

Abstract
Knowledge about the mechanism of impulse blockade by local anesthetics has evolved over the past four decades, from the realization that Na+ channels were inhibited to affect the impulse blockade to an identification of the amino acid residues within the Na+ channel that bind the local anesthetic molecule. Within this period appreciation has grown of the state-dependent nature of channel inhibition, with rapid binding and unbinding at relatively high affinity to the open state, and weaker binding to the closed resting state. Slow binding of high affinity for the inactivated state accounts for the salutary therapeutic as well as the toxic actions of diverse class I anti-arrhythmic agents, but may have little importance for impulse blockade, which requires concentrations high enough to block the resting state. At the molecular level, residues on the S6 transmembrane segments in three of the homologous domains of the channel appear to contribute to the binding of local anesthetics, with some contribution also from parts of the selectivity filter. Binding to the inactivated state, and perhaps the open state, involves some residues that are not identical to those that bind these drugs in the resting state, suggesting spatial flexibility in the “binding site”. Questions remaining include the mechanism that links local anesthetic binding with the inhibition of gating charge movements, and the molecular nature of the theoretical “hydrophobic pathway” that may be critical for determining the recovery rates from blockade of closed channels, and thus account for both therapeutic and cardiotoxic actions.
Keywords: local anesthetics, sodium channel, drug binding, modulated receptor
Historical Background
Although it had been known for more than 70 years that local anesthetics could produce numbness, and for about 30 years that this loss of sensation corresponded to a blockade of nerve impulses, the actual mechanism and site of action for this effect only emerged through modem electrophysiological and biochemical experiments. The first of these was the report of a strong, reversible inhibition of Na+ currents in the voltage-clamped squid giant axon by procaine [1]. Since the inward Na+ current was recognized as the quintessential feature of regenerative action potentials [2] this finding identified the major mode of action for impulse blockade by local anesthetics.
The squid axon continued to be a useful tool for studying the actions of local anesthetics, in a series of elegant experiments by Narahashi and colleagues in the 1970s, using the internal perfusion that, at that time, could only be accomplished with that large nerve structure. Internal perfusion allowed the direct application of the drug inside or outside the nerve's plasma membrane, and the control of the internal and external pH. In these experiments, Narahashi's team showed that the most potent action of tertiary amine local anesthetics occurred when the internally perfused drug was accompanied by a relatively acidic internal pH, suggesting that the cationic species acting from the intracellular side was most active in blocking the channel [3]. This conclusion was further supported by the observation that quaternary homologues of the smaller local anesthetics, such as the N-ethyl derivative of lidocaine, QX-314, were far more effective blockers when placed inside the axon rather than applied extracellularly [4]. Surprising later work, in frog muscle, showed that when a local anesthetic molecule was blocking the Na+ channel from the intercellular compartment, the protonation–deprotonation reaction of the tertiary amine (pK = 8–9 in solution) was dependent on the extracellular pH and virtually independent of the intracellular pH [5]! This observation, together with the control of the channel blocking and the unblocking rates by the open state of the channel [6–9] were consistent with a blocking site within the channel's pore. Some potency was still attributed to the neutral species however, based on the pH studies and on the findings that uncharged, non-ionizable derivatives of local anesthetics were able to inhibit Na+ currents [10–12].
Two seminal papers followed shortly after Narahashi's work, showing that the state transitions that underlay the gating (opening, inactivation) of Na+ channels were altered by local anesthetics [13] and that such gating was itself an essential modulator of local anesthetic block [6]. Both observations lead to the proposition that the Na+ channel had a direct interaction with local anesthetic molecules, rather than being inhibited by some non-direct membrane perturbing activity [14]. Subsequent studies showed that the “gating currents”, which resulted from the asymmetric movement of the charged regions of the Na+ channel that underlay and temporally preceded channel opening, were suppressed but not totally abolished by local anesthetics [15–18]. The channels, it thus appeared, were not simply inhibited by local anesthetic plugging of the ion-conducting pore, but rather were mechanically disrupted by the drug.
These observations formed the foundation for the Modulated Receptor Hypothesis, which viewed the interactions between channel and local anesthetic with the perspective of the dynamic binding of an enzyme-inhibitor complex, with reciprocal interactions between allosteric transitions of the enzyme and state-dependent binding of the inhibitor.
Modulated Receptor Hypothesis
As the preceding section has shown, block of Na+ currents by local anesthetics is most likely due to the existence of a local anesthetic receptor within the voltage-gated Na+ channels. The early framework regarding the local anesthetic receptor is best exemplified by the modulated receptor hypothesis put forward by Hille [19], initially proposed to explain the “use-dependent” block of sodium currents during repetitive pulses and the shift in apparent inactivation of the channel by local anesthetics [13,20]. This hypothesis envisions that (1) the local anesthetic receptor site is modulated by state transitions during membrane depolarization and (2) the open and inactivated states of voltage-gated Na+ channels have higher affinities toward LA drugs than that of the resting state. Such a hypothesis implies that the receptor site is movable during state transitions, flexible in its receptor configuration, and perhaps participating in an induced-fit with its ligands upon depolarization. More specifically, as the Na+ channel cycles from resting to open and inactivated states during a pulse, the local anesthetic receptor site also changes its configuration accordingly, which in turn displays higher binding affinities toward LA drugs because of its conformational changes. It is plausible that inactivated states are “stabilized” by LAs through high-affinity binding and may be “locked” in such states during repetitive pulses. If the drug remains bound with the inactivated channels because of its slow dissociation from the LA receptor, more and more receptors will be occupied by LA drugs until the drug-bound and drug-free receptor reach a steady state during repetitive pulses. This modulated receptor hypothesis readily accounts for the use-dependent phenotype of LA drugs during repetitive pulses. It is now apparent that the implications of Hille's hypothesis are far-reaching in the ion channel field. Numerous receptors for clinic drugs within ion channels are indeed modulated by ion channel gating.
The structure and function of S6 segments along with the LA receptor within voltage-gated Na+ channels. A number of mammalian voltage-gated sodium channel proteins were delineated during 1980 and 1990 by molecular cloning techniques. The voltage-gated Na+ channel in excitable membranes consists of one large α-subunit and one or two smaller auxiliary β-subunits [21]. To date, there are nine large α-sub-unit (Nav1.1–Nav1.9) and four β-subunit (β1–β4) isoforms found in mammals. The large α-subunit peptide alone can form a functional Na+ channel that carries Na+ currents when expressed in mammalian cells or in frog oocytes. Each α-subunit peptide contains four homologous domains (D1–D4), each with 6 transmembrane segments (S1–S6) (Fig. 1). The voltage sensor is located within S4 segment whereas the selectivity filter (DEKA locus) is situated within the S5–S6 linker (also known as the Pore region) in each of four homologous domains. The activation gate is probably located at the C-termini of the four S6 segments and the inactivation gate is likely formed by the IFM motif at the intracellular D3–D4 linker.
Fig. 1.
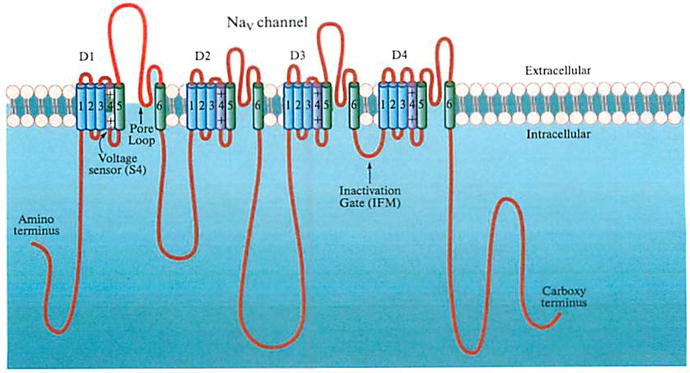
The topological diagram of the Na+ channel α-subunit peptide embedded in a lipid bilayer (kindly provided by Dr. Suzuki). The S5–S6 linkers are designated as the Pore Loop (P-loop) which contains a selectivity filter (the DEKA locus). The D3–D4 intracellular linker contains an IPM motif, which may act as the inactivation gale particle. The S4 segments contain multiple positive charges and likely act as the voltage sensor.
The structural aspects of the local anesthetic receptor have been inferred by site-directed mutagenesis so far. The receptor mapping of the local anesthetic binding site was first reported in 1994 by Catterall's group [22]. Using an alanine scanning technique, these authors found that the middle of the D4–S6 segment contains two aromatic residues (phenylalanine, position 13 and tyrosine, position 20) that are critical for local anesthetic binding (Fig. 2). In particular, the LA binding affinity toward the inactivated state of the sodium channel was drastically reduced when each of these two residues is substituted with alanine. The mutation, however, has lesser effects on the resting affinity toward LAs. It is interesting to note that these two aromatic residues are conserved in all 9 α-subunit isoforms. Further detailed studies have found that the phenylalanine residue is the most important residue for LA binding. Substitution of a lysine residue, which introduces a positive charge at this phenylalanine position, drastically reduced the LA binding affinities, probably due to the charge-charge repulsion between the lysine residue and the protonated amine moiety [23]. Incorporations with various unnatural phenylalanine derivatives at this position demonstrate further that this phenylalanine interacts with the tertiary amine moiety of LAs (protonated with a positive charge) through the π electron and positive charge interaction [24].
Fig. 2.
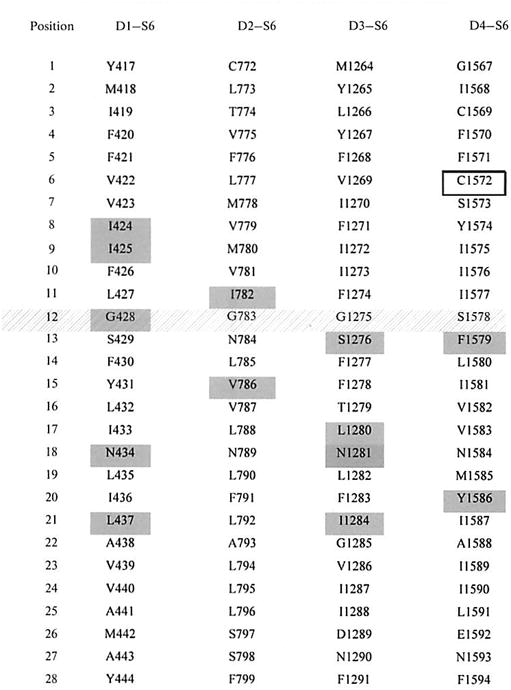
Amino acid sequences within S6 segments D1–S6, D2–S6, D3–S6, and D4–S6 of the rat skeletal muscle Na+ channel Nav1.4. Residues in gray are critical for LA binding. The putative gating hinge at position 12 containing a glycine/serine residue [30, 48] is underlined. This figure is modified from Nau and Wang [25].
The LA receptor site from the initial receptor mapping at the D4–S6 region suggests that additional S6 segments in the remaining three domains may also involve in LA binding. This notion is based on the possibility that the C-termini of all four S6 segments form an inverted teepee-like structure, which acts as an activation gate and must align in close proximity. Subsequent receptor mapping has identified a number of residues at D1–S6 and D3–S6 segments that are also critical for LA binding. A few residues at D2–S6 may also influence the binding affinities of LAs although the effects are not as drastic as S6 residues at Dl, D3, and D4. A summary of the involvement of various residues with LAs is provided in a diagram (Fig. 2) [25]. Overall, there is strong evidence that multiple S6 segments together form a single LA binding site within the Na+ channel and that residues in segments D3–S6 and D4–S6 arc the most prominent determinants of LA binding. The contact points are not likely to be static and might depend on both the state of the channel and the type of LA tested. There is also evidence that the LA binding site may not be limited to S6 segments. Residue Navl.4–K1237 in the putative domain D3 selectivity filter (DEKA locus) was shown to be involved in interactions with the hydrophilic part of lidocainc [26]. The selectivity filter may well be structurally adjacent to the LA binding site and may represent the more extracellular-located part of the binding site. Because of the limitation of site-direcled mutagenesis, structural details regarding the LA receptor remain ambiguous and can not be confirmed directly without the X-ray structure. Despite of this difficulty, several 3-D structures of the LA receptor have been constructed and they will undoubtedly provide a good starting point to visualize and to test the molecular interactions between LAs and their binding site [27, 28].
The inactivated-channel block and the LA receptor
LAs appear to bind to the fast inactivated state with a higher affinity as compared to the resting state. How the inactivation gating machinery enhances the LA binding affinities is not clear. Accumulated evidence now suggests that the S6 segments of voltage-gated Na+ channels contain crucial structural motifs that arc involved in channel opening, fast inactivation. and slow inactivation. The vast data on the block of Na+ currents by LAs also indicate the presence of a dynamic LA binding site situated at the middle of S6 segments, which likely encircle the inner cavity of the Na+ channel. Accordingly, the opening of the activation gate during the outward movement of S6 segments [21] should have a significant effect on the conformation of the LA receptor. In addition, it was first demonstrated that the phenylalanine (position 13 of D4–S6) at the LA receptor site was somewhat critical for the fast inactivation [29]. Substitutions of several residues at D1S6, D2S6, and D4S6 C-termini created mutants that generated inactivation-deficient Na+ currents during a prolonged depolarization. These findings together suggest a possibility that the receptor for the inactivation gate may be also located with in the region at multiple S6 C-termini [30, 31]. Lastly, both the P-region with in S5–S6 linkers and the S6 segments arc known to be critical for slow inactivation gating of voltage-gated sodium channels [32, 33]. Therefore, it would not be a surprise that the LA receptor within S6 segments is likewise modulated by slow inactivation gating of the Na+ channel [13].
The open channel block by LAs
Direct evidence that LAs block the open sodium channel with a higher affinity than that of the resting counterpart has been difficult to obtain. This is because the voltage-gated Na+ channel only opens once for a brief time of ∼0.5 ms and then is fast inactivated during depolarization. Attempts to prolong the open time using chemicals, such as chloramine-T, in order to study the open channel block, yield useful information regarding the high-affinity interaction between the open channel and LA drugs [34]. Unfortunately, such information contradicts the finding that QX-314 loses its use-dependent potency when measured using inactivation-deficient Na+ currents in the pronasetreated squid giant axon [35]. This loss-of-potency result is also found in the inactivation-deficient mutant Na+ channels (IFM/QQQ mutant) [36]. These contradictory findings have not been resolved by the recent evidence using other inactivation-deficient mutant Na+ channels. For example, lidocaine was found to block the open Na+ channels with a similar binding affinity as the inactivated Na+ channels (∼20 μM) (WCW mutant) [8, 37]. The phenotype of an apparent time-dependent block of inactivation-deficient Na+ currents signified a direct open-channel block of Na+ channels (Fig. 3). The peak currents were little blocked at various lidocaine concentrations whereas the maintained currents during the prolonged depolarization were highly sensitive to lidocaine even at low concentrations. The dose-response curve shows that the open-channel block by lidocaine has an IC50 value of ∼20 μM, which is an order more potent than that of the resting-channel block (∼300 μM). Furthermore, the block of open Na+ channels appears voltage-dependent which follows closely with the activation gating process [37].
Fig. 3.

Block of rNav1.4-WCW mutant currents by lidocaine. (a) Representative current traces are shown at various lidocaine concentrations. Cells were depolarized by a 50-ms test pulse at 30 mV. Pulses were delivered at 60 s intervals, (b) Peak currents (squares) and maintained late currents at the end of the test pulse (triangles) shown in (a) were measured at various lidocaine concentrations. Data were normalized to the control saline response and fitted with the Hill equation. The IC50 value for the peak current (n = 5) was 314 ± 25 μM (Hill coefficient, 0.8 ± 0.1). For the maintained late current (n = 5) the IC50 was 20.9 ± 3.3 μM (0.9 ± 0.1). This figure is taken from [37].
Since mammalian voltage-gated Na+ channels normally open for a short period of time and they predominantly only open once during depolarization, before inactivating, the significance of the open channel block by LAs is not easy to gauge for wild-type Na+ channels. According to a guarded receptor hypothesis [7, 38], the channel open conformation is viewed as controlling the flux of drugs as they diffuse between drug pools and the binding site. Under such a hypothesis, apparent variation in binding rates results from differences in the fraction of accessible sites and not in the variable-affinity receptor, i.e., the dynamic binding depends on the on-rate constant, modulated by availability of the reactive site, vs. the off-rate constant that reflects the energetics of the bound state [19,39]. For wild-type Na+ channels this guarded receptor hypothesis is able to adequately predict apparent changes in drug binding and apparent shifts in inactivation.
It is noteworthy that persistent late Na+ currents have been found in patients with neuropathic pain and with genetic disorders such as LQT-3 syndromes, familial erythromelalgia, familial periodic paralysis, and severe myoclonic epilepsy of infancy [14,40,42]. These abnormal Na+ currents generated by gain-of-function mutations are known to cause pain, myotonia, arrhythmia, epilepsy, and convulsion. A selective open-channel blocker may therefore be able to target these abnormal Na+ currents. Such blockers should be beneficial for patients with the syndromes mentioned above.
Hydrophilic and hydrophobic pathways for LAs
How do LA drugs reach their receptor sites after local injection? Since the LA receptor of Na+ channels is situated within the permeation pathway accessible by permanently charged LA derivatives only from the intracellular surface [6], clinic applications of LAs with a tertiary amine moiety require that LA drugs first penetrate the membrane phase in their neutral (unprotonatcd) form. The neutral form may enter and exit its receptor site via the transmembrane segments of Na+ channels from the lipid environment [12, 43]. Such a hydrophobic pathway through the membrane phase is not well defined and the significance of this direct hydrophobic pathway has not been evaluated but it may play an important role for a neutral LA drug, benzocaine. After partition through the membrane phase, neutral LAs may emerge at the intracellular surface and become protonated intracellularly. Protonation of the LA drug with a tertiary amine: allows its access to the LA receptor via the intracellular hydrophilic pathway. It has been shown that the protonated form of LAs is far more potent in the block of single Na+ channels than the neutral form [44].
Beside these two pathways for LAs, one additional hydrophilic pathway was found in the cardiac isoform from the external surface through the N-terminal ends of the S6 regions and the P-loops. For example, membrane-impermeant quaternary derivatives of lidocaine (QX-222 and QX-314) block cardiac Na+ channels when applied from either side of the membrane, but they block neuronal and skeletal muscle channels only from the inside. A unique threonine residue (T) at the D4S6 segment is critical for the extracellular access of QX-314 toward its LA receptor [45]. This residue (rH 1-Thrl755) is six residues away from the phenylalanine residue for the LA binding site (rHl-Phel762). Subsequent studies on this phenomenon further reveal that both the tyr residue at the P-loop (at D1) and the cys residue at D4S6 are responsible for the poor access of external QX-314 [46]. A reverse engineered mutant (rNav1.4-Y401C/C1572T) exhibits an excellent access of external QX-314 toward its receptor site [47].
Physical plug of the ion permeation pathway by LAs
Tetrodotoxin (TTX) is known to plug the Na+ channel from the external side of the permeation pathway adjacent to the narrow selectivity filter region. For decades, TTX was used as a molecular probe for the external side of Na+ channel permeation pathway. Like TTX, LAs were applied as probes to study the internal side of the ion permeation pathway. With the help of 3-D structural models several reports suggest that local anesthetics block the permeation pathway from the internal side by plugging the open Na+ channel between the selectivity filter and the inner cavity [27]. These models take into considerations of the possible involvement of multiple S6 segments in LA binding as suggested by site-directed mutagenesis. Experimental evidence also supports that the Phe residue at the D4S6 region is facing the pore and may function to bind Na+ ions and to stabilize the passage of cations within the permeation pathway [24]. A similar cation binding site was found in the K+ channel inner cavity where the quaternary ammonium ions are known to bind to the QA receptor and block the permeation of K+ ions.
Concluding Remarks
Our understanding of the mechanism by which local anesthetics cause impulse blockade has moved from the phcnomcnological to the molecular over the past 40 years. Sodium channels were identified as the causative target, direct binding of drug molecules to the channel was recognized, and a site within the channel's ion conducting pore has been resolved by mutagenesis experiments to reveal the actual amino acid residues involved in binding. But the major conceptual advance in this field has been the understanding that local anesthetic binding depends on the conformation of the channel and that transitions among the different conformations are modified by bound local anesthetics. This Modulated Receptor Model also explains the actions of many Class I anti-arrhythmic drugs. What still remains, however, is to understand how the binding site changes during gating, shifting the involved residues within the channel, and how the binding at this site, directly to residues in the S6 segments, influences the charge movements due to changes in the voltage-sensing S4 segments. In this regard, local anesthetics may be useful tools for understanding the basic gating mechanisms of Na+ channels.
Footnotes
The article was submitted by the authors in English.
References
- 1.Taylor RE. Effect of procaine on electrical properties of squid axon membrane. Am J Physiol. 1959;196:1071–1078. doi: 10.1152/ajplegacy.1959.196.5.1071. [DOI] [PubMed] [Google Scholar]
- 2.Hodgkin AL, Huxley AF. The components of membrane conductance in the giant axon of Loligo. J Physiol. 1952;116:473–496. doi: 10.1113/jphysiol.1952.sp004718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Narahashi T, Frazier X, Yamada M. The site of action and active form of local anesthetics. I. Theory and pH experiments with tertiary compounds. J Pharm Exp Therap. 1970;171:32–44. [PubMed] [Google Scholar]
- 4.Frazier DT, Narahashi X, Yamada M. The site of action and active form of local anesthetics. II. Experiments with quaternary compounds. J Pharm Exp Therap. 1970;171:45–51. [PubMed] [Google Scholar]
- 5.Schwarz W, Palade PX, Hille B. Local anesthetics. Effect of pH on use-dependent block of sodium channels in frog muscle. Biophys J. 1977;20:343–368. doi: 10.1016/S0006-3495(77)85554-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strichartz GR. The inhibition of sodium currents in myelinated nerve by quaternary derivatives of lidocaine. J Gen Physiol. 1973;62:37–57. doi: 10.1085/jgp.62.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Starmer CF, Grant AO. Phasic ion channel blockade. A kinetic model and parameter estimation procedure. Mol Pharmacol. 1985;28:348–356. [PubMed] [Google Scholar]
- 8.Chernoff DM. Kinetic analysis of phasic inhibition of neuronal sodium currents by lidocaine and bupivacaine. Biophys J. 1990;58(1):53–68. doi: 10.1016/S0006-3495(90)82353-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chernoff DM, Strichartz GR. Binding kinetics of local anesthetics to closed and open sodium channels: Relevance to antiarrhythmic actions. In: Hondeghem LM, Kisko Mt, editors. Molecular and Cellular Mechanisms of Anti-Arrhythmic Agents. N.Y.: Future Pub; 1989. pp. 307–335. [Google Scholar]
- 10.Neumcke B, Schwarz W, Stampfli R. Block of Na channels in the membrane of myelinated nerve by benzocaine. pflügers Arch. 1981;390:230–236. doi: 10.1007/BF00658267. [DOI] [PubMed] [Google Scholar]
- 11.Hille B. The pH-dependent rate of action of local anesthetics on the node of Ranvier. J Gen Physiol. 1977;69:475–496. doi: 10.1085/jgp.69.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chemoff DM, Strichartz GR. Tonic and phasic block of neuronal sodium currents by 5-hydroxy-hexano-2′,6′-xylide, a neutral lidocaine homologue. J Gen Physiol. 1989;93:1075–1090. doi: 10.1085/jgp.93.6.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khodorov BI, Shishkova L, Peganov E, Revenko S. Inhibition of sodium currents in frog Ranvier node treated with local anesthetics. Role of slow sodium inactivation. Biochim Biophys Acta. 1976;433:409– 435. [Google Scholar]
- 14.Elliott JR, Haydon DA, Hendry BM. The mechanisms of sodium current inhibition by benzocaine in the squid giant axon. Pflügers Arch. 1987;409:596–600. doi: 10.1007/BF00584659. [DOI] [PubMed] [Google Scholar]
- 15.Keynes RD, Rojas E. The temporal and steady-state relationships between activation of the sodium conductance and movement of the gating particles in the squid giant axon. J Physiol. 1976;255(1):157–189. doi: 10.1113/jphysiol.1976.sp011274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bekkers JM, Greeff NG, Keynes RD, Neumcke B. The effect of local anesthetics on the components of the asymmetry current in the squid giant axon. J Physiol. 1984;352:653–668. doi: 10.1113/jphysiol.1984.sp015315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cahalan MD, Aimers W. Interactions between quaternary lidocaine, the sodium channel gates, and tetrodotoxin. Biophys J. 1979;27:39–55. doi: 10.1016/S0006-3495(79)85201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khodorov BI. Sodium inactivation and drug-induced immobilization of the gating charge in nerve membrane. Progr Biophys Mol Biol. 1981;37:49–89. doi: 10.1016/0079-6107(82)90020-7. [DOI] [PubMed] [Google Scholar]
- 19.Hille B. Local anesthetics: Hydrophilic and hydrophobic pathways for the drug receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Courtney KR. Mechanism of frequency-dependent inhibition of sodium currents in frog myelinated nerve by the lidocaine derivative GEA. J Pharm Exp Therop. 1975;195:225–236. [PubMed] [Google Scholar]
- 21.Catterall WA. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 22.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- 23.Wright SN, Wang SY, Wang GK. Lysine point mutations in Na+ channel D4–S6 reduce inactivated channel block by local anesthetics. Mol Pharmacol. 1998;54:733–739. [PubMed] [Google Scholar]
- 24.Ahem CA, Eastwood AL, Dougherty DA, Horn R. Electrostatic contributions of aromatic residues in the local anesthetic receptor of voltage-gated sodium channels. Circ Res. 2008;102:86–94. doi: 10.1161/CIRCRESAHA.107.160663. [DOI] [PubMed] [Google Scholar]
- 25.Nau C, Wang GK. Interactions of local anesthetics with voltage-gated Na+ channels. J Membr Biol. 2004;201:1–8. doi: 10.1007/s00232-004-0702-y. [DOI] [PubMed] [Google Scholar]
- 26.Sunami A, Dudley SC, Fozzard HA. Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proc Natl Acad Sci USA. 1997;94:14126–14131. doi: 10.1073/pnas.94.25.14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipkind GM, Fozzard HA. Molecular modeling of local anesthetic drug binding by voltage-gated Na channels. Mol Pharmacol. 2005;68:1611–1622. doi: 10.1124/mol.105.014803. [DOI] [PubMed] [Google Scholar]
- 28.Tikhonov DB, Brunova I, Zhorov BS. Atomic determinants of state-dependent block of sodium channels by charged local anesthetics and benzocaine. FEBSLett. 2006;580:6027–6032. doi: 10.1016/j.febslet.2006.10.035. [DOI] [PubMed] [Google Scholar]
- 29.McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A critical role for transmembrane segment IVS6 of the sodium channel alpha subunit in fast inactivation. J Biol Chem. 1995;270:12025–12034. doi: 10.1074/jbc.270.20.12025. [DOI] [PubMed] [Google Scholar]
- 30.Yarov-Yarovoy V, McPhee JC, Idsvoog D, Pate C, Scheuer T, Catterall WA. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel α subunit in voltage-dependent gating and drug block. J Biol Chem. 2002;277:35393–35401. doi: 10.1074/jbc.M206126200. [DOI] [PubMed] [Google Scholar]
- 31.Wang SY, Bonner K, Russell C, Wang GK. Tryptophan scanning of D1S6 and D4S6 C-termini in voltage-gated sodium channels. Biophys J. 2003;85:911– 920. doi: 10.1016/S0006-3495(03)74530-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balser JR, Nuss HB, Chiamvimonvat N, Perez-Garcia MT, Marban E, Tomaselli GF. External pore residue mediates slow inactivation in μl rat skeletal muscle sodium channels. J Physiol. 1996;494:431–442. doi: 10.1113/jphysiol.1996.sp021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang SY, Wang GK. A mutation in segment I-S6 alters slow inactivation of sodium channels. Biophys J. 1997;72:1633–1640. doi: 10.1016/S0006-3495(97)78809-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang GK, Brodwick MS, Eaton DC, Strichartz GR. Inhibition of sodium currents by local anesthetics in chloramine-T treated squid axons: The role of channel activation. J Gen Physiol. 1987;89:645–667. doi: 10.1085/jgp.89.4.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cahalan MD. Local anesthetic block of sodium channels in normal and pronase-treated squid giant axons. Biophys J. 1978;23:285–311. doi: 10.1016/S0006-3495(78)85449-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennett PB, Vilenzuela C, Chen LQ, Kallen RG. On the molecular nature of the lidocaine receptor of cardiac Na+ channels: Modification of block by alterations in the α-subunit III-IV interdomain. Circ Res. 1995;77:584–592. doi: 10.1161/01.res.77.3.584. [DOI] [PubMed] [Google Scholar]
- 37.Wang SY, Mitchell J, Moczydlowski E, Wang GK. Block of inactivat ion-deficient Na+ channels by local anesthetics in stably transfected mammalian cells: Evidence for drug binding along the activation pathway. J Gen Physiol. 2004;124:691–701. doi: 10.1085/jgp.200409128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Starmer CF. Theoretical characterization of ion channel blockade: Competitive binding to periodically accessible receptors. Biophys J. 1987;52:405–412. doi: 10.1016/S0006-3495(87)83229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hondeghem LM, Katzung BG. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim Biophys Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- 40.Dib-Haij SD, Yang Y, Waxman SG. Genetics and molecular pathophysiology of Na(v)1.7-related pain syndromes. Adv Genet. 2008;63:85–110. doi: 10.1016/S0065-2660(08)01004-3. [DOI] [PubMed] [Google Scholar]
- 41.Cannon SC. Sodium channel defects in myotonia and periodic paralysis. Ann Rev Neurosci. 1996;19:141–164. doi: 10.1146/annurev.ne.19.030196.001041. [DOI] [PubMed] [Google Scholar]
- 42.George AL., Jr Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–1999. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheraoff DM, Strichartz GR. Kinetics of local anesthetic inhibition of neuronal sodium currents. pH and hydrophobicity dependence. Biophys J. 1990;58:69–81. doi: 10.1016/S0006-3495(90)82354-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nettleton J, Wing GK. pH-Dependent binding of local anesthetics in single batrachotoxin-activated Na+ channels. Cocaine vs quaternary compounds. Biophys J. 1990;58:95–106. doi: 10.1016/S0006-3495(90)82356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qu Y, Rogers J, Tanada T, Scheuer T, Catterall WA. Molecular determinants of drug access to the receptor site for antiarrhythmic drugs in the cardiac Na+ channel. Proc Natl Acad Sci USA. 1995;92:11839– 11843. doi: 10.1073/pnas.92.25.11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sunami A, Glaaser IW, Fozzard HA. Structural and gating changes of the sodium channel induced by mutation of a residue in the upper third of IVS6, creating an external access path for local anesthetics. Mot Pharmacol. 2001;59:684–691. doi: 10.1124/mol.59.4.684. [DOI] [PubMed] [Google Scholar]
- 47.Sunami A, Glaaser IW, Fozzard HA. A critical residue for isoform difference in tetrodotoxin affinity is a molecular determinant of the external access path for local anesthetics in the cardiac sodium channel. Proc Natl Acad Sci USA. 2000;97:2326–2331. doi: 10.1073/pnas.030438797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. The open pore conformation of potassium channels. Nature. 2002;417(6888):523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]