

Abstract
Objective
The goal of this study was to understand the molecular basis for how the amino acid substitution C112R that distinguishes human apolipoprotein (apo) E4 from apoE3 causes the more pro-atherogenic plasma lipoprotein cholesterol distribution that is known to be associated with expression of apoE4.
Methods and Results
Adeno-associated viruses, serotype 8 (AAV8) were used to express different levels of human apoE3, apoE4 and several C-terminal truncation and internal deletion variants in C57BL/6 apoE-null mice which exhibit marked dysbetalipoproteinemia. Plasma obtained from these mice two weeks after the AAV8 treatment was analyzed for cholesterol and triglyceride levels as well as for the distribution of cholesterol between the lipoprotein fractions. Hepatic expression of apoE3 and apoE4 induced similar dose-dependent decreases in plasma cholesterol and triglyceride to the levels seen in control C57BL/6 mice. Importantly, at the same reduction in plasma total cholesterol, expression of apoE4 gave rise to higher very low density lipoprotein-cholesterol (VLDL-C) and lower high density lipoprotein-cholesterol (HDL-C) levels relative to the apoE3 situation. The C-terminal domain, and residues 261-272 in particular, play a critical role because deleting them markedly affected the performance of both isoforms.
Conclusions
ApoE4 possesses enhanced lipid and VLDL binding ability relative to apoE3 which gives rise to impaired lipolytic processing of VLDL in apoE4-expressing mice. These effects reduce VLDL remnant clearance from the plasma compartment and decrease the amount of VLDL surface components available for incorporation into the HDL pool, accounting for the more pro-atherogenic lipoprotein profile (higher VLDL-C/HDL-C ratio) occurring in apoE4-expressing animals compared to their apoE3 counterparts.
Keywords: apolipoprotein E, cholesterol, high density lipoprotein, very low density lipoprotein, atherosclerosis
INTRODUCTION
Apolipoprotein (apo) E is a molecule of great biological and biomedical significance. Human apoE is polymorphic and the wild-type (apoE3) is associated with normal physiology which, among other functions, involves mediation of cholesterol transport in both the circulation and the central nervous system 1-3. In contrast, the substitution C112R which converts apoE3 to apoE4 is associated with pathological sequelae, namely increased risk of cardiovascular disease and Alzheimer’s disease 4. Consequently, there is an important need to understand the reasons for the different structure-function relationships of apoE3 and apoE4.
ApoE is a ligand for the low density lipoprotein (LDL) receptor (LDLR) and serves an anti-atherogenic function by mediating the clearance of very low density lipoprotein (VLDL) remnants, thereby decreasing plasma cholesterol 5-6. ApoE3 and apoE4 bind similarly to the LDLR 7 but, compared to apoE3, apoE4 reduces plasma cholesterol less in humans giving rise to a more pro-atherogenic lipoprotein-cholesterol distribution 8. The reasons for this paradox are not understood fully but are likely a consequence of the different lipoprotein distributions of apoE3 and apoE4 in plasma. When added to plasma, apoE3 binds preferentially to high density lipoprotein (HDL) and apoE4 binds more to VLDL 9. We have recently uncovered the molecular basis for this effect 10. ApoE4 exhibits better lipid binding ability than apoE3 and the strong lipid binding ability of apoE4 coupled with the VLDL particle surface being predominantly covered with lipid (in contrast to the primarily protein-covered surface of HDL particles) leads to apoE4 binding better than apoE3 to VLDL.
The human apoE molecule possesses a N-terminal helix bundle domain (residues 1-191) which contains the LDLR recognition site and a C-terminal domain (residues 192-299) which initiates lipid binding 11-15. The substitution C112R which differentiates apoE3 and apoE4 is located in the helix bundle domain and destabilizes it 16-17. As a consequence, the interactions between the N- and C-terminal domains are altered leading to different organization of the segment spanning residues 261-272 which plays a critical role in the interaction with lipid surfaces; this structural change is the basis for the preferential binding of apoE4 to VLDL 10, 18.
Here we apply the above understanding of apoE-lipoprotein interactions in vitro to an investigation of how the enhanced binding of apoE4 to VLDL alters lipoprotein metabolism, as compared to the effects of apoE3. Previously, we have employed an adeno-associated virus serotype 8 (AAV8) system to express human apoE in apoE-null mice 19 and now we extend this work by expressing apoE3, apoE4 and C-terminal deletion variants of both isoforms at different doses and measuring the effects on plasma cholesterol levels and lipoprotein cholesterol distributions. The results reveal which parts of the C-terminal domains of apoE3 and apoE4 determine the abilities of the two molecules to mediate clearance of plasma cholesterol. The two isoforms behave differently with respect to their abilities to mediate lipolytic processing of VLDL remnants and production of HDL particles, leading to the more pro-atherogenic VLDL-cholesterol/HDL-cholesterol distribution observed with apoE4 compared to apoE3.
METHODS
A detailed description of animals, reagents and methods used in these studies can be found in the Supplemental Materials available online at http://atvb.ahajournals.org. A brief description of the principal methods used is provided here.
Preparation of apoE adeno-associated virus vectors
The pseudo-typed AAV2/8-apoE3 used for expression of apoE3 and the control vector containing the LacZ gene packaged into serotype 8 have been described before 19. The cDNA of all the apoE3 and apoE4 variants used in this study cloned into the pAAV mcs shuttle plasmid were prepared with the Quikchange site-directed mutagenesis kit (Stratagene/Agilent) starting with apoE3 cDNA. The purified mutant plasmids were then submitted to the University of Pennsylvania Vector Core for use in creating the apoE AAV8, as described before 19-21.
Mouse study protocol
Male apoE-null mice on a C57BL/6 background (8-10 weeks old) were obtained from Jackson Laboratory (Bar Harbor, Maine) and maintained on a standard chow diet. Groups of 5 mice were injected intraperitoneally with the desired number of genome copies of either apoE AAV8 or null vector. Blood was obtained from the retro-orbital plexus after a 4 h fast one day prior to virus injection and 2 weeks after injection. Liver samples were collected after exsanguination. All mouse experiments were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Plasma from each mouse was analyzed for cholesterol, triglyceride (TG) and human apoE content using a Cobas Mira-L auto analyzer (Roche Diagnostics Systems, Inc.) 19. Human apoE mRNA in the mouse livers was assessed by real-time PCR. Pooled plasma samples were subjected to FPLC analysis on a Superose-6 column and cholesterol concentrations in the lipoprotein fractions were determined enzymatically using a Wako kit 19.
Statistics
Data are presented as means ± SEM. Statistical tests for significance were done using an unpaired t-test or 1-way ANOVA followed by a Dunnett test for multiple comparisons.
RESULTS
Effects of apoE3 and apoE4 on plasma cholesterol and lipoprotein levels
Fig. 1 shows how treatment of apoE-null mice with increasing doses of apoE3- and apoE4-AAV8 reduces plasma cholesterol and TG levels. Consistent with our earlier report with apoE3-AAV8 19, a dose of 10E10 gc AAV8 expressing either apoE3 or apoE4 is sufficient to eliminate the marked hyperlipidemia characteristic of apoE-null mice on a chow diet 22-23 and reduce the plasma lipid levels to those characteristic of control C57BL/6 mice. Importantly, unlike the situation when apoE is acutely over-expressed by the use of adenovirus vectors 24-25, there is no evidence of hypertriglyceridemia. The hepatic content of human apoE mRNA exhibits a hyperbolic dependence on AAV8 dose with the expression levels of apoE3 and apoE4 being the same at any given dose (Supplementary Fig. I). Consistent with prior work using different doses of AAV8 to express human apolipoproteins in mice 21, the hepatic content of human apoE mRNA determines the amount of apoE variant appearing in plasma. The plasma levels of human apoE3 and apoE4 are comparable and in mice two weeks after treatment with AAV8 doses of 1E10, 3E10 and 10E10 gc are approximately 3, 5 and 20 μg/ml, respectively. It is problematic to determine the distributions of apoE3 and apoE4 between VLDL and HDL because the highly efficient clearance of apoE from the plasma compartment leads to low apoE concentrations. Thus, at low AAV8 doses there is relatively more VLDL present but very little apoE expression and, conversely, at high AAV8 doses the increased apoE leads to clearance of practically all of the VLDL.
Figure 1.
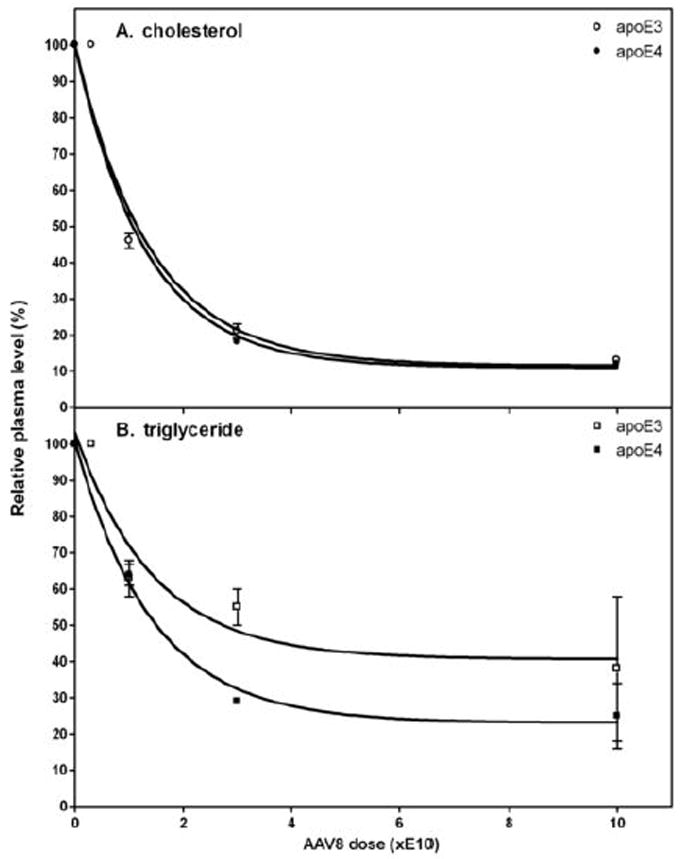
Influence of expression of human apoE3 and apoE4 on (A) plasma cholesterol and (B) triglyceride levels in apoE-null mice. The mice (5 per group) were treated with the indicated doses (gc) of AAV8 to express either apoE3 or apoE4. The mice were bled before and two weeks after administration of the AAV8 and samples of plasma were prepared for analysis of cholesterol and triglyceride levels. The lipid levels in the plasma samples are plotted relative to the values before treatment with AAV8. The 100% values were 618 ± 27 mg cholesterol/dL (mean ± SEM, n=23) and 114 ± 3 mg triglyceride/dL (mean ± SEM, n=25).
Although apoE3 and apoE4 reduce plasma total cholesterol levels to the same extent (Fig. 1A), the two apoE isoforms give rise to different lipoprotein-cholesterol distributions (Fig. 2). It is apparent that, at the same level of expression, apoE4 gives rise to a more pro-atherogenic lipoprotein profile with a higher level of cholesterol in VLDL/IDL/LDL and a lower level in HDL, relative to the apoE3 situation. The effects of different levels of expression of apoE3, apoE4 and apoE4 (1-260) on the lipoprotein cholesterol distribution are compared in Fig. 3A-C. In all cases, the progressive decrease in levels of apoB-containing lipoproteins (VLDL+IDL+LDL) with increasing apoE expression is apparent. Comparison of the elution volumes indicates that IDL and LDL particle size decreases at higher levels of apoE expression, consistent with TG removal by lipolysis. Fig.3D shows how the level of apoE expression modulates the lipolytic conversion of the VLDL-cholesterol (VLDL-C) fraction into the (IDL-C+LDL-C) fraction. In the case of increasing apoE3 expression, the fractional distribution of cholesterol between the (IDL+LDL) and VLDL fractions increases from the value of 0.4 seen in untreated mice to a value of 0.75 in mice treated with 10E10 gc AAV8. The latter value is essentially the same as the ratio of 0.77 seen for plasma of control C57BL/6 mice (data not shown). The equivalent increase in the fractional distribution with apoE4 expression is to a value of 0.54, reflecting relatively poor conversion of VLDL to IDL+LDL (Fig. 3D). Strikingly, deletion of residues 261-299 from apoE4 to give the 1-260 variant increases the fractional distribution to 0.72 at 10E10 gc AAV8, which is similar to the behavior of apoE3. It is apparent that removal of the C-terminal reduces the lipid-binding ability of apoE4 10, 18 but enhances its ability to mediate lipolytic conversion of VLDL into IDL + LDL.
Figure 2.
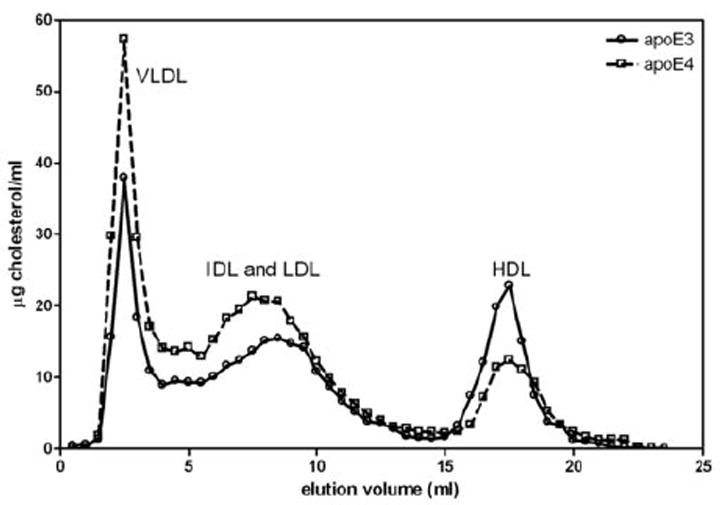
Distribution of lipoprotein cholesterol in apoE-null mice treated with AAV8 to express either human apoE3 or apoE4. Two weeks after administration of 1E10 gc AAV8, the mice (5 per group) were bled and samples of pooled plasma were fractionated by chromatography on a Superose 6 gel filtration column. The cholesterol levels in the lipoprotein fractions were determined using a Wako enzymatic kit.
Figure 3.

Influence of the level of expression of either apoE3, apoE4 or apoE4 (1-260) on the lipoprotein cholesterol distribution in apoE-null mice. The mice (5 per group) were treated with the indicated doses of AAV8 and 2 weeks later samples of pooled plasma were analyzed by gel filtration chromatography as described in the legend to Fig. 2. In panel A, the elution profile for plasma from untreated apoE-null mice is plotted with filled circles. In panels A-C, the profiles correspond to the following AAV8 doses (gc): (○) 1E10, (Δ) 3E10, (∇) 10E10. Panel D shows the influence of AAV8 dose on the distribution of lipoprotein cholesterol between the VLDL and (IDL+LDL) fractions. The ordinate shows the fractional distribution which represents the ratio (IDL-C + LDL-C) / [(IDL-C + LDL-C) + VLDL-C]. The VLDL-C and (IDL-C + LDL-C) values are the cholesterol masses that elute from the gel filtration column between 1-4.5 ml and 5-14 ml, respectively. The fractional distribution calculated in this fashion for untreated apoE-null mice (elution profile (●) in panel A) has a value of 0.4.
The apoE isoform effect on the conversion of VLDL into IDL + LDL is also associated with differences in HDL-C levels. The relatively high VLDL-C and low HDL-cholesterol (HDL-C) levels for apoE4 shown in Fig. 2 for an AAV8 dose of 1E10 gc occur at all AAV8 doses investigated (Supplementary Fig. II, Panels A and B). ApoE3 gives lower VLDL-C levels and higher HDL-C levels across the range of doses used. Greater expression of apoE3 increases the relative HDL-C value but this effect is not seen with apoE4 so that unlike the situation with apoE3, where high expression reduces the VLDL-C/HDL-C ratio to the very low level (~0.1) characteristic of control C57BL/6 mice, the ratio remains above one for apoE4-expressing mice (Supplementary Fig. II, Panel C). Fig. 4 shows the VLDL-C/HDL-C ratios for mice expressing different levels of apoE3 and apoE4 as a function of the relative plasma cholesterol level. The ratio is strongly dependent on total plasma cholesterol level and decreases progressively to a very low value as cholesterol is cleared from the plasma compartment by apoE3. The situation for apoE4 is similar at lower expression levels but when the relative plasma cholesterol level decreases below ~50%, the VLDL-C/HDL-C ratio does not decrease and remains in the range 1-2.
Figure 4.
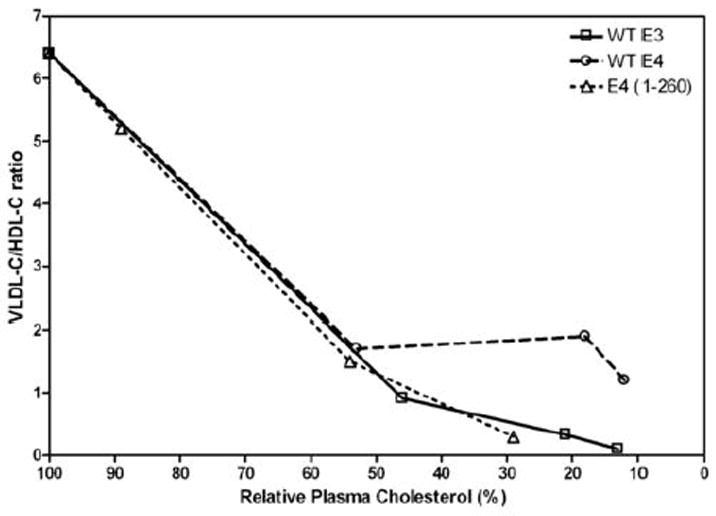
Influence of apoE structure on the distribution of lipoprotein cholesterol. Lipoprotein cholesterol profiles of the type depicted in Fig. 3 obtained using plasma from mice treated with different apoE AAV8 doses were analyzed for the distributions of VLDL-cholesterol (VLDL-C) and HDL-cholesterol (HDL-C) (elution volume = 15-20 ml). The VLDL-C/HDL-C ratios at each dose of apoE3 and apoE4 AAV8 are shown in Supplementary Figure II. The resultant VLDL-C/HDL-C ratios are plotted here as a function of the relative plasma cholesterol level in the mice treated with the different AAV8 doses (cf. Fig. 1, top panel). Comparison of the data for WT apoE4 and apoE4 (1-260) gives an indication of the influence of residues 261-299 in apoE4 on the VLDL-C/HDL-C ratio.
Influence of C-terminal domain on apoE functionality
The presence of the C-terminal domain (residues 192-299) is vital for the ability of apoE3 to reduce plasma cholesterol because expression of the isolated N-terminal helix bundle domain (residues 1-191) at very high dose fails to reduce plasma cholesterol (Supplementary Fig. III). In contrast, the same structural manipulation only partially decreases the ability of apoE4 to lower plasma cholesterol (Supplementary Fig. III). To understand the contribution of the C-terminal domain in more detail, we examined the effects of deleting segments of this domain of apoE3 and apoE4 on the abilities of the two isoforms to reduce plasma cholesterol levels when expressed in apoE-null mice. The results are summarized in Table 1. Deletion of either residues 273-299 or 192-260 in both apoE isoforms has little or no effect on the plasma cholesterol-lowering abilities. In marked contrast, removal of residues 261-272 has major effects. Thus, the effectiveness of the 1-260 and Δ261-272 variants is reduced, with the reduction being greater for apoE3 than for apoE4. The contribution of residues 261-272 to the cholesterol-lowering ability of apoE is emphasized by the fact that a three-times higher AAV8 dose of 1E11 gc is required to give the plasma cholesterol reductions for the Δ261-272 variants listed in Table I. This need for a higher expression level of the Δ261-272 variants is not due to reduced appearance of these proteins in the plasma compartment. At an AAV8 dose of 1E11gc, the plasma concentrations of apoE3 (Δ261-272) and apoE4 (Δ261-272) are 33.8 ± 13.4 (n=5) and 34.0 ± 2.0 μg/ml (n=3) (mean ± SEM), respectively; these values are similar to the concentration of ~ 20μg/ml observed with apoE3 and apoE4 at the same AAV8 dose. This result proves that removal of residues 261-272 has a direct effect on the functionality of apoE in the plasma compartment. The poor performance of apoE3 (Δ261-272) and apoE3 (1-260) in reducing plasma cholesterol is not due to a loss of ability of these molecules to bind to the LDLR (Supplementary Table I). All the apoE3 and apoE4 C-terminal variants can bind to the LDLR, as might be expected given that the receptor recognition site (residues 136-150) is located in the N-terminal helix bundle domain. The reductions in plasma total cholesterol induced by the apoE C-terminal variants (Table I) are accompanied by decreases in the VLDL-C/HDL-C ratio, with the dependence of this ratio on relative plasma cholesterol level being generally similar to that shown for apoE3 in Fig. 4. It is noteworthy that removal of residues 261-299 eliminates the aberrant behavior of apoE4 and allows the VLDL-C/HDL-C ratio to decrease to the low value observed with apoE3 (cf. data for apoE4 and apoE4 (1-260) in Fig. 4). This effect occurs because, as shown in Fig. 3D, the fractional conversion of VLDL to IDL + LDL is greater with apoE4 (1-260) than with apoE4 and similar to that seen with apoE3. As a consequence, VLDL-C is decreased and HDL-C is increased so that the VLDL-C/HDL-C ratio is reduced.
Table 1.
Comparison of effects of C-terminal domain alterations on plasma cholesterol-lowering abilities of apoE3 and apoE4
| ApoE variant* | % reduction in plasma cholesterol† | relative efficiency of chol. reduction‡ | ratio of apoE isoform efficiencies | ||
|---|---|---|---|---|---|
|
| |||||
| apoE3 | apoE4 | apoE3 | apoE4 | apoE3 / apoE4 | |
| 1 – 299 | 79 ± 4 | 82 ± 2 | 1.0 | 1.0 | 1.0 |
| 1 – 272 | 81 ± 5 | 84 ± 6 | 1.0 | 1.0 | 1.0 |
| 1 – 260 | 16 ± 6 | 46 ± 3 | 0.2 | 0.6 | 0.33 |
| Δ 261 - 272 | 48 ± 3 | 81 ± 18 | 0.6 | 0.9 | 0.67 |
| Δ 192 – 260 | 76 ± 20 | 72 ± 18 | 1.0 | 0.9 | 1.1 |
| 1 - 191 | 2 ± 7 | 27 ± 8 | 0 | 0.3 | - |
The AAV8 dose was 3E10 gc except for apoE Δ261-272 and apoE (1-191) where the doses were 1E11 and 1E12 gc, respectively.
Values (mean ± SEM) were obtained from independent experiments with 5 apoE-null mice/group. In some cases, multiple (n) independent experiments were performed: n=2 for apoE4 (1-299) and apoE3 (Δ261-272); n=3 for apoE4 (1-260); n=4 for apoE4 (1-299), apoE3 (1-191) and apoE4 (1-191). ANOVA followed by Dunnett’s multiple comparison test indicates that within the apoE3 variants the reductions in plasma cholesterol for 1-260, Δ261-272 and 1-191 are significantly different (p<0.05) from the 1-299 value. Similarly, for the apoE4 variants the values for 1-260 and 1-191 are significantly lower than the 1-299 value. Comparisons between apoE3 and apoE4 variants by unpaired t-test indicates that the plasma cholesterol reductions for the 1-260, Δ261-272 and 1-191 variants are significantly different.
The relative efficiencies of plasma cholesterol reduction are normalized to the value for wild-type apoE (apoE (1-299)). The reference value for the apoE (Δ261-272) variants where the AAV8 dose was 1E11 gc is the reduction in plasma cholesterol (87%) for apoE (1-299) at the same dose. The reference value for the apoE4 (1-191) calculation is the same; the reductions in plasma cholesterol for 1E11gc and 1E12gc AAV8 doses of apoE (1-299) are the same.
As mentioned above, apoE4 is exceptional in giving rise to a high VLDL-C/HDL-C ratio even when expressed at high doses (Fig. 4). Removal of C-terminal residues 273-299 to give the apoE4 (1-272) variant normalizes this behavior and the VLDL-C/HDL-C distribution becomes like that of apoE3 (Fig. 5). Importantly, deletion of these residues from apoE4 does not interfere with the ability to reduce plasma cholesterol (Table I) but only eliminates the more pro-atherogenic lipoprotein-cholesterol distribution.
Figure 5.
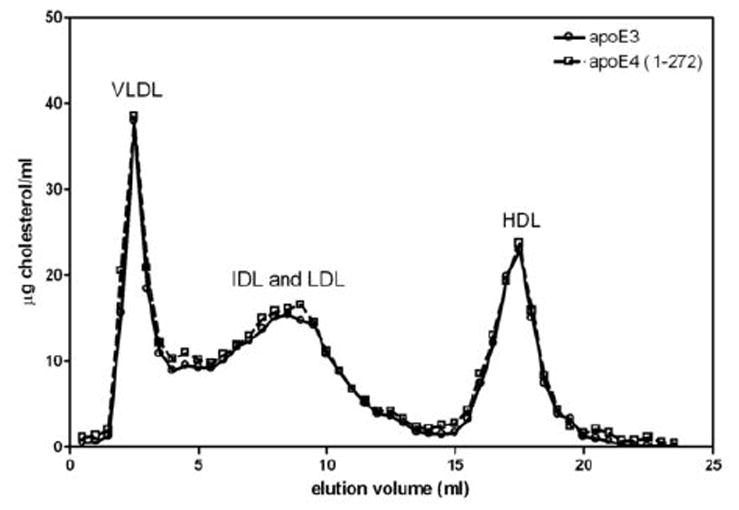
Distribution of lipoprotein cholesterol in apoE-null mice treated with AAV8 to express either human apoE3 or apoE4 (1-272). The experimental conditions were the same as those described in the legend to Fig. 2.
DISCUSSION
Plasma cholesterol-lowering abilities of apoE3 and apoE4
The finding that AAV8-mediated hepatic expression of human apoE3 and apoE4 corrects the hyperlipidemia in apoE-null mice is consistent with prior work with human apoE-transgenic mice 26. In both the AAV8-treated animals (Fig. 1) and the transgenic mice, the reduction in plasma cholesterol is the same with apoE3 and apoE4 expression. This observation is consistent with both isoforms binding similarly to the LDLR 7 and with this receptor being critical for VLDL remnant clearance in apoE-null mice 27. Since apoE has to bind appropriately to the VLDL remnants to mediate their uptake by the LDLR pathway, the low efficiency of plasma cholesterol reduction exhibited by apoE (1-191) variants which lack the lipid-binding C-terminal domain (Table 1) is unsurprising. Although both apoE3 (1-191) and apoE4 (1-191) can bind to VLDL, albeit poorly 10, expression of the former has no effect on plasma cholesterol whereas expression of the latter does. A possible explanation for the activity with apoE4 is that the lower stability of its helix bundle domain, due to the presence of R112 16, allows the bundle to open 12-13 permitting the receptor binding site (residues 136-150) 11 to interact with the LDLR. In contrast, the more stable helix bundle in apoE3 (1-191) does not open which eliminates LDLR binding and any reduction in plasma cholesterol.
The contributions of the helix bundle domain and various segments of the C-terminal domain in apoE3 and apoE4 to plasma cholesterol lowering are summarized in Fig. 6. It is apparent from the values of the fractional segmental contribution to cholesterol-lowering efficiency (F) that residues 261-272 make the largest contribution in both apoE isoforms. This effect of residues 261-272 is consistent with the ability of apoE to bind to lipoprotein surfaces being critical for plasma cholesterol-lowering because this segment, which forms a hydrophobic surface patch 13, is key for lipid emulsion and VLDL binding activity 10, 28. However, the plasma cholesterol-lowering ability of apoE variants does not always correlate simply with lipid and VLDL binding ability. The different F value for residues 261-272 in apoE3 and apoE4 is consistent with the structural organization of this segment being different in the two proteins 18 but it is not clear at this time why F is larger for apoE3. The finding that F = 0 for residues 273-299 (Fig. 5) indicates that the VLDL binding ability as measured in vitro and the ability to reduce plasma cholesterol in vivo do not correlate well because deletion of these residues significantly reduces VLDL binding in vitro 10 but does not impair cholesterol lowering in vivo. A possible explanation for this apparent discrepancy is that there is a threshold level of apoE on VLDL required for clearance by the LDLR and removal of residues 273-299 does not reduce the VLDL apoE content below the threshold.
Figure 6.
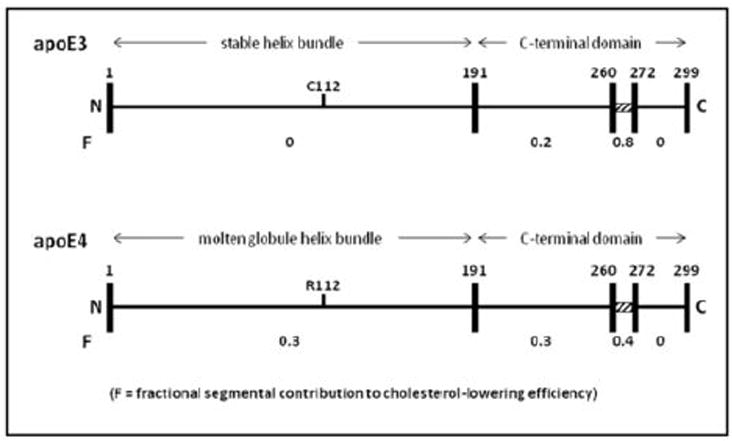
Contributions of different regions of the apoE3 and apoE4 molecules to their abilities to clear plasma cholesterol. The linear representations of the amino acid sequences show the position in the N-terminal helix bundle domain of the C112R substitution that distinguishes the two isoforms. The fractional contribution to plasma cholesterol-lowering (F) is indicated beneath the indicated segments of the apoE3 and apoE4 molecules. The F values are calculated from the relative efficiencies of plasma cholesterol reduction for the apoE3 and apoE4 C-terminal truncation variants (1-272), (1-260) and (1-191) listed in Table 1. For instance, the F value (0.8) of the segment spanning residues 261-272 in apoE3 is obtained by subtracting the cholesterol-lowering efficiency of apoE3 (1-260) (0.2) from that of apoE3 (1-272) (1.0). The F values derived in this fashion are different from the values inferred from the behavior of the apoE variants containing internal deletions (Δ192-260 and Δ261-272) probably because, compared to the simple removal of residues from the C-terminal end of the molecule, removal of these internal segments leads to greater structural reorganization of the entire C-terminal domain. The importance of the segment spanning residues 261-272 is highlighted by the hatched rectangle marking this section of the apoE molecule. See text for further details.
Differential effects of apoE3 and apoE4 on plasma lipoprotein cholesterol distribution
Besides being critical for the ability to reduce plasma cholesterol, the relative lipid- and lipoprotein-binding abilities of apoE3 and apoE4 have important consequences for the distribution of cholesterol between the VLDL and HDL fractions of plasma. The scheme in Fig. 7 presents the mechanistic basis for the higher VLDL-C/HDL-C ratio found in apoE4-expressing mice compared to animals expressing the same level of apoE3 and having the same plasma total cholesterol level (Fig. 2); this effect is not peculiar to AAV8-treated mice because the VLDL-C level is also higher in apoE4 transgenic mice compared to their apoE3 counterparts 26. The critical effect of the apoE content of VLDL remnants on LPL-mediated lipolysis is well established. This apoE content has to be sufficient to support LDLR binding but not too high otherwise lipolysis is inhibited 29-31 and HDL formation by release of excess surface components 32 is reduced. The inhibition of LPL activity probably occurs because the apoC-II cofactor is displaced from the surface of the VLDL particle 5, 33. ApoE3 binds better to HDL than to VLDL 9-10 and is apparently distributed appropriately between these lipoproteins in vivo. As a consequence, progress down the lipolytic cascade is optimal (Fig. 3D) so that there is efficient formation of small VLDL remnants possessing apoE3 content suitable for effective binding to the LDLR and removal from plasma 6. The result for apoE3-expressing mice is that a relatively low VLDL-C/HDL-C ratio is attained (represented by the ratio c/d in Fig. 7). The greater lipid-binding ability of apoE4 (due to the altered conformation of the segment spanning residues 261-272 18) increases the concentration of apoE4 on the VLDL surface so that more apoC-II is displaced and lipolysis is inhibited relative to the apoE3 situation. Thus, in apoE4-expressing animals the steady state VLDL-C/HDL-C distribution is relatively high (represented by the ratio a/b in Fig. 7). The critical role for the ability of apoE to partition appropriately between VLDL and HDL during the lipolytic cascade is supported by the observation that apoE4 (1-272) behaves more like apoE3 than apoE4 (Fig. 5). Removal of residues 273-299 from apoE4 reduces VLDL binding to a level below that of apoE3 10 so that the in vivo lipolytic processing of VLDL remnants is not impaired in mice expressing apoE4 (1-272). This result suggests that pharmacological intervention in apoE4 subjects to reduce the apoE content of VLDL could be beneficial therapeutically.
Figure 7.
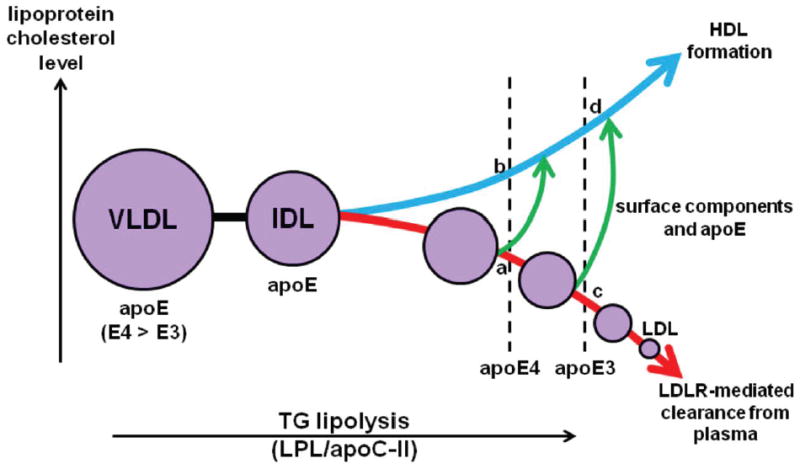
Schematic comparing the influence of apoE3 and apoE4 on the lipolysis cascade involved in the catabolism of VLDL particles. After secretion from the liver into the plasma compartment, the triglyceride (TG) in VLDL is hydrolyzed by lipoprotein lipase (LPL) with apoC-II as a cofactor leading to the creation of intermediate density lipoprotein (IDL) and progressively smaller remnant particles. ApoE bound to these particles mediates their interaction with the LDLR and clearance from plasma (the lower curved arrow shows the decrease in VLDL and remnant cholesterol levels). As the VLDL/IDL remnants shrink due to the removal of core TG, excess surface components (phospholipid, cholesterol and apoE) are released into the HDL pool (upper curved arrow shows the increase in HDL cholesterol level). ApoE3 partitions between the VLDL and HDL pools so that lipolysis, clearance of VLDL remnant cholesterol and HDL formation is optimal. In the diagram, points c and d represent the VLDL/IDL-cholesterol and HDL-cholesterol levels, respectively, when apoE3 is expressed. Relative to apoE3, apoE4 binds more to VLDL because of its higher lipid affinity leading to inhibition of lipolysis (probably because of displacement of apoC-II) so that, at the same apoE expression level, progression down the lipolysis cascade is relatively limited in the case of apoE4 (Fig. 3D). The ratio a/b represents the apoE4 VLDL-C/HDL-C ratio which is higher than the apoE3 ratio c/d (as seen in the experimental results in Fig. 2, 4, and Supplementary Fig. II). It should be noted that the alternate ABCA1 pathway for production of HDL is not included in the scheme presented here. However, this pathway cannot contribute to the different HDL levels found with apoE3 and apoE4 expression because both isoforms interact identically with ABCA1 and produce nascent HDL particles at the same rate 36. See text for further details.
In summary, relative to apoE3-expressing mice, the apoE4 mice exhibit a pro-atherogenic lipoprotein profile which explains the increased atherosclerosis seen in such animals 26. The mechanistic causes for this pathological effect associated with the presence of apoE4 are: (1) the structural change of the segment spanning residues 261-272 caused by the C112R substitution in apoE4, (2) the resultant increased lipid binding ability of apoE4 relative to apoE3 that leads to changes in the apoE content of VLDL, (3) impaired lipolytic processing, and (4) reduced VLDL remnant clearance from the plasma compartment and movement of excess VLDL surface components into the HDL pool. Knowledge of these molecular mechanisms that underlie the ability of apoE to reduce plasma cholesterol may aid in the future development of effective apoE-based gene therapy approaches to reducing atherosclerosis 34-35.
Supplementary Material
Acknowledgments
We thank Dawn Marchadier, David Nguyen, Margaret Nickel, Valeska Redon and Maosen Sun for expert assistance.
SOURCES OF FUNDING
This work was supported by National Institutes of Health Grant HL 56083 and the Gene Therapy Resource Program of the National Heart, Lung and Blood Institute.
Abbreviations
- Apo
apolipoprotein
- AAV
adeno-associated virus
- HDL
high density lipoprotein
- LDL
low density lipoprotein
- LDLR
low density lipoprotein receptor
- TG
triglyceride
- VLDL
very low density lipoprotein
Footnotes
DISCLOSURES
None.
References
- 1.Getz GS, Reardon CA. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J Lipid Res. 2009;50:S156–S161. doi: 10.1194/jlr.R800058-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lund-Katz S, Phillips MC. High density lipoprotein structure-function and role in reverse cholesterol transport. Subcell Biochem. 2010;51:183–227. doi: 10.1007/978-90-481-8622-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hauser PS, Narayanaswami V, Ryan RO. Apolipoprotein E: from lipid transport to neurobiology. Prog Lipid Res. 2011;50:62–74. doi: 10.1016/j.plipres.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: Structure determines function -from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. 2009;50:S183–S188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahley RW, Huang Y, Rall SC. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia): questions, quandaries, and paradoxes. J Lipid Res. 1999;40:1933–1949. [PubMed] [Google Scholar]
- 6.Fielding CJ, Fielding PE. Dynamics of lipoprotein transport in the circulatory system. In: Vance DE, Vance JE, editors. Biochemistry of Lipids, Lipoproteins and Membranes. Amsterdam: Elsevier; 2008. [Google Scholar]
- 7.Weisgraber KH, Innerarity TL, Mahley RW. Abnormal lipoprotein receptor-binding activity of the human E apoprotein due to cysteine-arginine interchange at a single site. J Biol Chem. 1982;257:2518–2521. [PubMed] [Google Scholar]
- 8.Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis. 1988;8:1–21. doi: 10.1161/01.atv.8.1.1. [DOI] [PubMed] [Google Scholar]
- 9.Weisgraber KH. Apolipoprotein E distribution among human plasma lipoproteins: role of the cysteine-arginine interchange at residue 112. J Lipid Res. 1990;31:1503–1511. [PubMed] [Google Scholar]
- 10.Nguyen D, Dhanasekaran P, Nickel M, Nakatani R, Saito H, Phillips MC, Lund-Katz S. Molecular basis for the differences in lipid and lipoprotein binding properties of human apolipoproteins E3 and E4. Biochemistry. 2010;49:10881–10889. doi: 10.1021/bi1017655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weisgraber KH. Apolipoprotein E: structure-function relationships. Adv Protein Chem. 1994;45:249–302. doi: 10.1016/s0065-3233(08)60642-7. [DOI] [PubMed] [Google Scholar]
- 12.Saito H, Lund-Katz S, Phillips MC. Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Prog Lipid Res. 2004;43:350–380. doi: 10.1016/j.plipres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Li Q, Wang J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc Natl Acad Sci U S A. 2011;108:14813–14818. doi: 10.1073/pnas.1106420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen D, Dhanasekaran P, Phillips MC, Lund-Katz S. Molecular mechanism of apolipoprotein E binding to lipoprotein particles. Biochemistry. 2009;48:3025–3032. doi: 10.1021/bi9000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizuguchi C, Hata M, Dhanasekaran P, Nickel M, Phillips MC, Lund-Katz S, Saito H. Fluorescence Analysis of the Lipid Binding-Induced Conformational Change of Apolipoprotein E4. Biochemistry. 2012;51:5580–5588. doi: 10.1021/bi300672s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, Weisgraber KH. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry. 2000;39:11657–11666. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 17.Morrow JA, Hatters DM, Lu B, Hocht P, Oberg KA, Rupp B, Weisgraber KH. Apolipoprotein E4 forms a molten globule. J Biol Chem. 2002;277:50380–50385. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- 18.Sakamoto T, Tanaka M, Vedhachalam C, Nickel M, Nguyen D, Dhanasekaran P, Phillips MC, Lund-Katz S, Saito H. Contributions of the carboxyl-terminal helical segment to the self-association and lipoprotein preferences of human apolipoprotein E3 and E4 isoforms. Biochemistry. 2008;47:2968–2977. doi: 10.1021/bi701923h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitajima K, Marchadier DH, Miller GC, Gao GP, Wilson JM, Rader DJ. Complete prevention of atherosclerosis in apoE-deficient mice by hepatic human apoE gene transfer with adeno-associated virus serotypes 7 and 8. Arterioscler Thromb Vasc Biol. 2006;26:1852–1857. doi: 10.1161/01.ATV.0000231520.26490.54. [DOI] [PubMed] [Google Scholar]
- 20.Alexander ET, Tanaka M, Kono M, Saito H, Rader DJ, Phillips MC. Structural and functional consequences of the Milano mutation (R173C) in human apolipoprotein A-I. J Lipid Res. 2009;50:1409–1419. doi: 10.1194/jlr.M800578-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexander ET, Vedhachalam C, Sankaranarayanan S, de la Llera-Moya M, Rothblat GH, Rader DJ, Phillips MC. Influence of Apolipoprotein A-I Domain Structure on Macrophage Reverse Cholesterol Transport in Mice. Arterioscler Thromb Vasc Biol. 2011;31:320–327. doi: 10.1161/ATVBAHA.110.216226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 23.Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 24.Tsukamoto K, Maugeais C, Glick JM, Rader DJ. Markedly increased secretion of VLDL triglycerides induced by gene transfer of apolipoprotein E isoforms in apoE-deficient mice. J Lipid Res. 2000;41:253–259. [PubMed] [Google Scholar]
- 25.Zannis VI, Chroni A, Kypreos KE, Kan HY, Cesar TB, Zanni EE, Kardassis D. Probing the pathways of chylomicron and HDL metabolism using adenovirus-mediated gene transfer. Curr Opin Lipidol. 2004;15:151–166. doi: 10.1097/00041433-200404000-00008. [DOI] [PubMed] [Google Scholar]
- 26.Knouff C, Hinsdale ME, Mezdour H, Altenburg MK, Watanabe M, Quarfordt SH, Sullivan PM, Maeda N. Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J Clin Invest. 1999;103:1579–1586. doi: 10.1172/JCI6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kypreos KE, Zannis VI. LDL receptor deficiency or apoE mutations prevent remnant clearance and induce hypertriglyceridemia in mice. J Lipid Res. 2006;47:521–529. doi: 10.1194/jlr.M500322-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Dong LM, Wilson C, Wardell MR, Simmons T, Mahley RW, Weisgraber KH, Agard DA. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J Biol Chem. 1994;269:22358–22365. [PubMed] [Google Scholar]
- 29.Rensen PC, van Berkel TJ. Apolipoprotein E effectively inhibits lipoprotein lipase-mediated lipolysis of chylomicron-like triglyceride-rich lipid emulsions in vitro and in vivo. J Biol Chem. 1996;271:14791–14799. doi: 10.1074/jbc.271.25.14791. [DOI] [PubMed] [Google Scholar]
- 30.Jong MC, Dahlmans VE, Hofker MH, Havekes LM. Nascent very-low-density lipoprotein triacylglycerol hydrolysis by lipoprotein lipase is inhibited by apolipoprotein E in a dose-dependent manner. Biochem J. 1997;328(Pt 3):745–750. doi: 10.1042/bj3280745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Y, Liu XQ, Rall SC, Jr, Taylor JM, von Eckardstein A, Assmann G, Mahley RW. Overexpression and accumulation of apolipoprotein E as a cause of hypertriglyceridemia. J Biol Chem. 1998;273:26388–26393. doi: 10.1074/jbc.273.41.26388. [DOI] [PubMed] [Google Scholar]
- 32.Patsch JR, Gotto AM, Jr, Olivercrona T, Eisenberg S. Formation of high density lipoprotein2-like particles during lipolysis of very low density lipoproteins in vitro. Proc Natl Acad Sci U S A. 1978;75:4519–4523. doi: 10.1073/pnas.75.9.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weisgraber KH, Mahley RW, Kowal RC, Herz J, Goldstein JL, Brown MS. Apolipoprotein C-I modulates the interaction of apolipoprotein E with beta-migrating very low density lipoproteins (beta-VLDL) and inhibits binding of beta-VLDL to low density lipoprotein receptor-related protein. J Biol Chem. 1990;265:22453–22459. [PubMed] [Google Scholar]
- 34.Rader DJ. Gene therapy for atherosclerosis. Int J Clin Lab Res. 1997;27:35–43. doi: 10.1007/BF02827240. [DOI] [PubMed] [Google Scholar]
- 35.Harris JD, Evans V, Owen JS. ApoE gene therapy to treat hyperlipidemia and atherosclerosis. Curr Opin Mol Ther. 2006;8:275–287. [PubMed] [Google Scholar]
- 36.Vedhachalam C, Narayanaswami V, Neto N, Forte TM, Phillips MC, Lund-Katz S, Bielicki JK. The C-terminal lipid-binding domain of apolipoprotein E is a highly efficient mediator of ABCA1-dependent cholesterol efflux that promotes the assembly of high-density lipoproteins. Biochemistry. 2007;46:2583–2593. doi: 10.1021/bi602407r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.