

The lumenal domain of Erv41p from S. cerevisiae has successfully been expressed, purified and crystallized. X-ray diffraction data were collected to a resolution of 2.0 Å.
Keywords: Saccharomyces cerevisiae, Erv41p, COPII, Erv proteins
Abstract
The membrane protein Erv41p is a major component of COPII-coated vesicles and is thought to play a role in the early secretory pathway in eukaryotic cells. In this study, the full lumenal domain of Erv41p from Saccharomyces cerevisiae (ScErv41p_LD) was recombinantly expressed in Sf9 insect cells and purified by nickel-affinity, ion-exchange and size-exclusion chromatography. ScErv41p_LD crystals were obtained using the sitting-drop vapour-diffusion method and native X-ray diffraction data were collected to 2.0 Å resolution. The crystals belonged to space group P21, with unit-cell parameters a = 49.8, b = 76.9, c = 65.1 Å, α = γ = 90.0, β = 104.8°.
1. Introduction
In the eukaryotic cell, all proteins destined for secretion to the extracellular environment, to organelles of the exocytotic pathway or to the plasma membrane depend on the secretory pathway for their correct localization. These proteins are synthesized on the rough endoplasmic reticulum (ER) and are simultaneously translocated into the ER lumen for folding and post-translational modification. At the point of ER exit, the secretory proteins are selectively packed into COPII-coated transport vesicles, which bud from the ER and fuse with the ER–Golgi intermediate compartment (ERGIC) in mammals or directly with the Golgi apparatus in lower eukaryotes such as yeast. Proteins that cycle between the compartments of the secretory pathway are then packaged into COPI-coated vesicles for retrograde transport, whereas the cargo proteins continue towards their final destinations. The traffic in the secretory pathway is controlled by a number of soluble or membrane-bound proteins which are resident in the ER or cycle between the ER, the ERGIC and the Golgi apparatus, as reviewed by, for example, Dancourt & Barlowe (2010 ▶).
Erv41p was first identified as a component of COPII-coated vesicles during a study that aimed to identify proteins involved in transport between the ER and Golgi in yeast (Otte et al., 2001 ▶). The protein is highly conserved across species, with an orthologue in all eukaryotes studied, and yeast Erv41p has a sequence identity of 30% to the corresponding human protein hErv41, also known as ERGIC2, (Welsh et al., 2006 ▶). Erv41p is an integral membrane protein with one predicted transmembrane helix close to each terminus of the sequence, a single large lumenal domain and short N- and C-terminal tails on the cytosolic side of the membrane (Otte et al., 2001 ▶). The C-terminal tail includes a hydrophobic sequence that controls sorting into COPII-coated vesicles for transport from the ER, but the sequence does not contain a signal for retrograde transport (Otte & Barlowe, 2002 ▶). In the cell, Erv41p is present in large heterogeneous complexes (Welsh et al., 2006 ▶). It is known to interact tightly with a related protein, Erv46p, which has a sequence identity of 28% to Erv41p and is predicted to have a similar overall topology. The Erv41p–Erv46p complex has also been shown to interact with Rot2p, a glucose-trimming enzyme localized to the ER (Welsh et al., 2006 ▶). The exact roles of Erv41p and Erv46p remain unknown, but yeast strains lacking either or both display cold-sensitivity and a reduction in ER-to-Golgi transport (Otte et al., 2001 ▶), and disruption of the Erv41p–Erv46p complex causes a mild glycoprotein-processing defect (Welsh et al., 2006 ▶).
Currently, no structural information is available for Erv41 or Erv46 from any species and they share low homology with proteins of known structure. In order to provide a better understanding of Erv41p and its potential roles in the cell, we have initiated structural studies of the protein. Here, we report the cloning, purification and crystallization of the lumenal domain of Erv41p from Saccharomyces cerevisiae (ScErv41p).
2. Experimental
2.1. Cloning
The domain topology of ScErv41p (Saccharomyces Genome Database entry YML067C) was previously reported by Otte & Barlowe (2002 ▶) and, based on this information together with secondary-structure predictions by the PSIPRED protein structure prediction server (Buchan et al., 2010 ▶), the borders of several different constructs of the lumenal part of the protein were assigned. The DNA sequences encoding these constructs were amplified by PCR using genomic S. cerevisiae S288C DNA as a template. Designed primers included NcoI and HindIII cleavage sites at the 5′ and 3′ ends, respectively. The PCR products were digested with the corresponding enzymes and ligated into the complementary digested pFHMSP-LIC N vector [a derivative of pFastBac HT A (Invitrogen); courtesy of SGC, Toronto, Canada], thus adding an N-terminal honeybee melittin signal sequence, a 6×His tag and a TEV protease cleavage site. The expression construct sequences were confirmed to be identical to YML067C by sequencing analysis. The longest construct, comprising residues 49–297 (ScErv41_LD; Fig. 1 ▶), could be expressed as soluble protein and this was used for subsequent crystallization trials.
Figure 1.

A graphical representation of full-length S. cerevisiae Erv41p with the membrane topology indicated. The lumenal domain is shown in dark grey, the transmembrane regions in light grey and the cytosolic tails in white. The construct of the lumenal domain used for crystallization, ScErv41p_LD, is shown below.
2.2. Expression and purification
ScErv41p_LD was expressed in Sf9 insect cells (Invitrogen) infected with baculovirus engineered by following the protocol previously described by Fitzgerald et al. (2006 ▶). Briefly, the DNA sequence of the pFHMSP-LIC N vector containing ScErv41p_LD flanked by LoxP sites was transposed into the baculovirus genome by transformation of Escherichia coli DH10Bac cells (Invitrogen). The baculovirus genome was isolated and 2 µg of DNA was used to transfect 0.75 × 106 Sf9 cells in 3 ml Insect-XPRESS Protein-free Insect Cell Medium with l-glutamine (Lonza) using X-tremeGENE HP DNA Transfection Reagent (Roche). First-generation virus (V0) was collected 3 d after transfection and amplified by infecting 25 ml of Sf9 (0.5 × 106 cells ml−1). Second-generation virus (V1) was collected 1 d after cell-proliferation arrest.
For protein production, Sf9 cells were grown in suspension in 2 l shaker flasks at 90 rev min−1 and 300 K using the same type of medium. To produce ScErv41p_LD, Sf9 cells (0.5 l, 0.5 × 106 cells ml−1) were infected with recombinant baculovirus (V1) and split every day to 0.5 × 106 cells ml−1 before reaching proliferation arrest. The secreted protein was collected from the medium 2 d after the day of proliferation arrest and was cleared from cells by centrifugation (30 min, 3300g, 277 K). The medium containing ScErv41p_LD was filtered (0.22 µm), concentrated using a Prep/Scale spiral-wound ultrafiltration module with 10 kDa cutoff (Millipore) followed by exchange of the buffer to binding buffer (50 mM sodium phosphate pH 8.0, 300 mM NaCl, 10 mM imidazole). The resulting solution was loaded onto a HisTrap HP column (GE Healthcare) equilibrated with binding buffer and the bound proteins were eluted with a linear imidazole gradient (10–125 mM in binding buffer). Fractions containing ScErv41p_LD were pooled and the signal sequence and 6×His tag were removed by overnight incubation with TEV protease and simultaneous dialysis against TEV protease cleavage buffer (25 mM Tris pH 8.0, 300 mM NaCl, 10 mM imidazole, 2 mM DTT). After TEV digestion ScErv41p_LD was further purified by Ni-affinity chromatography using a HisTrap HP column followed by anion-exchange chromatography on a HiTrap Q HP column (GE Healthcare) using a linear NaCl gradient (50–600 mM in 50 mM Tris pH 8.0, 2 mM DTT). Size-exclusion chromatography (SEC) was performed using a Superdex 75 16/600 column (GE Healthcare) equilibrated with SEC buffer (50 mM Tris pH 8.0, 300 mM NaCl, 5 mM DTT). All fractions containing ScErv41p_LD were pooled, dialysed against crystallization buffer (25 mM Tris pH 8.0, 50 mM NaCl, 2 mM DTT) and concentrated to 5 mg ml−1 for subsequent crystallization experiments.
2.3. Crystallization
Initial crystallization screening was performed by the sitting-drop vapour-diffusion method in 96-well plates using commercially available sparse-matrix crystal screens (Qiagen). Initial crystallization hits were established and optimized, and diffraction-quality crystals of ScErv41p_LD were ultimately grown at 277 K in sitting drops consisting of 0.6 µl protein solution mixed with 0.3 µl reservoir solution [0.2 M ammonium nitrate, 16%(w/v) PEG 3350] and equilibrated against 60 µl reservoir solution. Prior to data collection, crystals were transferred into a stabilizing cryo-solution consisting of 0.2 M ammonium nitrate, 16%(w/v) PEG 3350, 25%(v/v) PEG 400 and briefly incubated before being flash-cooled in liquid nitrogen.
2.4. Data collection and processing
Crystal screening and X-ray diffraction data collection were performed at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. High-resolution native data were collected from a single cryo-cooled crystal to a resolution of 2.0 Å on beamline ID14-1, which is equipped with an ADSC Q210 CCD detector. 192° of data were collected at a wavelength of 0.9334 Å using a 1.2° oscillation angle. All data were processed using XDS (Kabsch, 2010 ▶) and SCALA (Evans, 2006 ▶) from the CCP4 suite (Winn et al., 2011 ▶). Data-collection statistics are presented in Table 1 ▶.
Table 1. X-ray diffraction data-collection statistics for ScErv41p_LD.
Values in parentheses are for the outermost resolution shell.
| X-ray source | ID14-1, ESRF |
| Wavelength (Å) | 0.9334 |
| Temperature (K) | 100 |
| Resolution (Å) | 48.14–1.99 (2.10–1.99) |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 49.8, b = 76.9, c = 65.1, α = γ = 90.0, β = 104.8 |
| Total no. of reflections | 127463 |
| No. of unique reflections | 31843 |
| Multiplicity | 3.9 (3.2) |
| Completeness (%) | 98.8 (92.2) |
| Mean I/σ(I) | 19.4 (4.4) |
| R merge † (%) | 4.9 (27) |
R
merge = 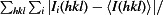
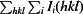 .
.
3. Results and discussion
Initial attempts to produce recombinant Erv41p in the expression host E. coli were not successful in yielding correctly folded protein, despite extensive trials with numerous constructs, and it was therefore decided to use a eukaryotic expression system instead. Multiple constructs of the gene encoding Erv41p from S. cerevisiae S288C were cloned and tested for expression in Sf9 insect cells, and ultimately a construct encoding the full lumenal domain (ScErv41p_LD) was selected because it was the longest that was successfully produced. The soluble recombinant protein was recovered from the culture medium and was purified to homogeneity using a protocol that included removal of the N-terminal signal sequence and 6×His tag, yielding 1–5 mg of purified protein per litre of culture medium (Fig. 2 ▶ a).
Figure 2.

(a) SDS–PAGE gel showing purified ScErv41p_LD in the right-hand lane. The left-hand lane contains the molecular-weight marker (PageRuler Prestained Protein Ladder, Thermo Scientific Ltd); the band slightly higher than ScErv41p_LD corresponds to 35 kDa. (b) Crystals of ScErv41p_LD.
Crystallization trials with ScErv41p_LD identified several closely related crystallization conditions with PEG 3350 as the precipitant and ammonium nitrate, sodium nitrate or sodium iodide as additives. Initial crystals were produced using protein that contained the intact signal sequence and a 6×His tag incorporated for purification purposes, but these small needle-shaped crystals proved impossible to optimize. It was later found that removal of the signal sequence and 6×His tag produced crystals with improved size and morphology, and the tag was therefore cleaved during purification of every subsequent protein batch. After optimization, diffraction-quality crystals of ScErv41p_LD were produced in 16%(w/v) PEG 3350 with 0.2 M ammonium nitrate as the additive. Even after optimization a large proportion of the crystals produced were very tightly clustered, resembling stacked plates, and could not be separated. These crystals diffracted to relatively high resolution but produced diffraction images with many overlapping lattices. However, a minority of the crystals were single and produced diffraction images of high quality. These crystals, which appeared after approximately 7 d of incubation at 277 K, had maximum dimensions of approximately 100 × 200 × 30 µm (Fig. 2 ▶ b). Many crystals were screened for diffraction at a synchrotron-radiation source in order to obtain the best possible resolution. Ultimately, a single crystal was used to collect a complete data set to a maximum resolution of 2.0 Å (Table 1 ▶, Fig. 3 ▶). Based on inspection of the diffraction images and on the auto-indexing routines of HKL-2000 (Otwinowski & Minor, 1997 ▶) and XDS (Kabsch, 2010 ▶), the data were indexed in space group P21, with unit-cell parameters a = 49.8, b = 76.9, c = 65.1 Å, α = γ = 90.0, β = 104.8°. The program MATTHEWS_COEF (Kantardjieff & Rupp, 2003 ▶) from the CCP4 suite (Winn et al., 2011 ▶) gave the most likely V M for the data set as 2.14 Å3 Da−1, which would correspond to the presence of two molecules in the asymmetric unit and a solvent content of 43% (Matthews, 1968 ▶). Screening of heavy-atom derivatives is in progress in order to enable determination of the structure.
Figure 3.

Diffraction pattern obtained from a ScErv41p_LD crystal. The resolution at the detector edge is 2.11 Å.
Acknowledgments
This work was supported by project grants to JEG from the Swedish Research Council (Vetenskapsrådet), Jeanssons Stiftelse, Åke Wibergs Stiftelse and Stiftelsen Lars Hiertas Minne, and by a Visiting Researcher Stipend for EIB from the Wenner–Gren Foundations. We would also like to acknowledge the European Synchrotron Radiation Facility (ESRF) for access to synchrotron beamlines.
References
- Buchan, D. W., Ward, S. M., Lobley, A. E., Nugent, T. C., Bryson, K. & Jones, D. T. (2010). Nucleic Acids Res. 38, W563–W568. [DOI] [PMC free article] [PubMed]
- Dancourt, J. & Barlowe, C. (2010). Annu. Rev. Biochem. 79, 777–802. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Fitzgerald, D. J., Berger, P., Schaffitzel, C., Yamada, K., Richmond, T. J. & Berger, I. (2006). Nature Methods, 3, 1021–1032. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Otte, S. & Barlowe, C. (2002). EMBO J. 21, 6095–6104. [DOI] [PMC free article] [PubMed]
- Otte, S., Belden, W. J., Heidtman, M., Liu, J., Jensen, O. N. & Barlowe, C. (2001). J. Cell Biol. 152, 503–518. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Welsh, L. M., Tong, A. H. Y., Boone, C., Jensen, O. N. & Otte, S. (2006). J. Cell Sci. 119, 4730–4740. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.