

Abstract
Insulin resistance is a major risk factor for type 2 diabetes. AMP-activated protein kinase (AMPK) is a drug target in the improvement of insulin sensitivity. Several insulin-sensitizing medicines are able to activate AMPK through inhibition of mitochondrial functions. These drugs, such as metformin and STZ, inhibit ATP synthesis in mitochondria to raise AMP/ATP ratio in the process of AMPK activation. However, chemicals that activate AMPK directly or by activating its upstream kinases have not been approved for treatment of type 2 diabetes in humans. In an early study, we reported that berberine inhibited oxygen consumption in mitochondria, and increased AMP/ATP ratio in cells. The observation suggests an indirect mechanism for AMPK activation by berberine. Berberine stimulates glycolysis for ATP production that offsets the cell toxicity after mitochondria inhibition. The study suggests that mitochondrial inhibition is an approach for AMPK activation. In this review article, literature is critically reviewed to interpret the role of mitochondria function in the mechanism of insulin resistance, which supports that mitochondria inhibitors represent a new class of AMPK activator. The inhibitors are promising candidates for insulin sensitizers. This review provides a guideline in search for small molecule AMPK activators in the drug discovery for type 2 diabetes.
Keywords: Insulin resistance, Mitochondria, Insulin sensitizer, Mitochondria inhibitor, Type 2 diabetes, Obesity
1. Introduction
Type 2 diabetes (T2D) is characterized by hyperglycemia coupled with hyperinsulinemia. The pathogenesis of T2D is insulin resistance, in which body has a low response to insulin although insulin remains at the normal or even higher levels in the blood. In compensation to insulin resistance, pancreatic β cells produce more insulin leading to insulin elevation. Long-term such compensation increases burden in β cells and often contributes to β cell failure at the end, which is responsible for hyperglycemia and diabetic complications in the late stage. Correction of insulin resistance has been a therapeutic approach in the treatment of type 2 diabetes. However, the medicines are very limited in the treatment of insulin resistance1. There is a strong demand for identification of new insulin sensitization drugs1.
Mechanism of insulin resistance is a focus in search for new diabetes medicines. There are several hypotheses regarding the cellular and molecular mechanisms of insulin resistance2,3 (Fig. 1). Those include mitochondrial dysfunction, endoplasmic reticulum (ER) stress, lipotoxicity, AMPK reduction, insulin elevation, oxidative stress, inflammation and adiponectin reduction. All of those mechanisms are proposed according to observations in the study of obesity, which represents energy surplus (fatty acids or glucose) in the body. Therefore, obese subjects have a high risk of insulin resistance. Fatty acids contain high density of energy and elevation of fatty acid in blood is a major risk factor for insulin resistance. In an early review, we proposed that fatty acids in combination with insulin is a major risk factor for insulin resistance in obesity3. In this review, we will extend this view point with a focus on mitochondrial function.
Figure 1.
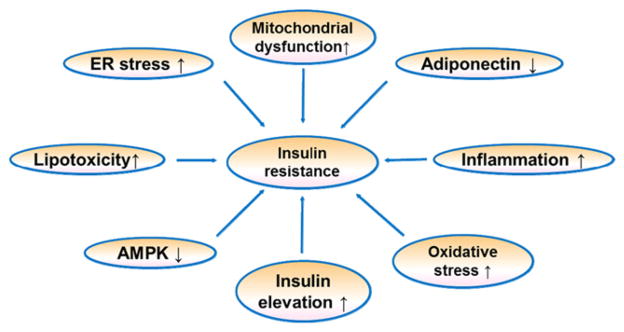
Mechanism of insulin resistance. There are several hypotheses about insulin resistance. These include mitochondrial dysfunction, endoplasmic reticulum (ER) stress, adiponectin reduction, inflammation, lipotoxicity, AMPK inactivation, oxidative stress, and insulin elevation.
Mitochondrion is a subcellular organ where fatty acids and glucose are used in the production of ATP through the oxidative phosphorylation process. The mitochondrial dysfunction is a hypothesis for insulin resistance4. The hypothesis suggests that when mitochondria cannot completely burn the fatty acids, the intermediate product of triglyceride such as diaglyceride (DAG) will accumulate in the cells to activate protein kinase C (PKC). In turn, PKC will induce insulin resistance by suppressing insulin signaling pathway through phosphorylation of insulin receptor substrate 1 (IRS-1)5. However, in two recent reviews, pharmacological and genetic evidence consistently suggest that mitochondrial dysfunction is not a cause of insulin resistance6,7. Instead, mitochondrial dysfunction is a consequence of insulin resistance.
2. Mitochondrial regulation
Mitochondria are the power-generating subcellular organ in cells and are required for ATP production in most cell types except red blood cells. The most important role of mitochondria is to produce ATP through oxidation of carbohydrate, fatty acids and amino acids. The key mitochondria enzymes include those such as pyruvate dehydrogenase, NADH dehydrogenase (Complex I), succinate dehydrogenase (Complex II), cytochrome bc1 (Complex III), and cytochrome c oxidase (Complex IV). In glucose catabolism, glucose is first converted into pyruvate through glycolysis in the cytoplasm. Pyruvate is converted into acetyl-CoA in mitochondria by pyruvate dehydrogenase, and then sent into TCA cycle to generate ATP, CO2 and H2O. This process is dependent on oxygen, and the activity is inhibited by hypoxia. In the absence of oxygen, pyruvate cannot be used in ATP production in mitochondria, but is converted into lactic acid in the cytoplasm in glycolysis (Fig. 2). In this condition, each molecule of glucose can only yield 2 molecules of ATP in net. In terms of fatty acid catabolism, long chain fatty acids are broken down into acetyl-CoA in mitochondria through β-oxidation, and acetyl-CoA is then used for ATP production in TCA cycle (Fig. 2). In hypoxia, TCA cycle is inhibited and thus, fatty acids cannot be used in ATP production. Fatty acid catabolism is completely dependent on oxygen supply.
Figure 2.

Glucose and fatty acids catabolism in cells. Glucose breaks down into pyruvate in the process of glycolysis. In the hypoxia condition, pyruvate becomes lactic acid in the cytoplasm and released out of cells. In the presence of oxygen, pyruvate is converted into acetyl-CoA by PDH in the mitochondria, and then used in TAC cycle for ATP production. The byproducts are water and carbon dioxide. Fatty acids breaks down into acetyl-CoA through β-oxidation in mitochondria, and then used in ATP production through TCA cycle. In hypoxia condition, fatty acid cannot be used in ATP production.
Mitochondrial function is regulated by several hormones and substrates. In the hormones, insulin regulates mitochondrial function through direct and indirect mechanisms. The direct mechanism includes insulin-induced gene expression and mitochondrial protein modification by phosphorylation. Indirectly, insulin increases substrate (glucose and fatty acid) supply to mitochondria through stimulation of the transporter activities. Glucose transporter 4 (GLUT4) and fatty acid transport protein (FATP) are activated by insulin leading to enhanced uptake of the substrates by cells. The gene expression, protein modification and substrate supply all promote mitochondrial function in ATP production. In obesity, glucose, fatty acids and insulin are all elevated in the blood. These factors promote mitochondrial function and contribute to the pathogenesis of insulin resistance. The mitochondria activation provides a mechanism for insulin resistance in the presence of insulin and fatty acids3.
Mitochondria function is determined by mitochondria number, size and quality. Those are controlled by mitochondrial biogenesis and mitochondria autophagy. The biogenesis is determined by gene expression and PGC-1α is a primary activator of the gene expression. Mitochondrial biogenesis is regulated by multiple factors. Insulin stimulates mitochondrial functions at multiple steps including oxidative phosphorylation, gene transcription and protein expression8. Mitochondrial recycle is regulated by autophagy, which removes damaged mitochondria from cells and uses them in energy production through recycling membrane lipids. In addition, mitochondrial function is also regulated by calcium homeostasis, reactive oxygen species (ROS), apoptosis and thermogenesis. These factors are influenced by substrates, nitric oxide, hypoxia, and hormones.
3. Mitochondrial dysfunction and insulin resistance
Mitochondrial dysfunction was first proposed as a mechanism of insulin resistance by Dr. Shulman’s group4. This hypothesis is supported by evidence in human and in animal. ADP is a substrate in the synthesis of ATP, and is able to stimulate mitochondria to produce ATP. This ADP activity is reduced in type 2 diabetes patients with obesity9. In animal studies, mitochondrial dysfunction is associated with insulin resistance10. All parameters of mitochondria functions are decreased in the adipose tissues of diabetic mice. Those include mitochondria number, mitochondria DNA content, and respiratory enzymes, oxidative phosphorylation (OXPHOS) and fatty acid β-oxidation. Mitochondrial defects were induced in murine C2C12 myotube cells using the respiratory chain inhibitors. In this cellular model, insulin actions such as insulin-stimulated glucose uptake and AKT activation in the insulin signaling pathway are decreased11. Inhibition of mitochondrial function by knocking down mitochondrial transcription factor A (mtTFA) suppressed insulin-stimulated glucose uptake. mtTFA is required for the replication and transcription of mitochondria DNA. These findings suggest that mitochondrial dysfunction from pharmacological treatment or genetic manipulation has close relationship with insulin resistance in adipocytes. In diabetic subjects, mitochondria number and mass are reduced in skeletal muscle 12,13. Several factors contribute to mitochondrial dysfunction.
3.1. Genetic factors
Mitochondrial biogenesis determines mitochondria mass and number. The biogenesis is regulated by mitochondrial DNA and genomic DNA. Mitochondrial DNA mutation presents in approximately 2% of patients with type 2 diabetes mellitus and in elderly individuals. In addition, a polymorphism of the mitochondrial coding region of the ND1 gene (a subunit of reduced NADH dehydrogenase) is associated with resting metabolic rate (RMR) in a large group of non-diabetic Pima Indians, a population with high risk of diabetes14. Mutation in mitochondrial DNA that encodes tRNA impairs insulin secretion in pancreatic β-cells15. Polymorphisms in the promoter DNA of UCP2 gene are associated with decreased incidence of obesity, reduced insulin secretion, and prevalence of diabetes16,17. In addition, nuclear DNA may determine mitochondrial function through encoding mitochondrial proteins. These association studies indicate that mitochondrial or nuclear DNA may influence mitochondrial function, and mitochondria may influence glucose metabolism through regulation of insulin sensitivity or insulin production. Nair et al.18 suggests that race/ethnicity determines the role of mitochondria in insulin resistance. In certain ethnic groups, there is dissociation between mitochondrial dysfunction and insulin resistance, and mitochondrial dysfunction cannot account for insulin resistance.
3.2. Oxidative stress
Mitochondrial function is dependent on oxygen during production of ATP. Derivatives of oxygen such as are generated during the process of ATP production. These intermediate products of oxygen are reactive oxygen species (ROS, a group of free radicals), and highly toxic for oxidation of lipids, proteins and DNA. Mitochondria usually prevent the toxicity by removal of those products through the antioxidant system, which convert the free radicals into H2O and CO2. If these intermediate products are not eliminated immediately in mitochondria, they will induce oxidative stress and damage protein, lipid and DNA functions through chemical reactions. In aging, oxidative stress is a factor for the reduced mitochondrial biogenesis. Oxidative stress is considered as risk factor for insulin resistance for inhibition of mitochondrial function. In high-fat diet models, H2O2 production is enhanced in mitochondria of human and rodent skeletal muscles, and redox-buffering capacity is reduced in the mitochondria19. When rodents were treated with a mitochondria- specific antioxidant or were genetically engineered to overexpress mitochondrial catalase, H2O2 emission was attenuated, and insulin sensitivity was preserved even in the face of a high-fat diet19.
Mitochondrial dysfunction increases the risk for oxidative stress, which in turn, activates various serine kinases (such as JNK, IKK, PKC, et al.). These serine kinases contribute to insulin resistance by phosphorylation of insulin receptor substrate (IRS) proteins. In hepatocytes, palmitate accelerated fatty acid β-oxidation and ROS generation, which inhibits insulin signal transduction through activation of JNK20. In addition, fatty acid metabolites such as DAG and long-chain fatty acyl-CoA (LCFA-CoA) may contribute to insulin resistance21,22. The intracellular accumulation of DAG activates PKCs, which increase serine phosphorylation of IRS proteins in the inhibition of insulin signaling. Enhanced oxidation of long-chain fatty acid in mitochondria is sufficient to reverse insulin resistance in liver and correct glucose intolerance in db/db obese mice23. In this study, expression of malonyl-CoA-insensitive carnitine palmitoyltransferase 1 (CPT1mt) in liver increases hepatic mFAO capacity and improves glucose tolerance in ob/ob mice. In the study, hepatic steatosis was not affected in CPT1mt mice, indicating dissociation between hepatic steatosis and insulin resistance. A recent research suggests that genetic deletion of the AMPK β1 subunit in mouse macrophages reduced fatty acid oxidation, mitochondrial content, and increased risk for insulin resistance. It suggests that increased fatty acid oxidation in macrophages by AMPK activation may represent a new therapeutic approach to the treatment of insulin resistance24.
Sedentary lifestyle and high-fat intake have deleterious impact on muscle mitochondrial oxidative capacity, and endurance exercise partly normalizes mitochondrial function and prevents age-associated insulin resistance25. Lim et al.26 reported that insulin resistance is associated with impairment of mitochondrial function. They demonstrated that chronic exposure to atrazine (ATZ) could block the electron transfer at Q site of electron transport chain and led to mitochondrial dysfunction, including morphological disruption, decreased activities of complexes I and III, and decreased oxygen consumption rate in the liver and skeletal muscle. Consequently, these resulted in insulin resistance in skeletal muscle by reducing AKT phosphorylation at Thr308 and Ser473 upon insulin stimulation. These findings indicate that ATZ may result in type 2 diabetes induced by impaired mitochondrial function.
Although a lot of articles suggest that mitochondrial dysfunction contributes to the pathogenesis of insulin resistance, more and more evidence suggests that inhibition of mitochondria actually improves insulin sensitivity. Mitochondrial dysfunction hypothesis is mainly based on the association studies, not cause/effect relationship studies. Recent studies using pharmacological and transgenic approaches have made a huge progress in the analysis of the cause/effect relationship for mitochondrial dysfunction and insulin resistance. The results consistently suggest that mitochondrial dysfunction is a consequence, but not cause of insulin resistance. Instead, mitochondrial over activation contributes to insulin resistance in obese conditions7. Following two points is supported by strong evidence: (I) mitochondrial dysfunction is a protection mechanism against insulin resistance; (II) mitochondrial inhibition is able to improve insulin sensitivity.
4. Mitochondrial inhibition as a strategy for insulin sensitization
Study of respiratory chain function in patients of type 2 diabetes suggests that mitochondrial dysfunction is not an intrinsic defect, but rather a consequence of the impaired insulin response27. In this study, the mitochondrial gene transcripts encoding proteins in the electric transport chain were expressed at higher levels in type 2 diabetic than in nondiabetic subjects. Increased insulin at postprandial stage caused an increase in mitochondrial ATP production rate in nondiabetic but not in type 2 diabetic subjects. Increased insulin reduced PGC-1, COX1, and citrate synthase expression in type 2 diabetic patients, but not in nondiabetic subjects27. It was reported that a reduction in oxidative phosphorylation in the liver or muscle of mice did not trigger the onset of diabetes but instead has the opposite effect, protecting mice against both diabetes and obesity28. The study suggests that the changes in respiratory chain function in insulin-resistant humans may be compensatory response in type 2 diabetes.
Mitochondrial dysfunction is not a cause of insulin resistance 6,7, but it may be a component of a “vicious cycle” exacerbating insulin resistance. Severe hyperglycemia can reversibly inhibit mitochondrial respiration in skeletal muscle cells29. Fatty acid also damages mitochondria functions30,31. Insulin resistance will reduce mitochondrial biogenesis12. In type 2 diabetes, hyperglycemia, hyperlipidimia and hyperinsulinemia together inhibits mitochondrial functions. In the mechanism, ROS may play a role, which inhibits mitochondrial proteins through oxidative stress and activation of stress-related signaling pathways.
In a time-course study, the relationship of insulin resistance and mitochondrial dysfunction was investigated in mice on a high-fat/high-sucrose diet32. Mice exhibited insulin resistance and evidence of oxidative stress in muscle at 1 month on the diet. Mitochondria impairment such as disrupted mitochondrial biogenesis, loss of mitochondrial structural integrity and respiratory functioning were found after prolonged exposure (>1 month) to the diet32. These results show that the mitochondrial dysfunction occurs after insulin resistance.
A growing body of evidence has revealed the importance of mitochondrial over activation in the pathogenesis of insulin resistance7. Transient inhibition of mitochondrial function is an approach in the improvement of insulin sensitivity. Many insulin sensitizing medicines and small molecules can transiently inhibit mitochondrial functions. Following pharmacological and genetic evidence supports that mitochondrial over activation contributes to insulin resistance and mitochondrial inhibition improves insulin sensitivity.
4.1. Pharmacological evidence
Pharmacological and genetic approaches have been used to test the relationship of mitochondrial dysfunction and insulin sensitivity. All of the clinically-approved medicines for insulin sensitization are able to inhibit mitochondrial function. Berberine, an herbal insulin sensitizer that is widely used in China, represents a class of herbal drug in the treatment of type 2 diabetes. Berberine down-regulates the expression of genes involved in lipogenesis and up-regulates those involved in energy expenditure in adipose tissue and muscle33. Besides, berberine treatment results in activation of AMPK, which stimulates oxidation of glucose and fatty acids in mitochondria. Berberine activates AMPK through inducing AMP/ATP ratio by suppressing mitochondrial function34.
The mitochondrial inhibition is transient and reversible in response to berberine. The mitochondrial function will be enhanced by AMPK after disappearance of the berberine-induced inhibition34. This study suggests that combination of inhibition and stimulation of mitochondrial function may contribute to improvement of insulin sensitivity. ATP depletion in response to berberine is associated with inhibition of gluconeogenesis and lipogenesis in the liver35. The study suggests that inhibition of mitochondrial function in liver may contribute to the therapeutic activities of berberine. In another study, insulin sensitizing medicines including berberine, thiazolidinediones (TZDs), and metformin all inhibited mitochondrial function36. The respiratory chain complex I is likely the target of those drugs in the inhibition of mitochondrial function37–40, while resveratrol and quercetin inhibit the ATP synthase 41.
Metformin is an effective anti-diabetic drug, which decreases hyperglycemia through insulin-like and insulin-sensitizing effects in liver and skeletal muscle cells. Metformin does not directly stimulate AMPK. It activates AMPK by inducing AMP/ATP ratio after inhibition of complex I of the electron transport chain in mitochondria. Metformin also enhances glucose utilization by uncoupling oxidative phosphorylation in isolated mitochondria42.
Thiazolidinediones (TZDs) are activators of PPAR and function as insulin sensitizers. Troglitazone is the first TZD-derived insulin sensitizer. Troglitazone significantly increased phosphorylation of AMPK and ACC at 5 mM in 15 min treatment. There was a transient increase in the AMP/ATP ratio. In adipose tissue, TZD promotes mitochondrial biogenesis 43,44. This activity may be related to AMPK activation. Release of adiponectin by adipocytes and inhibition of mitochondria may lead to AMPK activation. In addition, TZD may enhance mitochondrial biogenesis through induction of gene expression, such as PGC-1, which has strong activity in the mitochondrial biogenesis in vivo as shown in transgenic mice.
Resveratrol, a polyphenolic compound extracted from grape skins, has been demonstrated to increase the activity of SIRT145 as well as AMPK46, which in turn activates PGC-147. Resveratrol markedly attenuated weight gain in association with increment of mitochondrial biogenesis in muscle and adipose tissue in the high-fat-fed mice48. In parallel, the resveratrol-treated mice show enhanced endurance capacity, higher oxygen consumption as well as improved glucose tolerance48. Resveratrol inhibits the mitochondrial ATP synthase by binding to the subunit49, and leads to the activation of AMPK. This mechanism may contribute to insulin-sensitizing effect of resveratrol. The dose of resveratrol used in mice is not feasible in humans, although the effects of lower dose resveratrol was addressed in a human study50. Resveratrol analogs, such as combretastatin A-4 (CA-4), can function similarly as resveratrol and activate AMPK46.
4.2. Genetic evidence
In genetic study, inhibition of mitochondrial function by gene modification leads to protection of insulin sensitivity51. Several reports suggest mitochondrial hyperactivity and overload as major cause of insulin resistance. This point has been demonstrated in Asian Indian immigrants in the United States18 and in animal models of diabetes and obesity28,52,53. Previous genetic studies have shown that transient overexpression of the “master regulator” of mitochondrial biogenesis, PGC-1, increases mitochondrial content and insulin sensitivity in skeletal muscle cells54,55. However, persistent overexpression of PGC-1 in skeletal muscle induced systemic insulin resistance by reducing GLUT4 expression56. In contrast, skeletal muscle-restricted PGC-1 inactivation reduced mitochondrial content and protected mice from insulin resistance57,58. These mice show reduction in the mitochondrial biogenesis, low activity in TCA cycle and a shift from oxidative to glycolytic metabolism57,58.
When PGC-1 is reduced by knockdown, the skeletal muscle mitochondrial function was reduced in parallel with an increase in glucose uptake and glucose tolerance59. Similarly to the PGC-1 over expression mice, PPAR over-expressing mice have an increase in mitochondrial function and a decrease in insulin-induced glucose uptake in skeletal muscle 60. In these mice, insulin resistance is associated with diminished AMPK activity.
Mitochondrial flavoprotein apoptosis-inducing factor (AIF) is required to maintain the integrity of the mitochondrial respiratory apparatus, and this gene inactivation results in a progressive disruption of mitochondrial respiratory function with a long-term disruption in organ integrity61,62. Liver- and muscle-specific AIF ablation induces OXPHOS deficiency in mice, elevates glucose tolerance, reduces fat mass and increases insulin sensitivity28. Interestingly, the absence of PPAR in skeletal muscle results in down-regulation of mitochondrial biogenesis and function63. Moreover, these mice are resistant to high-fat-induced weight gain and have higher glucose tolerance even in the absence of exercise64.
A recent work on cardiolipin (CL) remodeling by ALCAT1 (a lyso-CL acyltransferase) suggests an alternative molecular mechanism by which mitochondrial hyperactivity causes insulin resistance51. ALCAT1 overexpression leads to CL deficiency and enrichment of docosahexaenoic acid (DHA) content, which is known to increase mitochondrial membrane potential, oxidative stress, and lipid peroxidation65,66. The ATP production rate was stimulated by ALCAT1 overexpression and insulin sensitivity was reduced in the condition. In opposite, insulin sensitivity was improved in muscle of knockout (ALCAT1−/−) mice. ALCAT1−/− mice are protected from diet-induced obesity and insulin resistance. In genetic obese db/db mice, ATP production rate is significantly higher in liver mitochondria relative to the control mice. Besides, ATP production rates were significantly lower in isolated mitochondria from liver and heart of ALCAT1−/− mice. These data support the notion that mitochondrial inhibition is able to improve insulin sensitivity. Muscle-specific deletion of the mitochondrial transcription factor Tfam does not induce insulin resistance in mice, suggesting that mitochondrial dysfunction in skeletal muscle is not a primary etiological event in type 2 diabetes59.
5. AMPK as a target for insulin sensitization
AMPK is a major cellular energy sensor and a master regulator of metabolic homeostasis. It is a heterotrimeric enzyme containing catalytic subunit and two regulatory subunits. AMPK is activated by two distinct signals: a Ca2+-dependent pathway mediated by CaMKK and an AMP-dependent pathway mediated by LKB167. AMPK serves as a unique metabolic control node as it senses cellular energy status through modulation of its activities via phosphorylation and allosteric activation by AMP. A number of physiological processes have been shown to stimulate AMPK, including conditions that lead to alterations of the intracellular AMP/ ATP ratio, which are hypoxia, glucose deprivation; and calcium concentration, as well as the action of various hormones, cytokines, and adipokines. The AMPK activation leads to the inhibition of energy-consuming biosynthetic pathways (such as fatty acid synthesis in liver and adipocytes, cholesterol synthesis in liver, protein synthesis in liver and muscle and insulin secretion from β-cells) and the activation of ATP-producing catabolic pathways (such as fatty acid uptake and oxidation in multiple tissue, glycolysis in heart and mitochondrial biogenesis in muscle). AMPK can also modulate transcription of specific genes involved in energy metabolism, thereby exerting long-term metabolic control. Activation of AMPK in the liver and muscle is expected to elicit a spectrum of beneficial metabolic effects with the potential to ameliorate the defects associated with insulin resistance. Because of its favorable effects on energy metabolism pathways, it is reasonable to consider AMPK as a potential therapeutic target in the prevention and the treatment of type 2 diabetes and insulin resistance.
There are a number of hormones and pharmacological agents reported to activate AMPK in vivo upon treatment of cells and tissues, such as metformin, TZDs, berberine, resveratrol, leptin, IL-6 and adiponectin. Changes in mitochondrial coupling and cellular energy state could account for the cellular AMPK activation. Metformin activates AMPK through reducing ATP levels after uncoupling oxidative phosphorylation in mitochondria in skeletal muscle cells; TZDs can activate AMPK by a mechanism inducing adiponectin expression in adipocytes68; adiponectin induces activation of AMPK in skeletal muscle and liver, increasing phosphorylation of ACC and fatty acid oxidation, enhancing glucose uptake and lactate production, and reducing glucose levels in vivo69, IL-6 can rapidly and robustly increased AMPK activity in myotubes, enhancing fatty acid oxidation as well as basal and insulin-stimulated glucose uptake70.
AMPK activation in the liver and skeletal muscle generates beneficial metabolic effects in the diabetic patients. However, the widespread AMPK activities make it hard to regulate the kinase activity in a tissue-specific manner. We need to keep in mind that AMPK activation could be beneficial for diabetic patients71,72.
6. Mitochondrial PDH and insulin resistance
Pyruvate dehydrogenase (PDH) is an enzyme with multiunit complex that catalyzes the conversion of pyruvate to acetyl-CoA in the glucose catabolism pathway. PDH is localized within the inner mitochondrial membrane, converting pyruvate to acetyl-CoA that is then oxidized in the trycarboxylic acid (TCA) cycle for ATP production. PDH activity is also required for fatty acid synthesis from glucose as acetyl-CoA is the substrate in fatty acid synthesis. When PDH activity is inhibited, glucose cannot be oxidized or converted into fatty acids. PDH activity is tightly regulated by reversible serine phosphorylation that is catalyzed by PDH kinase (PDK). Phosphorylation of the E1 catalytic subunit by the kinase inactivates the enzyme activity. The phosphorylation is catalyzed by four specific PDH kinases: PDK1, PDK2, PDK3 and PDK4. The activity of these kinases is enhanced by ATP, NADH and acetyl-CoA and inhibited by ADP, NAD+ and CoASH73,74. The activity of PDH is also regulated by Mg2+, Ca2+ and insulin.
PDH activity is reduced in type 2 diabetes condition, and the reduction is likely a result of insulin resistance. Insulin induces PDH activity through de-phosphorylation to enhance glucose utilization, which includes glycolysis, glucose oxidation, and fatty acid synthesis. PDH is required for glucose conversion into acetyl-CoA. Inhibition of PDH leads to reduction in ATP generation and fatty acid synthesis from glucose in most tissues. This point has been approved in transgenic mice with PDH inactivation by α subunit knockout. PDH inactivation in muscle, leads to heart hypertrophy in mice and on chow diet, the KO mice die from heart failure within a month after birth75. The inactivation in liver completely blocks fatty acid synthesis from glucose in liver76. However, glucose oxidation was not inhibited locally since liver expresses pyruvate carboxylase, which sends pyruvate into TCA cycle through oxaloacetate. In the liver-specific KO mice, insulin sensitivity is enhanced and body weight is reduced on chow diet76. A low PDH activity leads to a decrease in glucose utilization and an increase in fatty acid utilization in ATP production77. PDH activity is reduced in diabetes or obesity conditions in various tissues in animals or patients78,79. The reduced PDH activity is likely a result of insulin resistance as insulin induces PDH function.
However, an increase in PDH activity is beneficial in the control of blood glucose in obese condition. PDH activity is enhanced in PDK4 knockout mice, and is responsible for hypoglycemia during starvation of the mice80. The hypoglycemia is a result of decreased gluconeogenesis from lack of gluconeogenic substrate. PDH converts all pyruvates that come from amino acids or glycerol into acetyl-CoA, which is used in ATP production. However, this PDH activity protects mice from hyperglycemia and insulin intolerance in mice on HFD81. The KO mice have less weight gain on HFD, and this may contribute to the improved glucose tolerance. These studies suggest that an increase in PDH activity is beneficial in the control of blood glucose in obese condition. Current literature on genetic studies suggests that inhibition and activation of PDH activity both have beneficial effects in the control of insulin sensitivity in mice. More research is required to determine if PDH is a therapeutic target in the improvement of insulin sensitivity.
7. Mitochondrial inhibitors
Many mitochondrial inhibitors have been reported in literatures 82. Some of them are known to increase insulin sensitivity. These include berberine, metformin, TZDs, resveratrol, quercetin, curcumin and estrogen. Other inhibitors remain to be investigated for their activity in insulin sensitization. We list examples in Table 1.
Table 1.
Mitochondrial inhibitors and their function.
| Mitochondrial inhibitor | Function |
|---|---|
| Berberine | Inhibit mitochondrial respiration |
| Metformin | Inhibit mitochondrial respiration, uncoupling oxidative phosphorylation |
| TZDs | Inhibit respiratory chain complex I |
| Resveratrol | Inhibit mitochondrial ATP synthase |
| Piceatannol | Inhibit ATPase activity of mitochondrial ATP synthase |
| Diethylstilbestrol (DES) | Inhibit proton translocation activities of mitochondria |
| Quercetin | Inhibit ATPase activity of mitochondria |
| Genistein | Inhibit ATP hydrolysis and ATP synthesis activities of mitochondrial ATP synthase |
| Biochanin A | Inhibit ATPase activity of mitochondria |
| Epicatechin gallate (ECG) Epigallocatechin gallate (EGCG) | Inhibit ATP hydrolysis activity of ATP synthase |
| Curcumin | Inhibit ATPase activity of mitochondria |
| Phloretin, theaflavin, tannic acid | Inhibit ATPase activity of mitochondria |
| Estrogen | Inhibit ATPase activity of mitochondria |
| Oligomycin | Inhibit ATP synthase |
| Ossamycin | Inhibit both ATPase and oxidative phosphorylation activities of mitochondrial ATP synthase |
| Propranolol | Inhibit mitochondrial ATPase activity |
| Atrazine | Inhibit ATP synthesis activity of ATP synthase |
| Azide | Inhibit ATPase activity of mitochondria |
8. Conclusions
Mitochondrial dysfunction is not likely a risk factor for insulin resistance in type 2 diabetes. Instead, over activation of mitochondria is a potential risk for insulin resistance. A growing body of evidence has revealed the importance of mitochondrial over activation in the pathogenesis of insulin resistance. In obesity, substrate over-supply plus insulin over-production is a mechanism for the mitochondria over-activation. The mitochondria alteration leads to inhibition of AMPK. Insulin resistance occurs after AMPK activity is reduced (Fig. 3). These evidence-based views have advanced our understanding of mechanism of insulin resistance. Inhibition of mitochondrial function is an approach in the improvement of insulin sensitivity. Many insulin sensitizing medicines and small molecules inhibit mitochondrial functions. These activities of insulin sensitizing agents enforce our proposal that mitochondrial inhibitors may represent a new class of insulin sensitizer.
Figure 3.
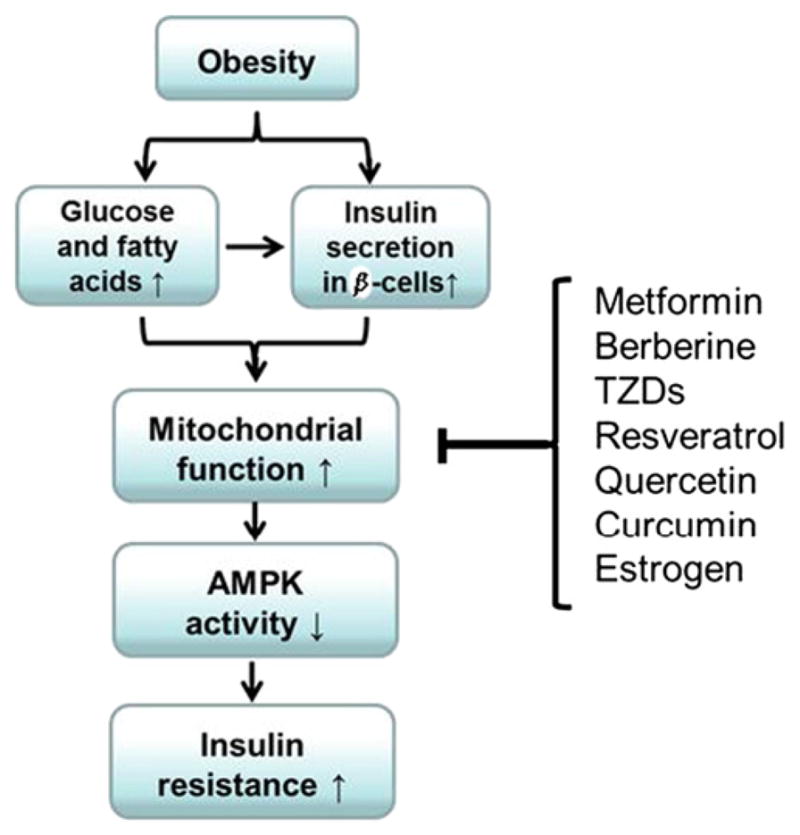
Mitochondrial inhibition as a new approach for insulin sensitization. In obesity, mitochondria over-activation by substrates and insulin leads to reduction in AMPK activity. Insulin signaling activity is inhibited in the presence of AMPK inactivation. Inhibition of mitochondrial function is an alternative pathway in AMPK activation.
Acknowledgments
Yong Zhang (postdoctoral fellow) and Jianping Ye (supervisor) wrote the manuscript. Dr. Ye is the guarantor of this work, had full access to all the data, and takes full responsibility for the integrity of data and the accuracy of data analysis. This work is supported by the National Institute of Health research projects (DK085495; DK068036).
Footnotes
Peer review under the responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Ye J. Challenges in drug discovery for thiazolidinedione substitute. Acta Pharm Sin B. 2011;1:137–42. doi: 10.1016/j.apsb.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ye J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int J Obes (Lond) 2009;33:54–66. doi: 10.1038/ijo.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ye J. Role of insulin in the pathogenesis of free fatty acid-induced insulin resistance in skeletal muscle. Endocr Metab Immune Disord Drug Targets. 2007;7:65–74. doi: 10.2174/187153007780059423. [DOI] [PubMed] [Google Scholar]
- 4.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–2. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–6. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schrauwen-Hinderling VB, Roden M, Kooi ME, Hesselink MK, Schrauwen P. Muscular mitochondrial dysfunction and type 2 diabetes mellitus. Curr Opin Clin Nutr Metab Care. 2007;10:698–703. doi: 10.1097/MCO.0b013e3282f0eca9. [DOI] [PubMed] [Google Scholar]
- 7.Pagel-Langenickel I, Bao J, Pang L, Sack MN. The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance. Endocr Rev. 2010;31:25–51. doi: 10.1210/er.2009-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stump CS, Short KR, Bigelow ML, Schimke JM, Nair KS. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mrna transcripts. Proc Natl Acad Sci USA. 2003;100:7996–8001. doi: 10.1073/pnas.1332551100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mogensen M, Sahlin K, Fernstrom M, Glintborg D, Vind BF, Beck-Nielsen H, et al. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2007;56:1592–9. doi: 10.2337/db06-0981. [DOI] [PubMed] [Google Scholar]
- 10.Choo HJ, Kim JH, Kwon OB, Lee CS, Mun JY, Han SS, et al. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia. 2006;49:784–91. doi: 10.1007/s00125-006-0170-2. [DOI] [PubMed] [Google Scholar]
- 11.Lim JH, Lee JI, Suh YH, Kim W, Song JH, Jung MH. Mitochondrial dysfunction induces aberrant insulin signalling and glucose utilisation in murine c2c12 myotube cells. Diabetologia. 2006;49:1924–36. doi: 10.1007/s00125-006-0278-4. [DOI] [PubMed] [Google Scholar]
- 12.Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 2005;115:3587–93. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 14.Jackman MR, Ravussin E, Rowe MJ, Pratley R, Milner MR, Willis WT. Effect of a polymorphism in the ND1 mitochondrial gene on human skeletal muscle mitochondrial function. Obesity. 2008;16:363–8. doi: 10.1038/oby.2007.40. [DOI] [PubMed] [Google Scholar]
- 15.Maassen JA, T Hart LM, Van Essen E, Heine RJ, Nijpels G, Jahangir TRS, et al. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004;53:S103–9. doi: 10.2337/diabetes.53.2007.s103. [DOI] [PubMed] [Google Scholar]
- 16.Esterbauer H, Schneitler C, Oberkofler H, Ebenbichler C, Paulweber B, Sandhofer F, et al. A common polymorphism in the promoter of UCP2 is associated with decreased risk of obesity in middle-aged humans. Nat Genet. 2001;28:178–83. doi: 10.1038/88911. [DOI] [PubMed] [Google Scholar]
- 17.Sesti G, Cardellini M, Marini MA, Frontoni S, D’Adamo M, Del Guerra S, et al. A common polymorphism in the promoter of UCP2 contributes to the variation in insulin secretion in glucose-tolerant subjects. Diabetes. 2003;52:1280–3. doi: 10.2337/diabetes.52.5.1280. [DOI] [PubMed] [Google Scholar]
- 18.Nair KS, Bigelow ML, Asmann YW, Chow LS, Coenen-Schimke JM, Klaus KA, et al. Asian indians have enhanced skeletal muscle mitochondrial capacity to produce ATP in association with severe insulin resistance. Diabetes. 2008;57:1166–75. doi: 10.2337/db07-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–81. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamura S, Takamura T, Matsuzawa-Nagata N, Takayama H, Misu H, Noda H, et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem. 2009;284:14809–18. doi: 10.1074/jbc.M901488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vankoningsloo S, Piens M, Lecocq C, Gilson A, De Pauw A, Renard P, et al. Mitochondrial dysfunction induces triglyceride accumulation in 3T3-l1 cells: role of fatty acid beta-oxidation and glucose. J Lipid Res. 2005;46:1133–49. doi: 10.1194/jlr.M400464-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Sato M, Ueda Y, Umezawa Y. Imaging diacylglycerol dynamics at organelle membranes. Nat Methods. 2006;3:797–9. doi: 10.1038/nmeth930. [DOI] [PubMed] [Google Scholar]
- 23.Monsénégo J, Mansouri A, Akkaoui M, Lenoir V, Esnous C, Fauveau V, et al. Enhancing liver mitochondrial fatty acid oxidation capacity in obese mice improves insulin sensitivity independently of hepatic steatosis. J Hepatol. 2012;56:632–9. doi: 10.1016/j.jhep.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 24.Galic S, Fullerton MD, Schertzer JD, Sikkema S, Marcinko K, Walkley CR, et al. Hematopoietic AMPK beta1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest. 2011;121:4903–15. doi: 10.1172/JCI58577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, et al. Endurance exercise as a countermeasure for aging. Diabetes. 2008;57:2933–42. doi: 10.2337/db08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim S, Ahn SY, Song IC, Chung MH, Jang HC, Park KS, et al. Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PLoS One. 2009;4:e5186. doi: 10.1371/journal.pone.0005186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asmann YW, Stump CS, Short KR, Coenen-Schimke JM, Guo Z, Bigelow ML, et al. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes. 2006;55:3309–19. doi: 10.2337/db05-1230. [DOI] [PubMed] [Google Scholar]
- 28.Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–91. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 29.Rabol R, Hojberg PM, Almdal T, Boushel R, Haugaard SB, Madsbad S, et al. Effect of hyperglycemia on mitochondrial respiration in type 2 diabetes. J Clin Endocrinol Metab. 2009;94:1372–8. doi: 10.1210/jc.2008-1475. [DOI] [PubMed] [Google Scholar]
- 30.Ghosh S, Kewalramani G, Yuen G, Pulinilkunnil T, An D, Innis SM, et al. Induction of mitochondrial nitrative damage and cardiac dysfunction by chronic provision of dietary omega-6 polyunsaturated fatty acids. Free Radic Biol Med. 2006;41:1413–24. doi: 10.1016/j.freeradbiomed.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 31.Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E. Peroxidative damage to cardiac mitochondria: cytochrome oxidase and cardiolipin alterations. FEBS Lett. 1998;424:155–8. doi: 10.1016/s0014-5793(98)00161-6. [DOI] [PubMed] [Google Scholar]
- 32.Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes. 2006;55:2256–64. doi: 10.2337/db06-0006. [DOI] [PubMed] [Google Scholar]
- 34.Yin J, Gao Z, Liu D, Liu Z, Ye J. Berberine improves glucose metabolism through induction of glycolysis. Am J Physiol Endocrinol Metab. 2008;294:E148–56. doi: 10.1152/ajpendo.00211.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia X, Yan J, Shen Y, Tang K, Yin J, Zhang Y, et al. Berberine improves glucose metabolism in diabetic rats by inhibition of hepatic gluconeogenesis. PLoS One. 2011;6:e16556. doi: 10.1371/journal.pone.0016556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11:554–65. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–8. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 38.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex I of the mitochondrial respiratory chain. Biochem J. 2000;348:607–14. [PMC free article] [PubMed] [Google Scholar]
- 39.Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes. 2004;53:1052–9. doi: 10.2337/diabetes.53.4.1052. [DOI] [PubMed] [Google Scholar]
- 40.Turner N, Li JY, Gosby A, To SW, Cheng Z, Miyoshi H, et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes. 2008;57:1414–8. doi: 10.2337/db07-1552. [DOI] [PubMed] [Google Scholar]
- 41.Zheng J, Ramirez VD. Inhibition of mitochondrial proton F1F0-ATPase/ATP synthase by polyphenolic phytochemicals. Br J Pharmacol. 2000;130:1115–23. doi: 10.1038/sj.bjp.0703397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martineau LC. Large enhancement of skeletal muscle cell glucose uptake and suppression of hepatocyte glucose-6-phosphatase activity by weak uncouplers of oxidative phosphorylation. Biochim Biophys Acta. 2012;1820:133–50. doi: 10.1016/j.bbagen.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 43.Boden G, Homko C, Mozzoli M, Showe LC, Nichols C, Cheung P. Thiazolidinediones upregulate fatty acid uptake and oxidation in adipose tissue of diabetic patients. Diabetes. 2005;54:880–5. doi: 10.2337/diabetes.54.3.880. [DOI] [PubMed] [Google Scholar]
- 44.Wilson-Fritch L, Nicoloro S, Chouinard M, Lazar MA, Chui PC, Leszyk J, et al. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest. 2004;114:1281–9. doi: 10.1172/JCI21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend saccharomyces cerevisiae lifespan. Nature. 2003;425:191–6. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 46.Zhang F, Sun C, Wu J, He C, Ge X, Huang W, et al. Combretastatin a-4 activates AMP-activated protein kinase and improves glucose metabolism in db/db mice. Pharmacol Res. 2008;57:318–23. doi: 10.1016/j.phrs.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 47.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–8. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 48.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–22. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 49.Gledhill JR, Montgomery MG, Leslie AG, Walker JE. Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proc Natl Acad Sci USA. 2007;104:13632–7. doi: 10.1073/pnas.0706290104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koo SH, Montminy M. In vino veritas: a tale of two SIRT1s? Cell. 2006;127:1091–3. doi: 10.1016/j.cell.2006.11.034. [DOI] [PubMed] [Google Scholar]
- 51.Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y, et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010;12:154–65. doi: 10.1016/j.cmet.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 53.Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes. 2006;55:2277–85. doi: 10.2337/db06-0062. [DOI] [PubMed] [Google Scholar]
- 54.Pagel-Langenickel I, Bao J, Joseph JJ, Schwartz DR, Mantell BS, Xu X, et al. PGC-1alpha integrates insulin signaling, mitochondrial regulation, and bioenergetic function in skeletal muscle. J Biol Chem. 2008;283:22464–72. doi: 10.1074/jbc.M800842200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, et al. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci USA. 2001;98:3820–5. doi: 10.1073/pnas.061035098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miura S, Kai Y, Ono M, Ezaki O. Overexpression of peroxisome proliferator-activated receptor gamma coactivator-1alpha down-regulates GLUT4 mRNA in skeletal muscles. J Biol Chem. 2003;278:31385–90. doi: 10.1074/jbc.M304312200. [DOI] [PubMed] [Google Scholar]
- 57.Handschin C, Chin S, Li P, Liu F, Maratos-Flier E, Lebrasseur NK, et al. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J Biol Chem. 2007;282:30014–21. doi: 10.1074/jbc.M704817200. [DOI] [PubMed] [Google Scholar]
- 58.Handschin C, Choi CS, Chin S, Kim S, Kawamori D, Kurpad AJ, et al. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J Clin Invest. 2007;117:3463–74. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wredenberg A, Freyer C, Sandstrom ME, Katz A, Wibom R, Westerblad H, et al. Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochem Biophys Res Commun. 2006;350:202–7. doi: 10.1016/j.bbrc.2006.09.029. [DOI] [PubMed] [Google Scholar]
- 60.Finck BN, Bernal-Mizrachi C, Han DH, Coleman T, Sambandam N, LaRiviere LL, et al. A potential link between muscle peroxisome proliferator-activated receptor-alpha signaling and obesity-related diabetes. Cell Metab. 2005;1:133–44. doi: 10.1016/j.cmet.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 61.Joza N, Oudit GY, Brown D, Benit P, Kassiri Z, Vahsen N, et al. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol. 2005;25:10261–72. doi: 10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–6. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 63.Schuler M, Ali F, Chambon C, Duteil D, Bornert JM, Tardivel A, et al. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006;4:407–14. doi: 10.1016/j.cmet.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 64.Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, et al. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hong MY, Chapkin RS, Barhoumi R, Burghardt RC, Turner ND, Henderson CE, et al. Fish oil increases mitochondrial phospholipid unsaturation, upregulating reactive oxygen species and apoptosis in rat colonocytes. Carcinogenesis. 2002;23:1919–25. doi: 10.1093/carcin/23.11.1919. [DOI] [PubMed] [Google Scholar]
- 66.Watkins SM, Carter LC, German JB. Docosahexaenoic acid accumulates in cardiolipin and enhances HT-29 cell oxidant production. J Lipid Res. 1998;39:1583–8. [PubMed] [Google Scholar]
- 67.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–48. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kubota N, Terauchi Y, Kubota T, Kumagai H, Itoh S, Satoh H, et al. Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin-dependent and -independent pathways. J Biol Chem. 2006;281:8748–55. doi: 10.1074/jbc.M505649200. [DOI] [PubMed] [Google Scholar]
- 69.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–95. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 70.Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, et al. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes. 2006;55:2688–97. doi: 10.2337/db05-1404. [DOI] [PubMed] [Google Scholar]
- 71.Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9:407–16. doi: 10.1016/j.cmet.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 72.Viollet B, Lantier L, Devin-Leclerc J, Hebrard S, Amouyal C, Mounier R, et al. Targeting the AMPK pathway for the treatment of type 2 diabetes. Front Biosci. 2009;14:3380–400. doi: 10.2741/3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329:191–6. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Baker JC, Yan X, Peng T, Kasten S, Roche TE. Marked differences between two isoforms of human pyruvate dehydrogenase kinase. J Biol Chem. 2000;275:15773–81. doi: 10.1074/jbc.M909488199. [DOI] [PubMed] [Google Scholar]
- 75.Sidhu S, Gangasani A, Korotchkina LG, Suzuki G, Fallavollita JA, Canty JM, Jr, et al. Tissue-specific pyruvate dehydrogenase complex deficiency causes cardiac hypertrophy and sudden death of weaned male mice. Am J Physiol Heart Circ Physiol. 2008;295:H946–52. doi: 10.1152/ajpheart.00363.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Choi CS, Ghoshal P, Srinivasan M, Kim S, Cline G, Patel MS. Liver-specific pyruvate dehydrogenase complex deficiency upregulates lipogenesis in adipose tissue and improves peripheral insulin sensitivity. Lipids. 2010;45:987–95. doi: 10.1007/s11745-010-3470-8. [DOI] [PubMed] [Google Scholar]
- 77.Montes M, Chicco A, Lombardo YB. The effect of insulin on the uptake and metabolic fate of glucose in isolated perfused hearts of dyslipemic rats. J Nutr Biochem. 2000;11:30–7. doi: 10.1016/s0955-2863(99)00068-6. [DOI] [PubMed] [Google Scholar]
- 78.Mondon CE, Jones IR, Azhar S, Hollenbeck CB, Reaven GM. Lactate production and pyruvate dehydrogenase activity in fat and skeletal muscle from diabetic rats. Diabetes. 1992;41:1547–54. doi: 10.2337/diab.41.12.1547. [DOI] [PubMed] [Google Scholar]
- 79.Alves TC, Befroy DE, Kibbey RG, Kahn M, Codella R, Carvalho RA, et al. Regulation of hepatic fat and glucose oxidation in rats with lipid-induced hepatic insulin resistance. Hepatology. 2011;53:1175–81. doi: 10.1002/hep.24170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jeoung NH, Wu P, Joshi MA, Jaskiewicz J, Bock CB, Depaoli-Roach AA, et al. Role of pyruvate dehydrogenase kinase isoenzyme 4 (PDHK4) in glucose homoeostasis during starvation. Biochem J. 2006;397:417–25. doi: 10.1042/BJ20060125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hwang B, Jeoung NH, Harris RA. Pyruvate dehydrogenase kinase isoenzyme 4 (PDHK4) deficiency attenuates the long-term negative effects of a high-saturated fat diet. Biochem J. 2009;423:243–52. doi: 10.1042/BJ20090390. [DOI] [PubMed] [Google Scholar]
- 82.Hong S, Pedersen PL. ATP synthase and the actions of inhibitors utilized to study its roles in human health, disease, and other scientific areas. Microbiol Mol Biol Rev. 2008;72:590–641. doi: 10.1128/MMBR.00016-08. [DOI] [PMC free article] [PubMed] [Google Scholar]