

The Cmr2–Cmr3 subcomplex from P. furiosus was co-crystallized with 3′-AMP. X-ray diffraction data for the crystals were collected to 2.6 Å resolution using a synchrotron-radiation source.
Keywords: Cmr2, Cmr3, CRISPR, Cas, RNA silencing
Abstract
Clustered, regularly interspaced, short palindromic repeat (CRISPR) loci, found in prokaryotes, are transcribed to produce CRISPR RNAs (crRNAs). The Cmr proteins (Cmr1–6) and crRNA form a ribonucleoprotein complex that degrades target RNAs derived from invading genetic elements. Cmr2dHD, a Cmr2 variant lacking the N-terminal putative HD nuclease domain, and Cmr3 were co-expressed in Escherichia coli cells and co-purified as a complex. The Cmr2dHD–Cmr3 complex was co-crystallized with 3′-AMP by the vapour-diffusion method. The crystals diffracted to 2.6 Å resolution using synchrotron radiation at the Photon Factory. The crystals belonged to the orthorhombic space group I222, with unit-cell parameters a = 103.9, b = 136.7, c = 192.0 Å. The asymmetric unit of the crystals is expected to contain one Cmr2dHD–Cmr3 complex with a Matthews coefficient of 3.0 Å3 Da−1 and a solvent content of 59%.
1. Introduction
Many prokaryotes employ a defence system against foreign genetic elements, such as plasmids and phages, which consists of clustered, regularly interspaced, short palindromic repeat (CRISPR) loci and CRISPR-associated (Cas) proteins (Marraffini & Sontheimer, 2008 ▶; van der Oost et al., 2009 ▶; Deveau et al., 2010 ▶; Karginov & Hannon, 2010 ▶; Terns & Terns, 2011 ▶). CRISPR loci are composed of conserved repeated sequences separated by variable spacer sequences which are identical to parts of the foreign DNAs. The primary transcripts from CRISPR loci are processed into short single-stranded RNA fragments (crRNAs) by the dedicated Cas proteins. The crRNAs and Cas proteins form effector complexes to cleave the invading genetic elements, in which the crRNAs act as guides for targeting mediated by base complementarity (Hale et al., 2009 ▶; Garneau et al., 2010 ▶; Jore et al., 2011 ▶; Semenova et al., 2011 ▶; Wiedenheft, Lander et al., 2011 ▶; Jinek et al., 2012 ▶).
There are two types of effector complexes in the CRISPR–Cas defence system: one targets DNA and the other targets RNA. In Escherichia coli, one crRNA and five protein subunits (CasA–E) form an effector complex known as Cascade (CRISPR-associated complex for antiviral defence; Brouns et al., 2008 ▶). The Cascade complex recognizes invading DNAs by using crRNA and subsequently induces the cleavage of invading DNAs by the Cas3 nuclease/helicase. Structural analyses of the Cascade complex and its individual protein subunits provided detailed insights into the mechanism by which the invading DNAs are cleaved (Jore et al., 2011 ▶; Wiedenheft, Lander et al., 2011 ▶). DNA-targeting effector complexes also exist in many prokaryotes, such as Bacillus halodurans, Pseudomonas aeruginosa, Streptococcus thermophilus and Staphylococcus epidermidis (Marraffini & Sontheimer, 2008 ▶; Garneau et al., 2010 ▶; Wiedenheft, Duijn et al., 2011 ▶; Nam et al., 2012 ▶).
RNA-targeting effector complexes have been found in Pyrococcus furiosus and Sulfolobus solfataricus (Hale et al., 2009 ▶; Zhang et al., 2012 ▶). The effector complex from P. furiosus consists of six Cmr proteins (Cmr1–6) and a 39- or 45-nucleotide (nt) crRNA (Hale et al., 2009 ▶, 2012 ▶). The crRNAs from P. furiosus contain an invariant 8 nt tag at the 5′ end, which is essential for the CRISPR-mediated RNA interference (Hale et al., 2009 ▶, 2012 ▶). The sequence at the 3′ end of the crRNA (31 or 37 nt, not including the 8 nt tag) is derived from the spacer sequence and functions as a guide for targeting the complementary RNA (Hale et al., 2009 ▶, 2012 ▶). The P. furiosus Cmr effector complex specifically cleaves the target RNA at a fixed distance (14 nt) from the 3′ end of the crRNA (Hale et al., 2009 ▶, 2012 ▶).
In contrast to the Cascade complex, the roles of the individual Cmr proteins in the Cmr effector complex have not been elucidated owing to a paucity of structural information. Recently, the crystal structure of a P. furiosus Cmr2 derivative (Cmr2dHD), which lacks its N-terminal HD nuclease domain, revealed that this protein contains two adenylyl cyclase-like domains that bind ADP and divalent metal ions at their domain boundary (Cocozaki et al., 2012 ▶). However, the amino-acid residues responsible for ADP- and metal-ion-binding are not involved in the crRNA-guided RNA-cleavage activity of the effector complex. Therefore, the role of Cmr2 in the complex has remained elusive. A biochemical study of the Cmr effector complex in S. solfataricus revealed that Cmr2 associates with Cmr3 (Zhang et al., 2012 ▶), but the precise interaction mechanism and the functional role of the Cmr2–Cmr3 complex in the activity have not been determined. We now report the crystallization and preliminary X-ray diffraction analysis of the complex of P. furiosus Cmr2dHD and Cmr3 bound with 3′-AMP, to clarify the crRNA-guided RNA-cleavage reaction by the Cmr effector complex.
2. Materials and methods
2.1. Protein preparation
The PCR-amplified genes encoding Cmr2dHD (residues 216–871) and Cmr3 were prepared as an operon construct, which was cloned into the pET28b expression vector. The recombinant Cmr2dHD, with an N-terminal His tag, and Cmr3 were co-expressed at 293 K in E. coli strain BL21-CodonPlus (DE3)-RIL. The harvested cells were resuspended in buffer A (20 mM Tris–HCl pH 8.0, 300 mM NaCl, 10 mM imidazole) supplemented with 1 mM PMSF and 1 mM benzamidine and disrupted by sonication. After centrifugation, the clarified lysate was incubated at 338 K for 20 min. The heat-treated lysate was centrifuged and the supernatant was applied onto a Ni–NTA (Qiagen) column previously equilibrated with buffer A. The Cmr2dHD–Cmr3 complex was eluted from the Ni–NTA column with buffer B (20 mM Tris–HCl pH 8.0, 300 mM NaCl, 250 mM imidazole). The eluate from the Ni–NTA column was dialysed against buffer C (20 mM Tris–HCl pH 7.0, 150 mM NaCl, 1 mM DTT) and was then loaded onto a HiTrap Heparin column (GE Healthcare) equilibrated with buffer C. A linear gradient was developed from 150 to 1000 mM NaCl in buffer C. The fractions rich in the Cmr2dHD–Cmr3 complex were combined and dialysed against buffer D (20 mM Tris–HCl pH 9.0, 100 mM NaCl, 1 mM DTT). The solution was then applied onto a RESOURCE Q column (GE Healthcare), which had previously been equilibrated with buffer D, and the elution was performed with a linear gradient of 100–1000 mM NaCl in buffer D. The fractions enriched in the Cmr2dHD–Cmr3 complex were combined and dialysed against buffer E (20 mM Tris–HCl pH 7.5, 200 mM NaCl, 1 mM DTT). The purified complex was concentrated to 11 mg ml−1, using Amicon Ultra-15 Centrifugal Filters (30 000 Da molecular-weight cutoff; Millipore).
2.2. Crystallization and X-ray data collection
Crystallization screening was performed by the sitting-drop vapour-diffusion method at 293 K. Sitting drops were prepared by mixing 1 µl reservoir solution with 1 µl protein solution (5 mg ml−1) containing 2 mM ADP and 2 mM MgCl2 and were equilibrated against 100 µl of reservoir solution. The crystals grew in reservoir consisting of 15% 2-propanol, 20 mM MgCl2, 50 mM MES pH 6.0. The crystallization conditions were then optimized in the presence of 2 mM 3′-AMP, instead of ADP, by the hanging-drop vapour-diffusion method and crystals suitable for X-ray diffraction analysis were obtained under conditions consisting of 12.5–15% 2-propanol, 18 mM MgCl2, 45 mM MES pH 6.0, 20 mM ammonium acetate, 10 mM sodium acetate, 3% PEG 4000.
The crystals were cryoprotected in the optimized crystallization conditions containing 25% glycerol and were flash-cooled in a nitrogen stream at 95 K. X-ray diffraction data were collected on beamline BL-17A of the Photon Factory, Ibaraki, Japan, using an ADSC Q270 CCD detector. The raw data were processed and scaled with HKL-2000 (Otwinowski & Minor, 1997 ▶). The processing statistics are summarized in Table 1 ▶.
Table 1. Data-collection statistics.
Values in parentheses are for the last shell.
| Space group | I222 |
| Unit-cell parameters (Å) | a = 109.9, b = 136.7, c = 192.0 |
| Wavelength (Å) | 0.98 |
| Resolution (Å) | 50–2.60 (2.64–2.60) |
| Measured reflections | 373906 |
| Unique reflections | 42206 |
| R merge † | 0.063 (0.333) |
| 〈I/σ(I)〉 | 50.5 (5.0) |
| Completeness (%) | 99.7 (99.5) |
| Multiplicity | 8.9 (7.0) |
R
merge = 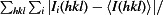
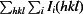 , where I
i(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity over symmetry-equivalent measurements.
, where I
i(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity over symmetry-equivalent measurements.
3. Results and discussion
The interaction between Cmr2 and Cmr3 has recently been suggested by Zhang et al. (2012 ▶). To elucidate the roles of the Cmr proteins in the RNA-silencing mechanism, we attempted to determine the crystal structure of the Cmr2–Cmr3 complex. Our first approach to prepare the individual P. furiosus Cmr2 and Cmr3 proteins was unsuccessful because only Cmr3 was produced as an inclusion body. To solve this problem, the genes encoding P. furiosus Cmr2dHD (residues 216–871), a Cmr2 protein lacking the putative HD nuclease domain, and full-length Cmr3 (residues 1–322) were co-expressed in E. coli cells by using an operon construct, with a single His tag at the N-terminus of Cmr2dHD. The deletion of the putative HD nuclease domain in P. furiosus Cmr2 reportedly had no effect on the RNA-cleavage activity by the Cmr effector complex (Cocozaki et al., 2012 ▶). Eventually, the co-expression of these proteins allowed us to obtain the soluble Cmr3 protein in complex with Cmr2dHD. The interaction between Cmr2dHD and Cmr3 is strong, and the proteins were purified as a complex through several chromatography steps. Analytic gel-filtration chromatography on a Superdex 200 HR 10/300 column revealed that the Cmr2dHD–Cmr3 complex migrated as a single peak corresponding to a molecular weight of 118 kDa (data not shown). Based on the molecular masses of Cmr2dHD (76 kDa) and Cmr3 (36 kDa), this result indicated that the Cmr2dHD–Cmr3 complex forms a 1:1 heterodimer.
The purified Cmr2dHD–Cmr3 complex was used for crystallization screening by the sitting-drop vapour-diffusion method at 293 K. We obtained crystals of the Cmr2dHD–Cmr3 complex in the presence of 2 mM ADP under conditions consisting of 15% 2-propanol, 20 mM MgCl2, 50 mM MES pH 6.0. The crystallization conditions were then optimized in the presence of 3′-AMP by the hanging-drop vapour-diffusion method. As a result, crystals suitable for X-ray diffraction grew using a reservoir consisting of 12.5–15% 2-propanol, 18 mM MgCl2, 45 mM MES pH 6.0, 20 mM ammonium acetate, 10 mM sodium acetate, 3% PEG 4000 and reached maximum dimensions of 200 × 300 × 300 µm within a week (Fig. 1 ▶). Electrophoretic analysis by SDS–PAGE demonstrated that the crystals contained both Cmr2dHD and Cmr3 (data not shown). The 3′-AMP-containing crystals diffracted to 2.6 Å resolution (Fig. 2 ▶) and belonged to the orthorhombic space group I222, with unit-cell parameters a = 103.9, b = 136.7, c = 192.0 Å. On the basis of the molecular masses of recombinant Cmr2dHD (76 kDa) and Cmr3 (36 kDa), the asymmetric unit of the crystal is expected to contain one Cmr2dHD–Cmr3 complex, with a Matthews coefficient of 3.0 Å3 Da−1 and a solvent content of 59% (Matthews, 1968 ▶). Very recently, the crystal structure of the Cmr2dHD–Cmr3 complex from P. furiosus was determined at 2.85 Å resolution (Shao et al., 2013 ▶). The crystals were grown in a reservoir containing 21–25% PEG 1500 at 303 K. Although their crystallization conditions are quite different from those reported here, both crystals belonged to the same space group with similar unit-cell parameters. However, the diffraction quality of our crystals was slightly higher (2.6 Å resolution) than that reported by Shao et al. (2013 ▶). Therefore, we expect that our analysis may provide more precise structural information. Moreover, in contrast to their crystals, which were complexed with ATP (Shao et al., 2013 ▶), we crystallized the Cmr2dHD–Cmr3 in complex with 3′-AMP and thus our crystals could reveal new details about the nucleotide-binding site. Attempts to solve the 3′-AMP-bound Cmr2dHD–Cmr3 complex structures by the molecular-replacement method are in progress.
Figure 1.

The crystal of the 3′-AMP-bound Cmr2dHD–Cmr3 complex.
Figure 2.

The diffraction patterns of the 3′-AMP-bound Cmr2dHD–Cmr3 complex.
Acknowledgments
We thank Y. Ishino for providing the P. furiosus genomic DNA for the experiment. We also thank the beamline staff at BL-17A of the Photon Factory, Ibaraki, Japan, for technical assistance during data collection. This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas from MEXT to TN, by a Grant-in-Aid for Young Scientists from JSPS to TN and by a Grant-in-Aid for JSPS Fellows to TO.
References
- Brouns, S. J., Jore, M. M., Lundgren, M., Westra, E. R., Slijkhuis, R. J., Snijders, A. P., Dickman, M. J., Makarova, K. S., Koonin, E. V. & van der Oost, J. (2008). Science, 321, 960–964. [DOI] [PMC free article] [PubMed]
- Cocozaki, A. I., Ramia, N. F., Shao, Y., Hale, C. R., Terns, R. M., Terns, M. P. & Li, H. (2012). Structure, 20, 545–553. [DOI] [PMC free article] [PubMed]
- Deveau, H., Garneau, J. E. & Moineau, S. (2010). Annu. Rev. Microbiol. 64, 475–493. [DOI] [PubMed]
- Garneau, J. E., Dupuis, M. È, Villion, M., Romero, D. A., Barrangou, R., Boyaval, P., Fremaux, C., Horvath, P., Magadán, A. H. & Moineau, S. (2010). Nature (London), 468, 67–71. [DOI] [PubMed]
- Hale, C. R., Majumdar, S., Elmore, J., Pfister, N., Compton, M., Olson, S., Resch, A. M., Glover, C. V., Graveley, B. R., Terns, R. M. & Terns, M. P. (2012). Mol. Cell, 45, 292–302. [DOI] [PMC free article] [PubMed]
- Hale, C. R., Zhao, P., Olson, S., Duff, M. O., Graveley, B. R., Wells, L., Terns, R. M. & Terns, M. P. (2009). Cell, 139, 945–956. [DOI] [PMC free article] [PubMed]
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. & Charpentier, E. (2012). Science, 337, 816–821. [DOI] [PMC free article] [PubMed]
- Jore, M. M. et al. (2011). Nature Struct. Mol. Biol. 18, 529–536. [DOI] [PubMed]
- Karginov, F. V. & Hannon, G. J. (2010). Mol. Cell, 37, 7–19. [DOI] [PMC free article] [PubMed]
- Marraffini, L. A. & Sontheimer, E. J. (2008). Science, 322, 1843–1845. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Nam, K. H., Haitjema, C., Liu, X., Ding, F., Wang, H., DeLisa, M. P. & Ke, A. (2012). Structure, 20, 1574–1584. [DOI] [PMC free article] [PubMed]
- Oost, J. van der, Jore, M. M., Westra, E. R., Lundgren, M. & Brouns, S. J. (2009). Trends Biochem. Sci. 34, 401–407. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Semenova, E., Jore, M. M., Datsenko, K. A., Semenova, A., Westra, E. R., Wanner, B., van der Oost, J., Brouns, S. J. & Severinov, K. (2011). Proc. Natl Acad. Sci. USA, 108, 10098–10103. [DOI] [PMC free article] [PubMed]
- Shao, Y., Cocozaki, A. I., Ramia, N. F., Terns, R. M., Terns, M. P. & Li, H. (2013). Structure, 21, 376–384. [DOI] [PMC free article] [PubMed]
- Terns, M. P. & Terns, R. M. (2011). Curr. Opin. Microbiol. 14, 321–327. [DOI] [PMC free article] [PubMed]
- Wiedenheft, B., van Duijn, E., Bultema, J. B., Waghmare, S. P., Zhou, K., Barendregt, A., Westphal, W., Heck, A. J., Boekema, E. J., Dickman, M. J. & Doudna, J. A. (2011). Proc. Natl Acad. Sci. USA, 108, 10092–10097. [DOI] [PMC free article] [PubMed]
- Wiedenheft, B., Lander, G. C., Zhou, K., Jore, M. M., Brouns, S. J., van der Oost, J., Doudna, J. A. & Nogales, E. (2011). Nature (London), 477, 486–489. [DOI] [PMC free article] [PubMed]
- Zhang, J., Rouillon, C., Kerou, M., Reeks, J., Brugger, K., Graham, S., Reimann, J., Cannone, G., Liu, H., Albers, S. V., Naismith, J. H., Spagnolo, L. & White, M. F. (2012). Mol. Cell, 45, 303–313. [DOI] [PMC free article] [PubMed]