necrosis factor-α and nuclear factor-κB levels; C, D: Cerulein induced AP increased malondialdehyde (MDA) levels in both pancreatic tissue and plasma compared to that of the RV group; E: Cerulein induced AP increased the peripheral white blood cell (WBC) count significantly compared to the RV group. HD: High-dose; MD: Medium-dose; LD: Low-dose; APC: Acute pancreatitis control.
Cerulein-induced AP increased MDA levels in both plasma and pancreatic tissue compared to that of the RV group (Figure 3C and D). ASA pretreatment at all three doses inhibited this increase. Cerulein-induced AP increased the peripheral WBC count (Figure 3E). Other antioxidant system parameters, including NO, SOD, HO-1 and CAT, were not affected by cerulein-induced AP and ASA treatment; there were no significant differences between the treatment, APC and RV groups regarding these parameters.
DISCUSSION
To the best of our knowledge, the present study is the first investigation of the effect of long-term ASA pretreatment on a cerulein-induced pancreatitis model. Our findings indicate that long term ASA pretreatment dose-dependently prevents or ameliorates certain hematological, serological and histological alterations caused by cerulein-induced AP.
Cerulein-induced pancreatitis is the most preferred animal model of AP, because it is non-invasive, easily applicable and highly reproducible[37]. The similarity of the cerulein-induced histopathology to human AP, especially in the early phase, has substantially increased the preference for this model[38]. Administration of cerulein, a cholecystokinin (CCK) analog, stimulates the pancreatic acinar cells via CCK receptors, which leads to prematuration of proteolytic enzymes[39]. The activation of proteases triggers an autodigestion of pancreatic tissue, causing vascular damage, edema, fibrosis and necrosis, which constitute the histopathological profile of AP[6]. The markedly higher edema, hemorrhage, necrosis and the total histopathological scores of the APC group compared to the RV group, observed in our present study, are the expected results of the cerulein-induced AP model and are consistent with the literature.
The serum amylase level has been the most widely used parameter for the diagnosis of AP[40], since 1929, when its diagnostic value was demonstrated for the first time[41]. The serum lipase level, another widely accepted marker of AP, rises after the onset of AP in parallel with the amylase level, although its rise starts slightly later and lasts longer than that of amylase[42]. The plasma levels of both enzymes have substantial sensitivity and specificity for the diagnosis of AP[43]. As expected, in the present study, both the amylase and lipase levels rose markedly in the AP group. In the pretreatment groups, ASA prevented the elevation of both enzyme levels. This observation constitutes additional evidence supporting the preventive effect of ASA against cerulein-induced AP.
There is a positive correlation between the severity of AP and the increase in the peripheral WBCs[44] and the WBC count is one of the parameters included in most of the scoring systems used for the assessment of the severity of AP[45]. The increased WBC in the AP group is an expected result of the inflammation induced by cerulein. Our observation that the WBC count of medium- and low-dose groups was significantly lower than that of the AP group and close to that of the RV animals, suggests that ASA pretreatment ameliorates the inflammation induced in the pancreas.
Cytokines are a group of low-molecular weight proteins that play a crucial role in induction and progression of inflammatory processes, including AP. Thus, they have been subjected to a wide range of studies in this context[8,46,47]. Consequently there is no doubt about the constitutive role of many cytokines in progression of local tissue damage and distant complications in AP[46].
Cytokines can be functionally divided into two groups: pro- and anti-inflammatory cytokines[46]. In the proinflammatory group TNF-α and IL-1β are especially prominent and are regarded as “first-line” cytokines[48]. IL-1β levels are elevated in the cerulein-induced models of AP[4,8,49]. Furthermore, a strong, positive correlation was found between the increase in IL-1β level and the severity of inflammation[46,50]. In the present study, the IL-1β level in pancreatic tissue increased nearly threefold in the AP group compared with the RV group, whereas the difference between plasma levels showed no statistical significance. This rise in the IL-1β level in the AP group is an expected result of cerulein-induced AP. In addition, the contrast observed between the tissue and plasma levels of IL-1β is consistent with previous reports[50,51]. Considering the tissue levels of IL-1β in the pretreatment groups, ASA pretreatment had a significant diminishing effect. This finding is consistent with the amylase, lipase and MDA levels. Thus, this represents further evidence of the protective effect of ASA pretreatment against AP. IL-6 is another proinflammatory cytokine that increases in cerulein-induced pancreatitis[52]. IL-6 levels correlate with the clinical scenario and severity of AP; therefore, IL-6 has been attributed as a marker of the disease[4,53]. Thus, the increase in the IL-6 level in the APC group in the present study is an expected result of cerulein-induced AP. ASA pretreatment in the medium- and high-dose groups decreased the pancreatic IL-6 level significantly. This effect is consistent with the other findings of our study. The numerical decrease in the low-dose group was not statistically significant.
Sanfey et al[54] suggested a possible involvement of reactive oxygen species (ROS) in the pathogenesis of AP and since then, observations from increasing numbers of experimental studies have supported this suggestion[55,56]. Consequently, there is currently no doubt about the detrimental role of oxidative stress in the pathogenesis of AP and this makes it a therapeutic target. Yu et al[52] reported that, in the cerulein-induced AP model, administration of cerulein increased ROS formation and oxidative stress, and caused an increase in IL-1β expression. In the present study, the MDA level, an indicator of oxidative stress, was elevated in the cerulein administered AP group, both in plasma and in pancreatic tissue. This high level of MDA in the AP group, taken together with increased pancreatic IL-1β expression, is a consequence of cerulein-induced AP and these data are consistent with the findings of Yu et al[52]. ASA reduces oxidative stress by exerting free radical scavenging activity and antioxidant efficacy[57-60]. The findings of Shi et al[61] support ASA’s free radicals scavenging efficacy and also suggest that it is more potent than several well established antioxidants, such as ascorbate, glutathione and cysteine, with respect to the reaction rate. In the present study, oxidative stress, indicated by the MDA levels, decreased in all the ASA treated groups compared with the AP-induced groups, in both the plasma and pancreatic tissue homogenates. Moreover, considering the plasma, these levels were even below the levels of the reference group, which was fed with commercial diet without ASA supplementation. These findings can be explained by potent antioxidant effect of ASA and are in accordance with the other results presented in this study. Nevertheless, parameters related to the enzymatic antioxidant system, including NO, SOD, HO-1 and CAT, showed no significant changes. Thus, the reduced oxidative stress induced by ASA in the treatment groups seems not to involve the classic enzymatic antioxidant system and could be attributed to alternative mechanisms[60].
When examining the histopathological scores numerically, the ASA pretreatment generally attenuated the alterations caused by AP. Nevertheless, these numerical changes could not be confirmed statistically for all treatment doses or for all histopathological parameters. Only the reducing effect of the medium dose (150 mg/kg) on edema, necrosis and total score values was found to be statistically significant (P = 0.002, P = 0.007 and P < 0.001, respectively). These histopathological scoring results seem to be inconsistent with the previously discussed data for amylase, lipase, MDA and IL-1β, which were reduced significantly by ASA pretreatment at all three doses. Although there is evidence showing that serum amylase and lipase levels do not correlate with the histopathological alterations and severity of AP[62-64], we believe the aforementioned inconsistency resulted from the high variance of our data set and the small sample size and should be considered as a limitation of our study.
The analgesic, antipyretic, antiplatelet and antiinflammatory effects of ASA have been known for a long time, and most of these effects have been attributed to its COX-inhibitory activity[14]. However, it has been speculated that the therapeutic potential of ASA cannot be completely explained by COX inhibition[15,65]. Previous results from a wide variety of studies revealed different mechanisms of action of ASA, including inhibition of proinflammatory cytokines, such as IL-1β[65], scavenging of ROS[59], triggering the production of antiinflammatory mediators, such as ATL[21,22], and inhibition of transcription factors, such as NF-κB[66].
In conclusion, our findings indicate that the ASA pretreatment exerted preventive and/or ameliorating effects against AP by normalizing some of the hematological and biochemical indicators of the disease to close to those of the reference value group. These beneficial effects of the pretreatment were confirmed partially by the histopathological findings.
Our findings suggest that these beneficial effects of ASA can be explained by its free radical scavenging efficacy and the inhibitory effect on proinflammatory cytokines IL-1β and IL-6. The ATL pathway, involving the stimulation of NO and HO-1 expression, seemed not to play a role in this preventive effect, as there was no difference between groups with respect to the ATL levels.
Beside its conventional use as an analgesic, antiinflammatory and antipyretic agent, daily intake of ASA is recommended by a large group of physicians as a preventive therapy against cardiovascular diseases[67,68]. Furthermore, several studies have indicated the efficacy of long-term ASA use in prevention of colorectal cancer[69], and the long-term use of ASA as a chemopreventive agent against other cancer types has attracted substantial research interest[70]. Our results may provide another perspective on the effects of long-term ASA pretreatment.
ACKNOWLEDGMENTS
We thank Turgay Cakmak, Gizem Kutun, Dilara Yilmaz and Hayriye Guler Tarinci for their assistance in laboratory work.
COMMENTS
Background
Acute pancreatitis (AP) is an inflammatory disease with broad clinical variation, ranging from a mild and self-limiting condition to a severe, life-threatening necrotizing inflammation. Aspirin (acetylsalicylic acid, ASA) is the oldest and most widely used non-steroidal anti-inflammatory drug. In addition to its conventional effects, ASA is effective in the prevention of a wide range of diseases, including gastrointestinal cancer, ischemic stroke, myocardial infarction and Alzheimer’s disease. This broad range of effectiveness has led to the daily intake of aspirin being recommended by a wide group of physicians as a preventive therapy for the aforementioned diseases. Considering the early events in pathophysiology of AP and the broad variety of aspirin’s mechanisms of action, it is reasonable to hypothesize that long term aspirin pretreatment can effectively prevent AP.
Research frontiers
An estimated 80000 cases of AP occur each year in the United States. Much research interest has focused on prevention strategies because there is no specific cure for AP.
Innovations and breakthroughs
In the present study, the effects of ASA pretreatment on a pancreatitis model were investigated for the first time. The findings indicate that long term ASA pretreatment dose-dependently prevents or ameliorates some hematological, serological and histological alterations caused by cerulein-induced AP.
Applications
The experimental data obtained in the present study point out another aspect of aspirin’s preventive effectiveness and could be used in further studies of preventive strategies against AP.
Peer review
This is a very interesting, well-structured and original study, being the first reported study on this topic. It is methodologically well planned and performed with well-designed cohorts. The paper is well written and clear.
Footnotes
Supported by The Istanbul University Department of Scientific Research Projects, Grant No. 3101
P- Reviewers Dambrauskas Z, Ko SBH, Pan WS, Rerknimitr R S- Editor Song XX L- Editor Stewart G E- Editor Xiong L
References
- 1.Frossard JL, Steer ML, Pastor CM. Acute pancreatitis. Lancet. 2008;371:143–152. doi: 10.1016/S0140-6736(08)60107-5. [DOI] [PubMed] [Google Scholar]
- 2.Waldthaler A, Schütte K, Malfertheiner P. Causes and mechanisms in acute pancreatitis. Dig Dis. 2010;28:364–372. doi: 10.1159/000319416. [DOI] [PubMed] [Google Scholar]
- 3.Clancy TE, Benoit EP, Ashley SW. Current management of acute pancreatitis. J Gastrointest Surg. 2005;9:440–452. doi: 10.1016/j.gassur.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 4.Granger J, Remick D. Acute pancreatitis: models, markers, and mediators. Shock. 2005;24 Suppl 1:45–51. doi: 10.1097/01.shk.0000191413.94461.b0. [DOI] [PubMed] [Google Scholar]
- 5.Sakorafas GH, Tsiotou AG. Etiology and pathogenesis of acute pancreatitis: current concepts. J Clin Gastroenterol. 2000;30:343–356. doi: 10.1097/00004836-200006000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Bhatia M, Wong FL, Cao Y, Lau HY, Huang J, Puneet P, Chevali L. Pathophysiology of acute pancreatitis. Pancreatology. 2005;5:132–144. doi: 10.1159/000085265. [DOI] [PubMed] [Google Scholar]
- 7.Nagar AB, Gorelick FS. Acute pancreatitis. Curr Opin Gastroenterol. 2002;18:552–557. doi: 10.1097/00001574-200209000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Makhija R, Kingsnorth AN. Cytokine storm in acute pancreatitis. J Hepatobiliary Pancreat Surg. 2002;9:401–410. doi: 10.1007/s005340200049. [DOI] [PubMed] [Google Scholar]
- 9.Cianferoni A, Schroeder JT, Kim J, Schmidt JW, Lichtenstein LM, Georas SN, Casolaro V. Selective inhibition of interleukin-4 gene expression in human T cells by aspirin. Blood. 2001;97:1742–1749. doi: 10.1182/blood.v97.6.1742. [DOI] [PubMed] [Google Scholar]
- 10.Cuzick J, Otto F, Baron JA, Brown PH, Burn J, Greenwald P, Jankowski J, La Vecchia C, Meyskens F, Senn HJ, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009;10:501–507. doi: 10.1016/S1470-2045(09)70035-X. [DOI] [PubMed] [Google Scholar]
- 11.De Cristóbal J, Cárdenas A, Lizasoain I, Leza JC, Fernández-Tomé P, Lorenzo P, Moro MA. Inhibition of glutamate release via recovery of ATP levels accounts for a neuroprotective effect of aspirin in rat cortical neurons exposed to oxygen-glucose deprivation. Stroke. 2002;33:261–267. doi: 10.1161/hs0102.101299. [DOI] [PubMed] [Google Scholar]
- 12.Wolff T, Miller T, Ko S. Aspirin for the primary prevention of cardiovascular events: an update of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2009;150:405–410. doi: 10.7326/0003-4819-150-6-200903170-00009. [DOI] [PubMed] [Google Scholar]
- 13.in t’ Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- 14.Vane JR, Botting RM. Mechanism of action of aspirin-like drugs. Semin Arthritis Rheum. 1997;26:2–10. doi: 10.1016/s0049-0172(97)80046-7. [DOI] [PubMed] [Google Scholar]
- 15.Tegeder I, Pfeilschifter J, Geisslinger G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J. 2001;15:2057–2072. doi: 10.1096/fj.01-0390rev. [DOI] [PubMed] [Google Scholar]
- 16.Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265:956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- 17.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109 Suppl:S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 19.Parkinson JF. Lipoxin and synthetic lipoxin analogs: an overview of anti-inflammatory functions and new concepts in immunomodulation. Inflamm Allergy Drug Targets. 2006;5:91–106. doi: 10.2174/187152806776383125. [DOI] [PubMed] [Google Scholar]
- 20.Clària J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci USA. 1995;92:9475–9479. doi: 10.1073/pnas.92.21.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fierro IM, Serhan CN. Mechanisms in anti-inflammation and resolution: the role of lipoxins and aspirin-triggered lipoxins. Braz J Med Biol Res. 2001;34:555–566. doi: 10.1590/s0100-879x2001000500002. [DOI] [PubMed] [Google Scholar]
- 22.Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids. 2005;73:141–162. doi: 10.1016/j.plefa.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Paul-Clark MJ, Van Cao T, Moradi-Bidhendi N, Cooper D, Gilroy DW. 15-epi-lipoxin A4-mediated induction of nitric oxide explains how aspirin inhibits acute inflammation. J Exp Med. 2004;200:69–78. doi: 10.1084/jem.20040566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grosser N, Abate A, Oberle S, Vreman HJ, Dennery PA, Becker JC, Pohle T, Seidman DS, Schröder H. Heme oxygenase-1 induction may explain the antioxidant profile of aspirin. Biochem Biophys Res Commun. 2003;308:956–960. doi: 10.1016/s0006-291x(03)01504-3. [DOI] [PubMed] [Google Scholar]
- 25.Morse D, Choi AM. Heme oxygenase-1: the “emerging molecule” has arrived. Am J Respir Cell Mol Biol. 2002;27:8–16. doi: 10.1165/ajrcmb.27.1.4862. [DOI] [PubMed] [Google Scholar]
- 26.Wagener FA, Volk HD, Willis D, Abraham NG, Soares MP, Adema GJ, Figdor CG. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol Rev. 2003;55:551–571. doi: 10.1124/pr.55.3.5. [DOI] [PubMed] [Google Scholar]
- 27.Nascimento-Silva V, Arruda MA, Barja-Fidalgo C, Villela CG, Fierro IM. Novel lipid mediator aspirin-triggered lipoxin A4 induces heme oxygenase-1 in endothelial cells. Am J Physiol Cell Physiol. 2005;289:C557–C563. doi: 10.1152/ajpcell.00045.2005. [DOI] [PubMed] [Google Scholar]
- 28.Gilroy DW. New insights into the anti-inflammatory actions of aspirin-induction of nitric oxide through the generation of epi-lipoxins. Mem Inst Oswaldo Cruz. 2005;100 Suppl 1:49–54. doi: 10.1590/s0074-02762005000900009. [DOI] [PubMed] [Google Scholar]
- 29.Kapetanovic IM, Bauer KS, Tessier DM, Lindeblad MO, Zakharov AD, Lubet R, Lyubimov A. Comparison of pharmacokinetic and pharmacodynamic profiles of aspirin following oral gavage and diet dosing in rats. Chem Biol Interact. 2009;179:233–239. doi: 10.1016/j.cbi.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 30.FitzGerald GA. Prostaglandins, aspirin and other NSAIDs. In: Goldman L, Ausiello D, editors. Cecil Textbook of Medicine. Philadelphia: Saunders; 2004. pp. 155–161. [Google Scholar]
- 31.Zheng L, Howell SJ, Hatala DA, Huang K, Kern TS. Salicylate-based anti-inflammatory drugs inhibit the early lesion of diabetic retinopathy. Diabetes. 2007;56:337–345. doi: 10.2337/db06-0789. [DOI] [PubMed] [Google Scholar]
- 32.KLEIBER M. Body size and metabolic rate. Physiol Rev. 1947;27:511–541. doi: 10.1152/physrev.1947.27.4.511. [DOI] [PubMed] [Google Scholar]
- 33.Hau J. Animal Models. In: Hau J, Van Hoosier GL, editors. Handbook of Laboratory Animal Science. 2nd. CRC Press;; 2003. pp. 8–15. [Google Scholar]
- 34.Ip SP, Tsang SW, Wong TP, Che CT, Leung PS. Saralasin, a nonspecific angiotensin II receptor antagonist, attenuates oxidative stress and tissue injury in cerulein-induced acute pancreatitis. Pancreas. 2003;26:224–229. doi: 10.1097/00006676-200304000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 36.Gülçubuk A, Sönmez K, Gürel A, Altunatmaz K, Gürler N, Aydin S, Oksüz L, Uzun H, Güzel O. Pathologic alterations detected in acute pancreatitis induced by sodium taurocholate in rats and therapeutic effects of curcumin, ciprofloxacin and metronidazole combination. Pancreatology. 2005;5:345–353. doi: 10.1159/000086534. [DOI] [PubMed] [Google Scholar]
- 37.Chan YC, Leung PS. Acute pancreatitis: animal models and recent advances in basic research. Pancreas. 2007;34:1–14. doi: 10.1097/01.mpa.0000246658.38375.04. [DOI] [PubMed] [Google Scholar]
- 38.Dabrowski A, Konturek SJ, Konturek JW, Gabryelewicz A. Role of oxidative stress in the pathogenesis of caerulein-induced acute pancreatitis. Eur J Pharmacol. 1999;377:1–11. doi: 10.1016/s0014-2999(99)00421-5. [DOI] [PubMed] [Google Scholar]
- 39.Saluja AK, Bhagat L, Lee HS, Bhatia M, Frossard JL, Steer ML. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am J Physiol. 1999;276:G835–G842. doi: 10.1152/ajpgi.1999.276.4.G835. [DOI] [PubMed] [Google Scholar]
- 40.Takács T, Szabolcs A, Hegyi P, Rakonczay Z, Farkas G. [Changes in diagnostic and therapeutic standards of acute pancreatitis in clinical practice. Epidemiologic analysis of data from a regional center of internal medicine and surgery] Orv Hetil. 2008;149:645–654. doi: 10.1556/OH.2008.28265. [DOI] [PubMed] [Google Scholar]
- 41.Elman R, Arneson N, Graham EA. Value of blood amylase estimations in the diagnosis of pancreatic disease: a clinical study. Arch Surg. 1929;19:943–967. [Google Scholar]
- 42.Smotkin J, Tenner S. Laboratory diagnostic tests in acute pancreatitis. J Clin Gastroenterol. 2002;34:459–462. doi: 10.1097/00004836-200204000-00018. [DOI] [PubMed] [Google Scholar]
- 43.Yadav D, Agarwal N, Pitchumoni CS. A critical evaluation of laboratory tests in acute pancreatitis. Am J Gastroenterol. 2002;97:1309–1318. doi: 10.1111/j.1572-0241.2002.05766.x. [DOI] [PubMed] [Google Scholar]
- 44.Jacobs ML, Daggett WM, Civette JM, Vasu MA, Lawson DW, Warshaw AL, Nardi GL, Bartlett MK. Acute pancreatitis: analysis of factors influencing survival. Ann Surg. 1977;185:43–51. doi: 10.1097/00000658-197701000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pavlidis TE, Pavlidis ET, Sakantamis AK. Advances in prognostic factors in acute pancreatitis: a mini-review. Hepatobiliary Pancreat Dis Int. 2010;9:482–486. [PubMed] [Google Scholar]
- 46.Rau BM, Krüger CM, Schilling MK. Anti-cytokine strategies in acute pancreatitis: pathophysiological insights and clinical implications. Rocz Akad Med Bialymst. 2005;50:106–115. [PubMed] [Google Scholar]
- 47.Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg. 1998;175:76–83. doi: 10.1016/s0002-9610(97)00240-7. [DOI] [PubMed] [Google Scholar]
- 48.Yamauchi J, Shibuya K, Sunamura M, Arai K, Shimamura H, Motoi F, Takeda K, Matsuno S. Cytokine modulation in acute pancreatitis. J Hepatobiliary Pancreat Surg. 2001;8:195–203. doi: 10.1007/s005340170016. [DOI] [PubMed] [Google Scholar]
- 49.Norman JG, Fink GW, Denham W, Yang J, Carter G, Sexton C, Falkner J, Gower WR, Franz MG. Tissue-specific cytokine production during experimental acute pancreatitis. A probable mechanism for distant organ dysfunction. Dig Dis Sci. 1997;42:1783–1788. doi: 10.1023/a:1018886120711. [DOI] [PubMed] [Google Scholar]
- 50.Fink GW, Norman JG. Specific changes in the pancreatic expression of the interleukin 1 family of genes during experimental acute pancreatitis. Cytokine. 1997;9:1023–1027. doi: 10.1006/cyto.1997.0260. [DOI] [PubMed] [Google Scholar]
- 51.Mayer J, Rau B, Gansauge F, Beger HG. Inflammatory mediators in human acute pancreatitis: clinical and pathophysiological implications. Gut. 2000;47:546–552. doi: 10.1136/gut.47.4.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu JH, Lim JW, Namkung W, Kim H, Kim KH. Suppression of cerulein-induced cytokine expression by antioxidants in pancreatic acinar cells. Lab Invest. 2002;82:1359–1368. doi: 10.1097/01.lab.0000032377.09626.c7. [DOI] [PubMed] [Google Scholar]
- 53.Bhatia M, Brady M, Shokuhi S, Christmas S, Neoptolemos JP, Slavin J. Inflammatory mediators in acute pancreatitis. J Pathol. 2000;190:117–125. doi: 10.1002/(SICI)1096-9896(200002)190:2<117::AID-PATH494>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 54.Sanfey H, Bulkley GB, Cameron JL. The role of oxygen-derived free radicals in the pathogenesis of acute pancreatitis. Ann Surg. 1984;200:405–413. doi: 10.1097/00000658-198410000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petrov MS. Therapeutic implications of oxidative stress in acute and chronic pancreatitis. Curr Opin Clin Nutr Metab Care. 2010;13:562–568. doi: 10.1097/MCO.0b013e32833b64b9. [DOI] [PubMed] [Google Scholar]
- 56.Kim H. Cerulein pancreatitis: oxidative stress, inflammation, and apoptosis. Gut Liver. 2008;2:74–80. doi: 10.5009/gnl.2008.2.2.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berger C, Xia F, Schabitz WR, Schwab S, Grau A. High-dose aspirin is neuroprotective in a rat focal ischemia model. Brain Res. 2004;998:237–242. doi: 10.1016/j.brainres.2003.11.049. [DOI] [PubMed] [Google Scholar]
- 58.Podhaisky HP, Abate A, Polte T, Oberle S, Schröder H. Aspirin protects endothelial cells from oxidative stress--possible synergism with vitamin E. FEBS Lett. 1997;417:349–351. doi: 10.1016/s0014-5793(97)01307-0. [DOI] [PubMed] [Google Scholar]
- 59.Tauseef M, Shahid M, Sharma KK, Fahim M. Antioxidative action of aspirin on endothelial function in hypercholesterolaemic rats. Basic Clin Pharmacol Toxicol. 2008;103:314–321. doi: 10.1111/j.1742-7843.2008.00277.x. [DOI] [PubMed] [Google Scholar]
- 60.Wu R, Lamontagne D, de Champlain J. Antioxidative properties of acetylsalicylic Acid on vascular tissues from normotensive and spontaneously hypertensive rats. Circulation. 2002;105:387–392. doi: 10.1161/hc0302.102609. [DOI] [PubMed] [Google Scholar]
- 61.Shi X, Ding M, Dong Z, Chen F, Ye J, Wang S, Leonard SS, Castranova V, Vallyathan V. Antioxidant properties of aspirin: characterization of the ability of aspirin to inhibit silica-induced lipid peroxidation, DNA damage, NF-kappaB activation, and TNF-alpha production. Mol Cell Biochem. 1999;199:93–102. doi: 10.1023/a:1006934612368. [DOI] [PubMed] [Google Scholar]
- 62.Matull WR, Pereira SP, O’Donohue JW. Biochemical markers of acute pancreatitis. J Clin Pathol. 2006;59:340–344. doi: 10.1136/jcp.2002.002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nordestgaard AG, Wilson SE, Williams RA. Correlation of serum amylase levels with pancreatic pathology and pancreatitis etiology. Pancreas. 1988;3:159–161. doi: 10.1097/00006676-198804000-00008. [DOI] [PubMed] [Google Scholar]
- 64.Schmidt J, Lewandrowski K, Fernandez-del Castillo C, Mandavilli U, Compton CC, Warshaw AL, Rattner DW. Histopathologic correlates of serum amylase activity in acute experimental pancreatitis. Dig Dis Sci. 1992;37:1426–1433. doi: 10.1007/BF01296014. [DOI] [PubMed] [Google Scholar]
- 65.Vervoordeldonk MJ, Pineda Torra IM, Aarsman AJ, van den Bosch H. Aspirin inhibits expression of the interleukin-1beta-inducible group II phospholipase A2. FEBS Lett. 1996;397:108–112. doi: 10.1016/s0014-5793(96)01148-9. [DOI] [PubMed] [Google Scholar]
- 66.Jung KJ, Kim JY, Zou Y, Kim YJ, Yu BP, Chung HY. Effect of short-term, low dose aspirin supplementation on the activation of pro-inflammatory NF-kappaB in aged rats. Mech Ageing Dev. 2006;127:223–230. doi: 10.1016/j.mad.2005.09.029. [DOI] [PubMed] [Google Scholar]
- 67.Hennekens CH, Buring JE, Sandercock P, Collins R, Peto R. Aspirin and other antiplatelet agents in the secondary and primary prevention of cardiovascular disease. Circulation. 1989;80:749–756. doi: 10.1161/01.cir.80.4.749. [DOI] [PubMed] [Google Scholar]
- 68.Fuster V, Dyken ML, Vokonas PS, Hennekens C. Aspirin as a therapeutic agent in cardiovascular disease. Special Writing Group. Circulation. 1993;87:659–675. doi: 10.1161/01.cir.87.2.659. [DOI] [PubMed] [Google Scholar]
- 69.Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, Meade TW. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376:1741–1750. doi: 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
- 70.Jacobs EJ. Will an aspirin a day help keep fatal cancer away? Lancet. 2011;377:3–4. doi: 10.1016/S0140-6736(10)62301-X. [DOI] [PubMed] [Google Scholar]