

Crystals of E. coli triosephosphate isomerase were obtained as a contaminant and its structure was determined to 1.85 Å resolution.
Keywords: crystallization contaminants, triosephosphate isomerase, TIM
Abstract
Attempts to crystallize several mammalian proteins overexpressed in Escherichia coli revealed a common contaminant, triosephosphate isomerase, a protein involved in glucose metabolism. Even with triosephosphate isomerase present in very small amounts, similarly shaped crystals appeared in the crystallization drops in a number of polyethylene glycol-containing conditions. All of the target proteins were His-tagged and their purification involved immobilized metal-affinity chromatography (IMAC), a step that was likely to lead to triosephosphate isomerase contamination. Analysis of the triosephosphate isomerase crystals led to the structure of E. coli triosephosphate isomerase at 1.85 Å resolution, which is a significant improvement over the previous structure.
1. Introduction
Protein crystallization depends on numerous factors. The crystallizability of a given protein is still difficult to predict, although protein purity is considered to be an important factor in obtaining diffracting crystals of a particular protein. The introduction of expression tags greatly improves the purity of proteins produced for structural studies through the application of affinity columns. However, the presence of even small amounts of contaminants that crystallize easily may result in crystals of the contaminants instead of the target proteins. Structure determination of these crystals significantly detours projects, diverting resources and human time. There are several examples in the literature of the crystallization of contaminants of membrane proteins (Veesler et al., 2008 ▶; Psakis et al., 2009 ▶; Panwar et al., 2012 ▶), but relatively few documented examples of the crystallization of contaminants of soluble proteins (Lohkamp & Dobritzsch, 2008 ▶). The description of common crystallization contaminants will help to identify crystals of these contaminants during the early stages of a project, saving a significant amount of time and laboratory resources.
Here, we found the same type of crystals during crystallization trials of four different proteins. Structure determination at 1.85 Å resolution identified the crystallized protein as E. coli triosephosphate isomerase (TIM). TIM is a glycolytic enzyme that catalyzes the isomerization of dihydroxyacetone phosphate (DHAP) to d-glyceraldehyde 3-phosphate (GAP) (for a recent review, see Wierenga et al., 2010 ▶). In all cases, TIM crystallized in a range of polyethylene glycol (PEG)-containing crystallization conditions in the same crystal form.
2. Materials and methods
2.1. Cloning, protein expression and purification
The 46.7 kDa mouse calreticulin (residues 18–416) was cloned into pET15b (Novagen) and expressed in E. coli BL21(DE3) in rich (LB) medium as an N-terminally His-tagged fusion protein. Bacterial cultures were grown at 310 K with shaking until the absorbance measured at 600 nm reached 0.7. Protein expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside and the cultures were shaken for 4 h at 303 K.
The 26.8 kDa N-terminal fragment of human PDIR (residues 29–256) was cloned into pET15b (Novagen) and expressed in E. coli BL21(DE3) in rich (LB) medium as an N-terminally His-tagged fusion protein. Bacterial cultures were grown at 310 K with shaking until the absorbance measured at 600 nm reached 0.7. Protein expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside and the cultures were shaken for 4 h at 303 K.
The 58.3 kDa human PDIR (residues 22–519) was cloned into pET23b (Novagen) and expressed in E. coli Rosetta-gami 2 in rich (LB) medium as an N-terminally His-tagged fusion protein. Bacterial cultures were grown at 310 K with shaking until the absorbance measured at 600 nm reached 0.7. Protein expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside and the cultures were shaken overnight at 289 K.
The 52.5 kDa human endo-α-1,2-mannosidase (residues 27–462) was cloned into pColdI vector (Takara) and expressed in E. coli Rosetta-gami 2 in rich (LB) medium as an N-terminally His-tagged fusion protein. Bacterial cultures were grown at 310 K with shaking until the absorbance measured at 600 nm reached 0.7. Protein expression was induced with 0.2 mM isopropyl β-d-1-thiogalactopyranoside and the cultures were shaken overnight at 288 K.
After centrifugation, the pellets were resuspended in binding buffer [50 mM HEPES, 0.5 M NaCl, 5%(v/v) glycerol, 5 mM imidazole pH 7.4] containing 1 mM phenylmethylsulfonyl fluoride, 0.1 mg ml−1 lysozyme and 0.1%(v/v) β-mercaptoethanol and lysed using sonication. Cell debris was removed by centrifugation and the fusion protein was bound to Ni2+-charged Chelating Sepharose (GE Healthcare) or Ni–NTA Agarose (Qiagen) beads, washed with binding buffer containing 30 mM imidazole and eluted with buffer containing 250 mM imidazole.
The His tag was cleaved from PDIR (29–256) with thrombin (GE Healthcare) overnight at 277 K. The resulting PDIR (29–256) construct contained an additional N-terminal Gly-Ser-His-Met sequence. The tag was left uncleaved for the other three proteins. The proteins were additionally purified by size-exclusion chromatography using HPLC buffer (10 mM HEPES, 100 mM NaCl, 5 mM dithiothreitol pH 7.0 for the PDIR constructs, 20 mM Tris, 100 mM NaCl, 3 mM CaCl2 pH 7.5 for calreticulin and 10 mM MES, 100 mM NaCl, 3 mM CaCl2 pH 6.0 for endo-α-1,2-mannosidase).
2.2. Crystallization, data collection and processing
Crystallization conditions were identified in hanging-drop vapour-diffusion experiments using the commercial screens The Classics II, PACT and JCSG+ Suites (Qiagen). Crystals were obtained at 294 K by equilibrating a drop consisting of 0.6 µl target protein solution (2–12 mg ml−1) in HPLC buffer mixed with 0.6 µl reservoir solution (Table 1 ▶) against 0.6 ml reservoir solution. The solution used for cryoprotection consisted of the reservoir solution with the addition of 20%(v/v) glycerol. For data collection, crystals were picked up in a nylon loop and flash-cooled in an N2 cold stream (Oxford Cryosystem). The best data set was collected using an ADSC Quantum 210 CCD detector (Area Detector Systems Corp.) on beamline A1 at the Cornell High-Energy Synchrotron Source (CHESS) using a single-wavelength (0.9767 Å) regime (Table 2 ▶). Data processing and scaling were performed with HKL-2000 (Otwinowski & Minor, 1997 ▶).
Table 1. Crystallization conditions for the observed TIM crystals.
| Crystallization conditions | Diffraction resolution (Å) |
|---|---|
| 0.1 M bis-tris pH 6.5, 20% PEG 5000 MME | 1.85 |
| 0.1 M bis-tris pH 6.5, 25% PEG 3350 | 2.1 |
| 0.1 M bis-tris pH 5.5, 25% PEG 3350 | — |
| 0.1 M ammonium acetate, 0.1 M bis-tris pH 5.5, 17% PEG 10 000 | 2.5 |
| 0.2 M lithium chloride, 0.1 M MES pH 6.0, 20% PEG 6000 | 3.0 |
| 0.01 M zinc chloride, 0.1 M Tris pH 8.0, 20% PEG 6000 | — |
| 0.2 M MES pH 6.4, 22% PEG 6000† | 2.6 |
From Noble et al. (1993 ▶).
Table 2. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| X-ray wavelength (Å) | 0.9767 |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 46.47, b = 67.45, c = 150.35 |
| Resolution range (Å) | 50.0–1.85 (1.88–1.85) |
| No. of reflections | 38754 |
| Completeness (%) | 99.5 (93.7) |
| Multiplicity | 5.2 (3.1) |
| 〈I/σ(I)〉 | 21.0 (3.3) |
| R merge † | 0.085 (0.404) |
| Model refinement | |
| R work/R free ‡ | 0.172/0.206 |
| No. of non-H atoms | |
| Protein | 3827 |
| Water | 474 |
| Average B factor (Å2) | |
| Protein | 11.2 |
| Water | 31.0 |
| R.m.s. deviations from ideal geometry | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.04 |
| Ramachandran plot | |
| Favoured (%) | 93.5 |
| Allowed (%) | 6.5 |
| Outliers (%) | 0.0 |
R
merge = 
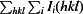 , where I
i(hkl) is the intensity of the ith observation of reflection hkl,
, where I
i(hkl) is the intensity of the ith observation of reflection hkl, 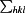 is the sum over all reflections and
is the sum over all reflections and 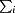 is the sum over i measurements of reflection hkl.
is the sum over i measurements of reflection hkl.
R = 
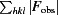 ∑, where R
free is calculated for a randomly chosen 5% of reflections which were not used for structure refinement and R
work is calculated for the remaining reflections.
∑, where R
free is calculated for a randomly chosen 5% of reflections which were not used for structure refinement and R
work is calculated for the remaining reflections.
2.3. Structure determination and refinement
The search model for molecular replacement was identified by wide-search molecular replacement (Stokes-Rees & Sliz, 2010 ▶). The E. coli triosephosphate isomerase structure (PDB entry 1tre; Noble et al., 1993 ▶) was the best hit and provided the starting phases when used as a search model in Phaser (McCoy et al., 2007 ▶). The resulting model was extended manually with the help of the program Coot (Emsley & Cowtan, 2004 ▶) and was improved by several cycles of refinement using REFMAC (Murshudov et al., 2011 ▶) followed by translation–libration–screw (TLS) refinement (Winn et al., 2003 ▶). The final model has good stereochemistry, with no outliers in the Ramachandran plot computed using PROCHECK (Laskowski et al., 1993 ▶). Figures were produced with PyMOL (http://www.pymol.org). The coordinates and structure factors have been deposited in the RCSB Protein Data Bank (entry 4iot).
3. Results and discussion
We performed crystallization screening of four different proteins (mouse calreticulin, human endo-α-1,2-mannosidase and two constructs of human PDIR) over a period of two years. In all of these cases we obtained similar-looking crystals with the shape of long thin rods (Fig. 1 ▶ a). There were typically very few crystals in the drop, which is likely to reflect a low amount of available protein. In some cases the crystals were surrounded by precipitate. We were able to acquire diffraction data sets ranging from 1.85 to 3.0 Å resolution and we attempted to solve the structures by molecular replacement using search models of higher then 35% sequence identity to the target protein. Unexpectedly, molecular replacement did not yield a solution even in cases in which a fragment of the same protein (100% identity) was used as a search model. Eventually, comparison of the unit-cell parameters and space groups of the collected data sets made it apparent that the crystals had the same identity and were likely to contain a protein from E. coli, the expression host. Notably, all of these crystallization conditions contained PEGs of different molecular weights as a precipitant, while there was no dependence on the buffer or salts present in the crystallization drops (Table 1 ▶).
Figure 1.

(a) Triosephosphate isomerase crystals. The figure represents a portion of the crystallization drop in the PDIR crystallization screen. The typical length of the TIM crystals is 100–400 µm. (b) SDS–PAGE of PDIR (22–519) and endo-α-1,2-mannosidase crystallization samples after two-step purification. The main bands correspond to the expected molecular weights of the target proteins. The protein band at 27 kDa corresponds to triosephosphate isomerase. Lane 1 contains molecular-weight markers (labelled in kDa).
Owing to the very few crystals that were obtained in each case, we could not analyze the crystals themselves by mass spectrometry. Thus, we analyzed the protein sample that was used for the crystallization of endo-α-1,2-mannosidase. The results revealed the presence of several E. coli proteins as impurities, including the 60 kDa chaperonin GroEL, glutamyl-tRNA synthetase, glucosamine–fructose-6-phosphate aminotransferase and ATP synthase subunit α. Unfortunately, using the structures of these proteins as search models did not result in a molecular-replacement solution either. Finally, the solution was found using wide-search molecular replacement (WSMR), a brute-force method that uses up to 100 000 domains present in the Protein Data Bank as search models for molecular replacement (Stokes-Rees & Sliz, 2010 ▶). WSMR yielded more than 100 hits, with the best being the triosephosphate isomerase from E. coli (PDB entry 1tre; Noble et al., 1993 ▶), with an LLG of 268 and a TFZ score of 23.5. We applied crystallization samples of the PDIR constructs and endo-α-1,2-mannosidase to SDS–PAGE and observed a common band at approximately 27 kDa that corresponds to the molecular weight of TIM (Fig. 1 ▶ b). We used the relative intensity of the bands to estimate the concentration of TIM in the crystallization drops, which ranged from approximately 0.1 mg ml−1 in the PDIR (22–519) sample to as high as 1 mg ml−1 in the PDIR (29–256) sample. The time required for the appearance of the crystals strongly depended on the concentration of TIM. In cases in which the target protein solution was heavily contaminated with TIM (0.5–1 mg ml−1), the crystals appeared in a matter of few days. When the concentration of TIM was very low (approximately 0.1 mg ml−1), it typically took more than a month for the crystals to appear.
Understanding how contamination with TIM occurred when purifying these particular protein targets is necessary in order to eliminate it from future crystallization experiments. TIM is not implicated in protein–protein interactions and the target proteins have very different structures that would not provide a similar scaffold for interaction with TIM. However, all of the target proteins contained a His tag to facilitate their purification, and a recent study has identified TIM as a common contaminant in immobilized metal-affinity chromatography (IMAC) purification (Tiwari et al., 2010 ▶). Thus, it is very likely that TIM was enriched during the purification step on Ni2+-charged Sepharose beads. Notably, a common trait of the target proteins used in this work is their tendency to aggregate, resulting in their partial precipitation during concentration prior to the crystallization trial. On the other hand, TIM is a well behaved and stable protein, which would lead to its enrichment in the protein sample. There was no apparent dependence of the presence of TIM on the expression conditions or on the E. coli strains (BL21 and Rosetta-gami 2) used in this work. Mass spectrometry performed on one of the crystallized samples (endo-α-1,2-mannosidase) did not detect TIM. It is possible that the stable TIM-barrel fold is resistant to trypsin digestion, which prevented its detection.
This work shows that a single size-exclusion chromatography step following IMAC purification is often insufficient to separate TIM from target proteins and that additional steps of purification would be beneficial to eliminate TIM and other impurities. For instance, we envision ion exchange to be effective in the purification of positively charged proteins, as E. coli TIM is an acidic protein with a theoretical pI of 5.64.
E. coli TIM crystallized as a dimer with two molecules in the asymmetric unit. The structure shows the typical TIM-barrel fold with eight β-strand–α-helix segments, in which the β-strands form a barrel in a parallel arrangement and the eight α-helices form the outer layer of the fold (Fig. 2 ▶ a). The dimer contains two independent active sites, in which all of the catalytic residues in each active site are from a corresponding protomer. The only available structure of wild-type E. coli TIM in the PDB is at 2.6 Å resolution (Noble et al., 1993 ▶). The structure obtained in this work improves the resolution of the E. coli enzyme; the two structures are highly similar, with an r.m.s.d. of 0.3 Å. Superposition of the structures shows conformational variations in several loops. The most significant difference occurs in the loop following the catalytic residue Glu167. This is the loop that closes on the substrate upon its binding and the observed differences underline the dynamic nature of this loop. There was unidentified density in one of the active sites of triosephosphate isomerase. The density is positioned in a position that is usually occupied by a phosphate moiety of the substrate (Fig. 2 ▶ b). In the refined structure we modelled this density as a sulfate ion with half occupancy and a water molecule. It is possible that this density is from the sulfonic acid moiety of 2-(N-morpholino)ethanesulfonic acid (MES), which was buffering the protein solution at a 10 mM concentration.
Figure 2.

Structure of E. coli triosephosphate isomerase. (a) View of the dimer comprised of TIM-barrel folds. (b) Enlarged view of the active sites of overlaid E. coli TIM structures from this work (yellow) and the previously determined structure (PDB entry 1tre; cyan). Loop 6 adopts a different conformation in the structures. Residues that are involved in the catalytic activity of TIM are shown as sticks.
A number of proteins are known to be common contaminants in IMAC-mediated purification (for a review, see Bolanos-Garcia & Davies, 2006 ▶). However, this list is incomplete and the effect of the presence of these impurities in crystallization experiments has not been studied. In this work, we report TIM to be a common crystallization contaminant of His-tagged target proteins produced in E. coli. Notably, TIM crystallized in the same P212121 space group with very similar unit-cell parameters; initial indexing should help in the identification of TIM crystals in future crystallization trials. When sporadic crystals are obtained and molecular replacement with the expected protein fails, triosephosphate isomerase should be one of the first proteins to try as a search model for molecular replacement. It is important to identify contaminating TIM crystals early in a crystallization project in order to avoid the unnecessary and time-consuming optimization and experimental phasing of these crystals.
Supplementary Material
PDB reference: triosephosphate isomerase, 4iot
Acknowledgments
RV acknowledges a fellowship from the Natural Sciences and Engineering Research Council of Canada (NSERC). We thank Dr Akira Seko (ERATO Ito Glycotrilogy Project, Japan) for the gift of the endo-α-1,2-mannosidase plasmid and Dr Lloyd Ruddock (University of Oulu, Finland) for the gift of the PDIR (22–519) plasmid. This work was funded by Canadian Institutes of Health Research (CIHR) Genomics grant GSP-48370. Data were acquired at the Macromolecular Diffraction (MacCHESS) facility at the Cornell High Energy Synchrotron Source (CHESS). CHESS is supported by the NSF and NIH/NIGMS via NSF award DMR-0225180 and the MacCHESS resource is supported by NIH/NCRR award RR-01646.
References
- Bolanos-Garcia, V. M. & Davies, O. R. (2006). Biochim. Biophys. Acta, 1760, 1304–1313. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lohkamp, B. & Dobritzsch, D. (2008). Acta Cryst. D64, 407–415. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Noble, M. E. M., Zeelen, J. P., Wierenga, R. K., Mainfroid, V., Goraj, K., Gohimont, A.-C. & Martial, J. A. (1993). Acta Cryst. D49, 403–417. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Panwar, P., Deniaud, A. & Pebay-Peyroula, E. (2012). Acta Cryst. D68, 1272–1277. [DOI] [PubMed]
- Psakis, G., Polaczek, J. & Essen, L.-O. (2009). J. Struct. Biol. 166, 107–111. [DOI] [PubMed]
- Stokes-Rees, I. & Sliz, P. (2010). Proc. Natl Acad. Sci. USA, 107, 21476–21481. [DOI] [PMC free article] [PubMed]
- Tiwari, N., Woods, L., Haley, R., Kight, A., Goforth, R., Clark, K., Ataai, M., Henry, R. & Beitle, R. (2010). Protein Expr. Purif. 70, 191–195. [DOI] [PubMed]
- Veesler, D., Blangy, S., Cambillau, C. & Sciara, G. (2008). Acta Cryst. F64, 880–885. [DOI] [PMC free article] [PubMed]
- Wierenga, R. K., Kapetaniou, E. G. & Venkatesan, R. (2010). Cell. Mol. Life Sci. 67, 3961–3982. [DOI] [PMC free article] [PubMed]
- Winn, M. D., Murshudov, G. N. & Papiz, M. Z. (2003). Methods Enzymol. 374, 300–321. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: triosephosphate isomerase, 4iot