

Abstract
The heterodimeric hypoxia inducible factor-1 (HIF-1) complex is composed of the hypoxia inducible factor-1 alpha (HIF-1α) and the aryl hydrocarbon receptor nuclear translocator (ARNT). Activation of the HIF-1 function is essential for tumor growth and metastasis. We previously showed that transfection of a plasmid containing an ARNT-interacting peptide (Ainp1) cDNA suppresses the HIF-1 signaling in Hep3B cells. Here we generated TAT fusion of the Ainp1 peptide (6His-TAT-Ainp1) to determine whether and how the Ainp1 peptide suppresses the HIF-1 function. The bacterially expressed 6His-TAT-Ainp1 was purified under denatured condition and then refolded by limited dialysis. The refolded 6His-TAT-Ainp1 interacts with the helix-loop-helix (HLH) domain of ARNT in a similar fashion as the native 6His-Ainp1. 6His-TAT-Ainp1 colocalizes with ARNT in the nucleus of HeLa and Hep3B cells after protein transduction. The transduced protein reaches the maximum intracellular levels within 2 h while remains detectable up to 96 h in HeLa cells. At 2 µM concentration, 6His-TAT-Ainp1 is not cytotoxic in HeLa cells but suppresses the cobalt chloride-activated, hypoxia responsive enhancer-driven luciferase expression in a dose-dependent manner. In addition, it decreases the cobalt chloride-dependent induction of the HIF-1 target genes at both the message (vascular endothelial growth factor and aldolase C) and protein (carbonic anhydrase IX and glucose transporter 1) levels. The protein levels of HIF-1α and ARNT are not altered in the presence of 6His-TAT-Ainp1. In summary, we provided evidence to support that the Ainp1 peptide directly suppresses the HIF-1 function by interacting with the ARNT HLH domain, and in turn interfering with the heterodimerization of HIF-1α and ARNT.
Keywords: ARNT, HIF-1α, ARNT-interacting peptide, cell-penetrating peptide
1. Introduction
ARNT (aka HIF-1β) and HIF-1α contain homologous bHLH and two in-tandem PAS (PAS-A and PAS-B) domains. These domains serve as dimerization motifs to form the HIF-1 heterodimer, which in turn binds to hypoxia inducible elements in the regulatory regions of the corresponding target genes [1, 2]. Under normoxia, HIF-1α is hydroxylated by oxygen-dependent prolyl hydroxylase, and the hydroxylated HIF-1 is subjected for proteasomal degradation. Hypoxia, therefore, stabilizes HIF-1α by limiting the hydroxylase activity [3, 4]. The upregulated HIF-1 activity alters the expression of more than 60 downstream target genes, and many of these genes are related to cancer progression involving metabolic adaption, apoptosis resistance, angiogenesis, and metastasis [5]. The correlation between the high intratumoral HIF-1α protein levels and the increased mortality rates of cancer patients has been reported [6, 7]. Thus, any mechanism to suppress the HIF-1α function could potentially be an effective approach for new anticancer drug development.
The protein transduction domains (PTDs) have been shown to effectively deliver a wide range of “cargos” – such as peptides, proteins, polyanionic oligonucleotides, and liposomes – across the mammalian cell membrane in both size and concentration-independent manners [8]. For example, the transactivator of transcription (TAT) PTD is a short arginine-rich 11 amino acid peptide (YGRKKRRQRRR), which is amino acid 47–57 of the human immunodeficiency virus TAT protein. TAT PTD strongly interacts with the cell surface because of its positive-charged arginine-rich sequence. After penetrating into cells through lipid raft-mediated endocytosis, the TAT fusion that survives through lysosomal degradation escapes from endosomes to cytoplasm to elicit function [9].
Previously we discovered Ainp1, which is an ARNT-interacting peptide of 59 amino acids in length, using a phage display method [10]. We observed that transfected Ainp1 suppresses the HIF-1α function by retaining ARNT in the cytoplasm of Hep3B cells [11]. In this study, we characterized the interaction between Ainp1 and ARNT, revealing that hindrance at the HLH domain of ARNT would prohibit its dimerization with HIF-1α. Our evidence supports that the HLH domain of ARNT could be a potential target for suppression of the HIF-1 function. In addition, we utilized the TAT-mediated peptide delivery to unambiguously prove that the Ainp1 peptide reaches the cell nucleus and suppresses the HIF-1 signaling in cell culture experiments.
2. Materials and methods
2.1. Reagents
All cell culture were grown at 37 °C and 5% CO2. HeLa and MCF-7 cells were grown in DMEM (Sigma-Aldrich, St Louis, MO) supplemented with 10% Hyclone FBS, 2mM GlutaMAX-I, 100 units/ml of penicillin, and 0.1 mg/ml of streptomycin. Hep3B cells were grown in Advanced MEM (Gibco, Carlsbad, CA) supplemented with 5% Hyclone FBS, 2mM GlutaMAX-I, 100 units/ml of penicillin, and 0.1 mg/ml of streptomycin. Cell culture reagents, if not specified, were purchased from Invitrogen (Carlsbad, CA). Cobalt chloride, the hypoxia-mimicking agent, was purchased from Sigma-Aldrich. Anti-Thio monoclonal mouse IgG was purchased from Invitrogen (Carlsbad, CA). His-probe monoclonal mouse IgG (sc-8036), mouse IgG (sc-2025), Trx monoclonal mouse IgG (sc-13526), and anti-CAIX polyclonal rabbit IgG (sc-25599) were purchased from Santa Cruz Biotechonology. Anti-Glut1 polyclonal rabbit IgG (NB300–666) was purchased from Novus Biologicals (Littleton, CO). Anti-HIF1α monoclonal rabbit IgG (2015-1) was purchased from Epitomics (Burlingame, CA). Anti-Ainp1 polyclonal mouse IgG was generated using the bacterially expressed 6His-Ainp1 as previously described [11]. Alexa Fluor 555 goat anti-mouse IgG, Alexa Fluor 488 goat anti-rabbit IgG, and DAPI were generous gifts from Dr. Lisa Wrischnik (University of the Pacific). Oligonucleotides were purchased from Invitrogen (Carlsbad, CA). pThioHis-ARNT CΔ418 plasmid containing the thioredoxin fusion of C-terminal 418-amino-acid deletion of human ARNT cDNA was generated as previously described [11]. The thioredoxin fusion of ARNT deletion plasmids – pThioHis-D1, pThioHis-D2, pThioHis-D1A, pThioHis-D1B, pThioHis-D1C, pThioHis-basic and pThioHis-HLH – were generated by amplifying the corresponding cDNAs with the specific primers (Table 1) using PCR and then cloning the cDNAs into BglII and XbaI sites of the pThioHis C plasmid (Invitrogen, Carlsbad, CA). Expression and affinity purification of thioredoxin fusion proteins were previously described [10]. The pQE80-TAT plasmid was generated by cloning the annealed OL460 (sense, 5’-GGAGGCTACGGCCGCAAGAAACGCCGCCAGCGCCGCCGCGGTGGAGGTAC-3’) and OL461 (antisense, 5’-CTCCACCGCGGCGGCGCTGGCGGCGTTTCTTGCGGCCGTAGCCTCCGCATG-3’) into the SphI and KpnI sites of the pQE-80 plasmid (Qiagen, Valencia, CA). The TAT fusion of Ainp1 plasmid (pQE80-TAT-Ainp1) was generated by amplifying the Ainp1 cDNA [11] with OL472 (sense, 5’-AAAACTGCAGCCCAGACACATGCAGACACACAC-3’) and OL442 (antisense, 5’-CCCAAGCTTCTATGAGTGTGTCTGTTTGTGTGTCTG-3’) using PCR and then cloning the Ainp1 cDNA into the PstI and HindIII sites of the pQE80-TAT plasmid. The pQE80-TAT-ΔGFP plasmid was generated by amplifying the N-terminal 58 amino acids of GFP with OL584 (sense, 5’-AAAACTGCAGCCGTGAGCAAGGGCGAGGAGCTG-3’) and OL585 (antisense, 5’-CCCAAGCTTCTAGGGCCAGGGCACGGGCAGCTT-3’) using PCR and then cloning the ΔGFP cDNA into PstI and HindIII sites of the pQE80-TAT plasmid. The pGL3-Epo luciferase reporter plasmid was generated as previously described [12]. The β-galactosidase plasmid pCH110 was purchased from Amersham Pharmacia (Piscataway, NJ). RT-qPCR primers for ARNT are OL144 (sense: 5’-GAATTGGACA0]TGGTACCAGG-3’) and OL145 (5’-AAGCTGATGGCTGGACAATG-3’). RT-qPCR primers for 18S, VEGF, and adolase C were previously published [12]. CellTiter 96 non-radioactive cell proliferation assay kit was purchased from Promega (Madison, WI). Dual-Light luciferase and β-galactosidase reporter assay was purchased from Applied Biosystems (Foster City, CA). All western analyses were performed using a LI-COR Odyssey imaging system (Lincoln, NE). The western protocol using a near-infrared detection method was described previously [11].
Table 1.
A summary of all PCR primers used to generate human ARNT deletions.
| D1 (1–160 aa) | Forward: OL535 5’-GAAGATCTGCGGCGACTACTGCCAACCCC-3’ Reverse: OL536 5’-GCTCTAGACTAAAAGCCATCTGCTGCCTCCAA-3’ |
| D2 (127–356 aa) | Forward: OL537 5’-GAAGATCTTTGCGGGGAACTGGCAACACA-3’ Reverse: OL540 5’-GCTCTAGACTAACCCTCAATGTTGTGTCGGGA-3’ |
| D1A (1–75 aa) | Forward: OL535 5’-GAAGATCTGCGGCGACTACTGCCAACCCC-3’ Reverse: OL548 5’-GCTCTAGACTAGGCAAACCGCTCCTTATCGTT-3’ |
| D1B (70–140 aa) | Forward: OL549 5’-GAAGATCTGATAAGGAGCGGTTTGCCAGG-3’ Reverse: OL550 5’-GCTCTAGACTACTTATAGGAGCCATCAGTGGA-3’ |
| D1C (130–160 aa) | Forward: OL551 5’-GAAGATCTACTGGCAACACATCCACTGAT-3’ Reverse: OL536 5’-GCTCTAGACTAAAAGCCATCTGCTGCCTCCAA-3’ |
| basic (75–87) aa | Forward: OL601 5’-GATCTGCCAGGGAAAATCACAGTGAAATTGAACGGCGGCGACGGTAAT-3’ Reverse: OL602 5’-CTAGATTACCGTCGCCGCCGTTCAATTTCACTGTGATTTTCCCTGGCA-3’ |
| HLH (88–128 aa) | Forward: OL603 5’-GAAGATCTAACAAGATGACAGCCTACATC-3’ Reverse: OL604 5’-GCTCTAGATTACCGCAAGGACTTCATGTGAGA-3’ |
2.2. Denatured purification and refolding of TAT fusions
Overnight LB culture (JM109, 100 ml) carrying either pQE80-TAT-Ainp1 or pQE80-TAT-ΔGFP plasmid in the presence of ampicillin (100 µg/ml) was added to 500 ml of fresh LB media containing IPTG (1 mM) and ampicillin (100 µg/ml). After 6 h induction at 37°C with shaking at 225 rpm, the bacteria were collected by centrifugation at 3,100 g for 20 min. After washing with PBS once, the bacteria were incubated with 15 ml of lysis buffer (20 mM HEPES, pH 8.0, 8 M urea, 100 mM NaCl) at room temperature with rotation for 1 h. The bacteria lysate was sonicated on ice for 5 min and then centrifuged at 16,000 g for 30 min. The supernatant was combined with 0.5 ml of pre-equilibrated TALON resin (Clontech, Mountain View, CA) and incubated with rotation at room temperature overnight. The resulting protein-resin suspension was poured into the poly-prep column (Bio-Rad, Hercules, CA). The resin was washed with 5 ml of lysis buffer, followed by a second wash of 5 ml of lysis buffer containing 10 mM imidazole before elution with 1.5 ml of lysis buffer containing 0.5 M imidazole. The eluant containing denatured recombinant protein was subjected for protein refolding by sequential limited dialysis as follows: 1.5 ml of eluant was dialyzed against 100 ml of five dialysis buffers (all containing PBS and 10% glycerol) with different ingredients in the following order: 6 M urea, 4 M urea, 2 M urea, 1 M arginine, and finally with PBS containing 10% glycerol. Each dialysis was performed at 4 °C for 2–4 h. The refolded protein (6His-TAT-Ainp1 or 6His-TAT-ΔGFP) was filtered through 0.2 µm syringe filter (GE Healthcare, Waukesha, WI) before being used for cell culture experiment.
2.3. Co-immunoprecipitation assay
We performed the co-immunoprecipitation assay as previously described with modification [11]. Bacterially expressed 6His-Ainp1, 6His-TAT-Ainp1 or 6His-TAT-ΔGFP (40 µg) was incubated with bacterially expressed thioredoxin fusion of D1, D2, D1A, D1B, D1C, HLH or basic (40 µg) in 400 µl of HEDG buffer (25 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM DTT and 10% glycerol) containing 0.1 M KCl at 30 °C for 30 min. For Ainp1∷ARNT interaction study, bacterially expressed 6His-Ainp1, 6His-TAT-Ainp1 or 6His-TAT-ΔGFP (40 µg) and HeLa whole cell lysate (1 mg) was used instead. HeLa whole cell lysate was prepared by four cycles of freeze-thaw in lysis buffer (HEDG buffer containing 0.4 M KCl), followed by centrifugation at 16,000g for 20 min to obtain the supernatant. Each supernatant was quickly diluted with HEDG buffer to 0.1 M final concentration of KCl to become whole cell lysate. Pre-equilibrated Dynabeads protein G (2 µl, Invitrogen, Carlsbad, CA) was added to each sample and the resulting samples were incubated at 4 °C with rotation for pre-clearing. After 30 min, samples were separated from the beads using Dynamag-2 magnet (Invitrogen, Carlsbad, CA) and then either anti-Ainp1 mouse IgG or mouse IgG (1 µg) was incubated with the corresponding samples at 4 °C. In some cases, anti-thioredoxin mouse IgG (1 µg) was used instead. After 4 h, pre-equilibrated Dynabeads (5 µl) was added to each sample, followed by incubation with rotation at 4 °C. After 1.5 h, the beads were washed four times with 600 µl of wash buffer (HEDG buffer containing 0.4% Tween-20, 5 mM imidazole and 10 mM 2-mercaptoethanol). Samples were retrieved from beads by boiling in SDS-PAGE sample denaturing buffer (20 µl) and then subjected for western analysis.
2.4. TAT-fusion protein transduction
HeLa cells (1× 105) were grown in each well of a 24-well plate. Cells were treated with Opti-MEM (400µl) containing 6His-TAT-Ainp1 (2 µM). At each time point, cells were washed once with TrypLE Express (150 µl, Invitrogen, Carlsbad, CA) and then incubated with TrypLE Express (150 µl) at 37 °C. After 3 min, cells were gently washed three times by cold PBS (500 µl) and then lysed with SDS-PAGE sample denaturing buffer (30 µl) for western analysis. To study how long the TAT-fusion protein would remain in HeLa cells, HeLa cells were incubated with either 6His-TAT-Ainp1 or 6His-TAT-ΔGFP (2 µM) at 37 °C. After 2 h, cells were washed three times with 500 µl of medium and allowed to grow in the medium at 37 °C. To harvest, cells in each well were resuspended with 50 µl of RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.1% SDS, 0.25% sodium deoxycholate, 1% Triton X-100, 1 mM PMSF, 2 µM leupeptin). The cell suspension was incubated with rotation for 1 h at 4 °C, followed by centrifugation at 16,000g for 10 min. The supernatant (30 µl) was subjected for western analysis.
2.5. Immunofluorescence staining study
HeLa cells were treated with either 6His-TAT-Ainp1 or 6His-TAT-ΔGFP (2 µM) when cells reached 50% confluence in a 6-cm tissue culture plate. After 4 h, the cells were washed twice with PBS, fixed using 4% formaldehyde for 15 min, and then incubated in blocking buffer (PBS containing 5% BSA and 0.3% Triton X-100) at room temperature. After 30 min, the cells were incubated with the primary antibodies (blocking buffer containing anti-Ainp1 mouse IgG and anti-ARNT rabbit IgG, both were 1 µg/ml) at 4 °C overnight. The cells were washed (5 min) twice with PBS and then twice with blocking buffer, followed by incubation with the secondary antibodies (blocking buffer containing Alexa Fluor 555 goat anti-mouse IgG and Alexa Fluor 488 goat anti-rabbit IgG, both at 1:5000 dilution) at room temperature. After 2 h, the cells were washed (5 min) with PBS twice and water once. After wash, cells were incubated with DAPI (100 ng/ml in water) at room temperature for 10 min. Immunofluorescence staining was observed using a Nikon Eclipse TE200 fluorescence microscope.
2.6. Cell viability studies
HeLa cells (8 × 103) were grown in each well of a 96-well plate overnight. Cells were treated with 80 µl of Opti-MEM containing 2 µM 6His-TAT-ΔGFP or 6His-TAT-Ainp1 or vehicle (PBS containing 10% glycerol). After 2 h at 37 °C, 20 µl of growth media containing cobalt chloride (100 µM) or water was added into each well. Cell viability was determined every 24 h using the CellTiter assay kit (Promega, Madison, WI). For microscopic imaging, either HeLa or Hep3B cells (1 × 105) were grown in each well of a 24-well plate overnight, then cells were incubated in 400 µl of Opti-MEM containing 2 µM 6His-TAT-ΔGFP or 6His-TAT-Ainp1 or vehicle (PBS containing 10% glycerol). After 2 h at 37 °C, media was aspirated and 100 µl of growth media was added.
After 48 h at 37 °C, pictures were taken under a Nikon light microscope.
2.7. Luciferase assay
HeLa cells (1 × 105) were grown in each well of a 24-well plate. Cells were transfected with 0.6 µl of Fugene HD (Promega, Madison, WI) and 0.2 µg of reporter plasmids (175 ng of pGL3-Epo and 25 ng of pCH110) in growth media (500 µl) at 37 °C, 5% CO2. After 24 h, media was aspirated and cells were treated with 400 µl of Opti-MEM containing different concentrations of 6His-TAT-ΔGFP and 6His-TAT-ΔGFP (0–2 µM) in the presence or absence of cobalt chloride. After 16 h, cells were analyzed using the Dual-Light luciferase kit (Applied Biosystems, Foster City, CA).
2.8. Real-time qPCR
HeLa cells (6 × 105) were grown in each well of a 6-well plate. Cells were treated with 1 ml of Opti-MEM containing 6His-TAT-ΔGFP or 6His-TAT-Ainp1 (2 µM) or vehicle (PBS containing 10% glycerol). After 2 h, growth media (200 µl) containing cobalt chloride (100 µM) or water was added to each well. After 16 h, mRNA was extracted using MasterPure RNA purification kit (Epicentre, Madison, WI). The purified mRNA (1 µg) was amplified by 0.5 µg of random primer (Promega, Madison, WI) to generate cDNA using MMLV reverse transcriptase (Epicentre, Madison, WI). The reverse transcription product (1 µl) was mixed with iQ SYBR Green Supermix (Bio-Rad, Hercules, CA) for RT-qPCR analysis of target gene expression, whereas 1 µl of 1:10,000 dilution was used for 18S standard determination. A Bio-Rad iCycler was used for real-time PCR analysis. The PCR conditions were: 90 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s (40 cycles). The RT-qPCR product was analyzed on an agarose gel to make sure that only one DNA product was amplified. The data was analyzed using 2−ΔΔCT method and 18S was used as the standard [13].
2.9. Statistical analysis
GraphPad Prism 5 software was utilized for statistical analysis. The significant difference was determined by performing one-way (Fig. 3A, 4A) or two-way (Fig. 5A–D, F) ANOVA followed by post hoc Bonferroni’s multiple comparison test to determine the statistical significance with 95% confidence intervals.
Fig. 3.
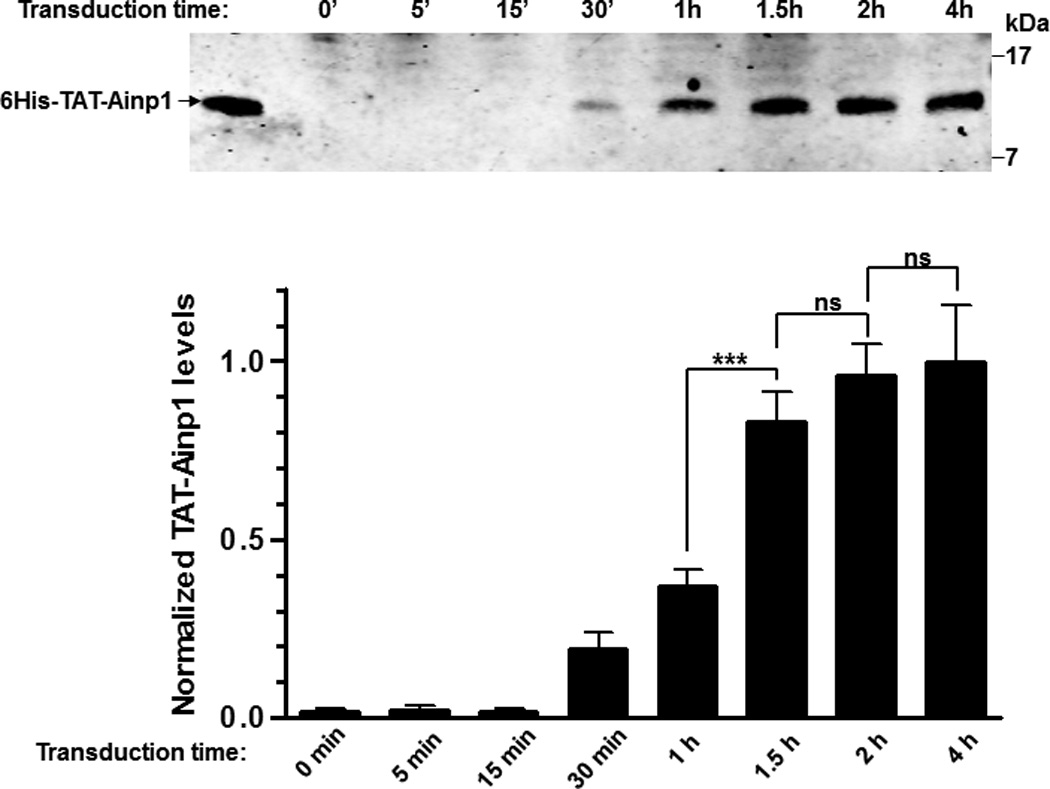
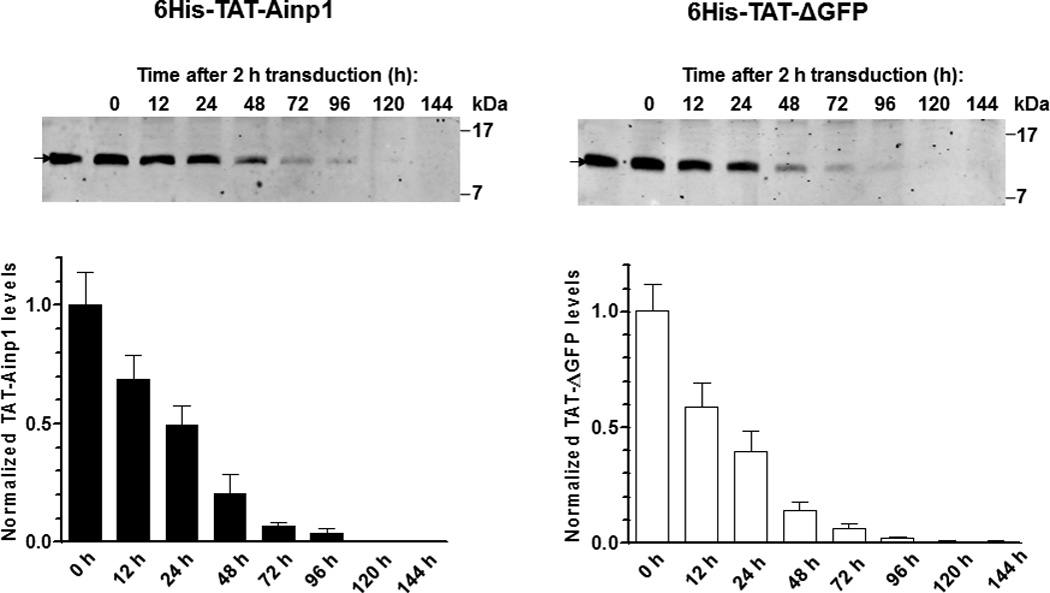
Timecourse study of protein transduction of 6His-TAT-Ainp1. (A) Western showing the intracellular 6His-TAT-Ainp1 peptide levels within 4 h of 2 µM treatment in HeLa cells. (B) Western showing the timecourse of the intracellular 6His-TAT-Ainp1 or 6His-TAT-ΔGFP peptide levels from 0–144 h. Fresh media was exchanged 2 h after 2 µM treatment. 16% tricine gel was used for analysis [39]. Arrows indicate the bands of interest. Error bars indicate the variations of the means (mean ± SD, n = 3). *** p < 0.001; ns, not significant. The experiments (A and B) were repeated twice with similar results.
Fig. 4.
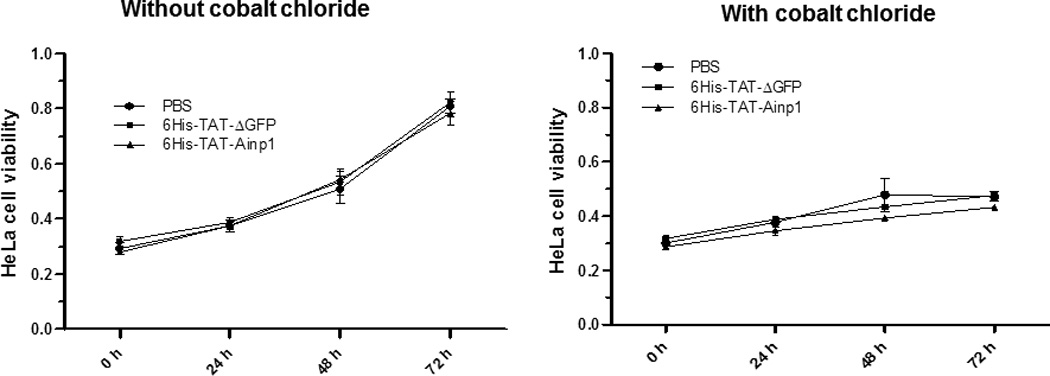
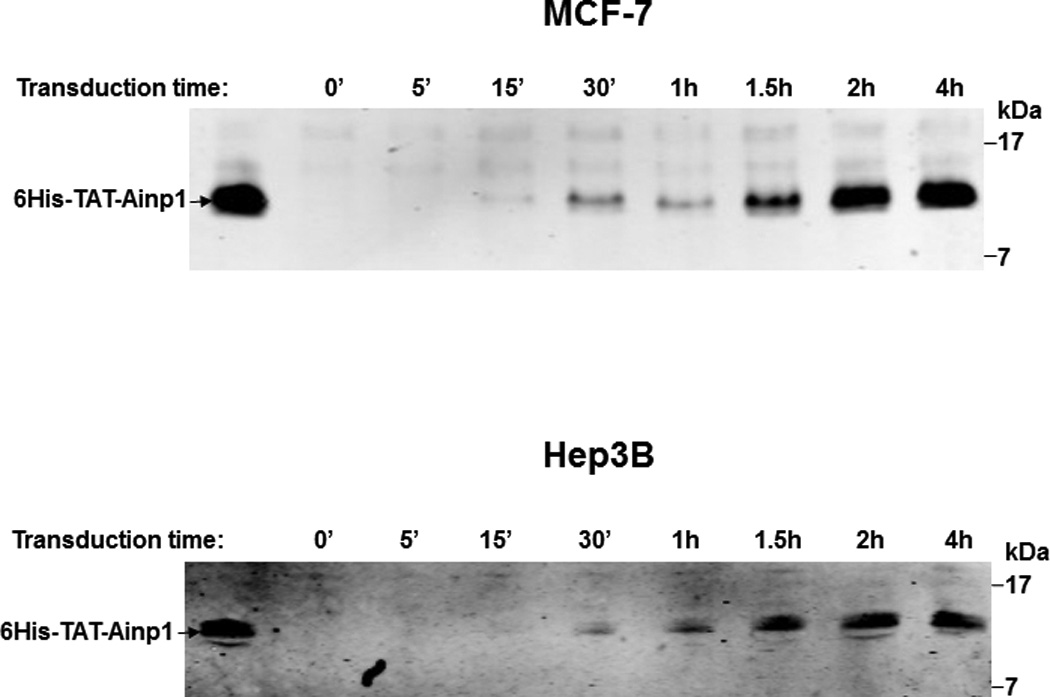
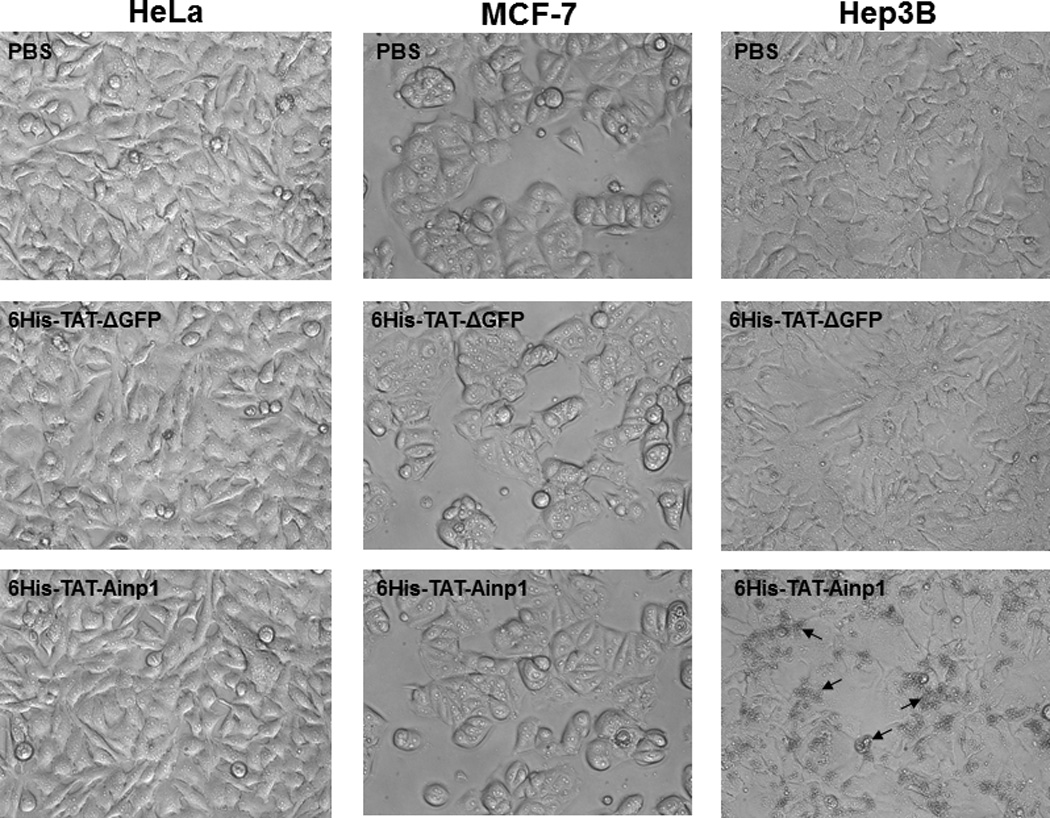
Cell viability assay of 6His-TAT-Ainp1 treatment. (A) Cell viability was measured every 24 h in HeLa cells treated with PBS, 6His-TAT-ΔGFP (2 µM) or 6His-TAT-Ainp1 (2 µM) ± 100 µM cobalt chloride. Error bars indicate the variations of the means (mean ± SD, n = 3). This experiment was repeated once with similar results. (B) Western showing the intracellular 6His-TAT-Ainp1 levels within 4 h of 6His-TAT-Ainp1 (2 µM) treatment in MCF-7 and Hep3B cells. 16% tricine gel was used for analysis [39]. (C) Light microscopy images showing the cell morphology after 24 h of PBS, 6His-TAT-ΔGFP (2 µM) or 6His-TAT-Ainp1 (2 µM) treatment in HeLa, MCF-7 and Hep3B cells. Arrows indicate examples of dead Hep3B cells.
Fig. 5.
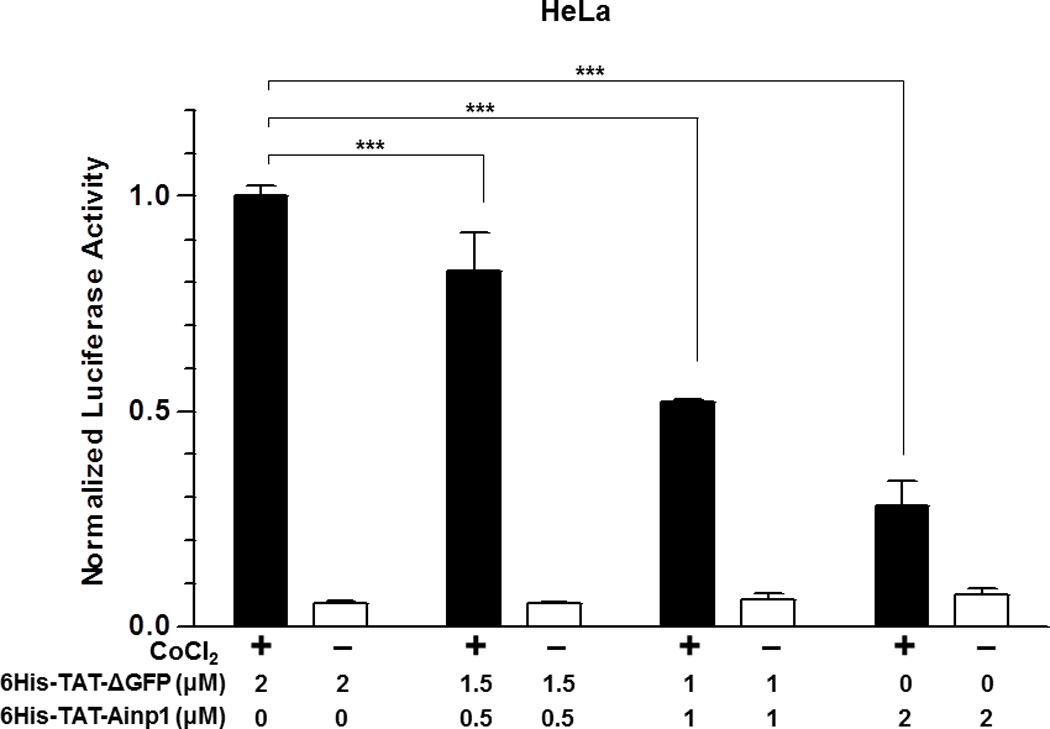
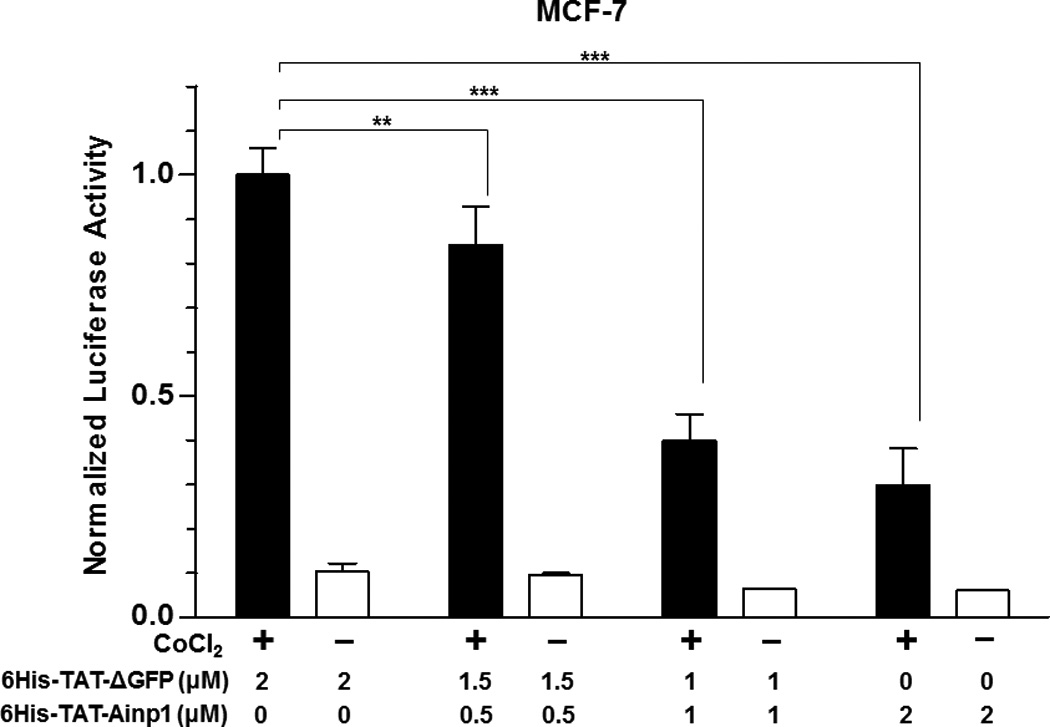
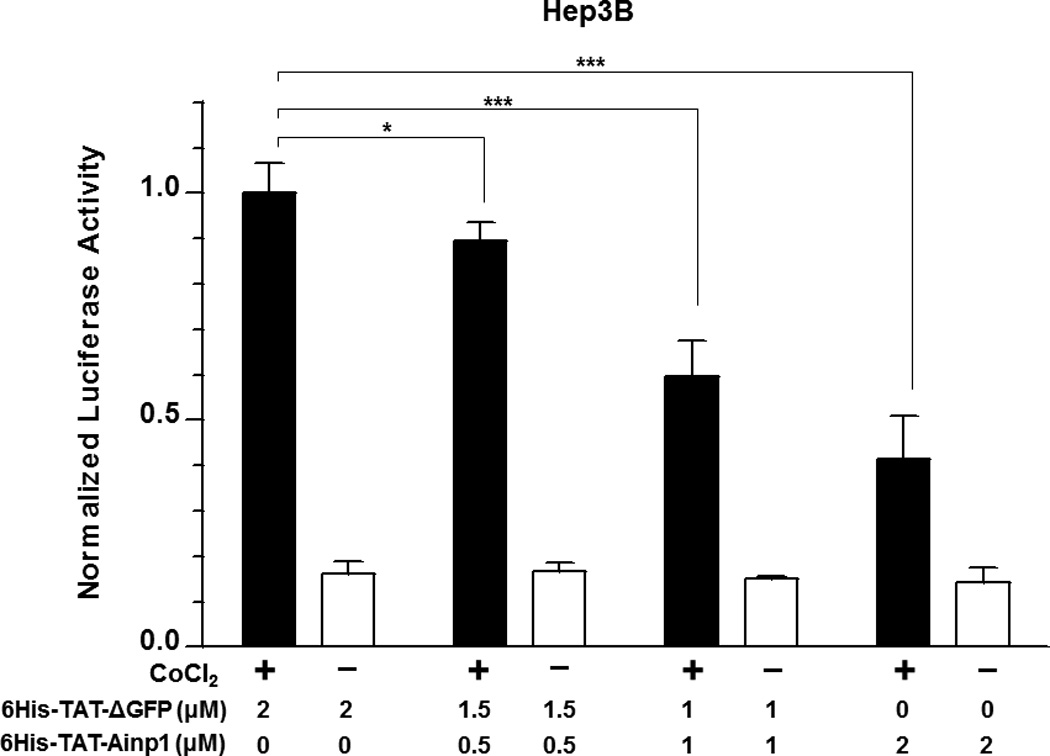
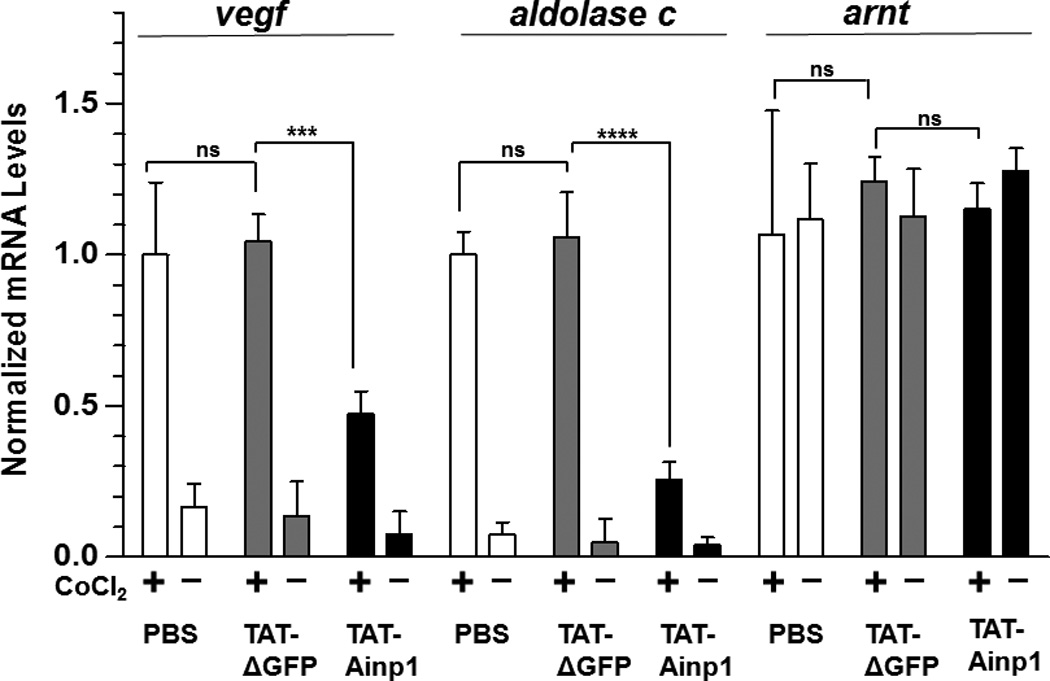
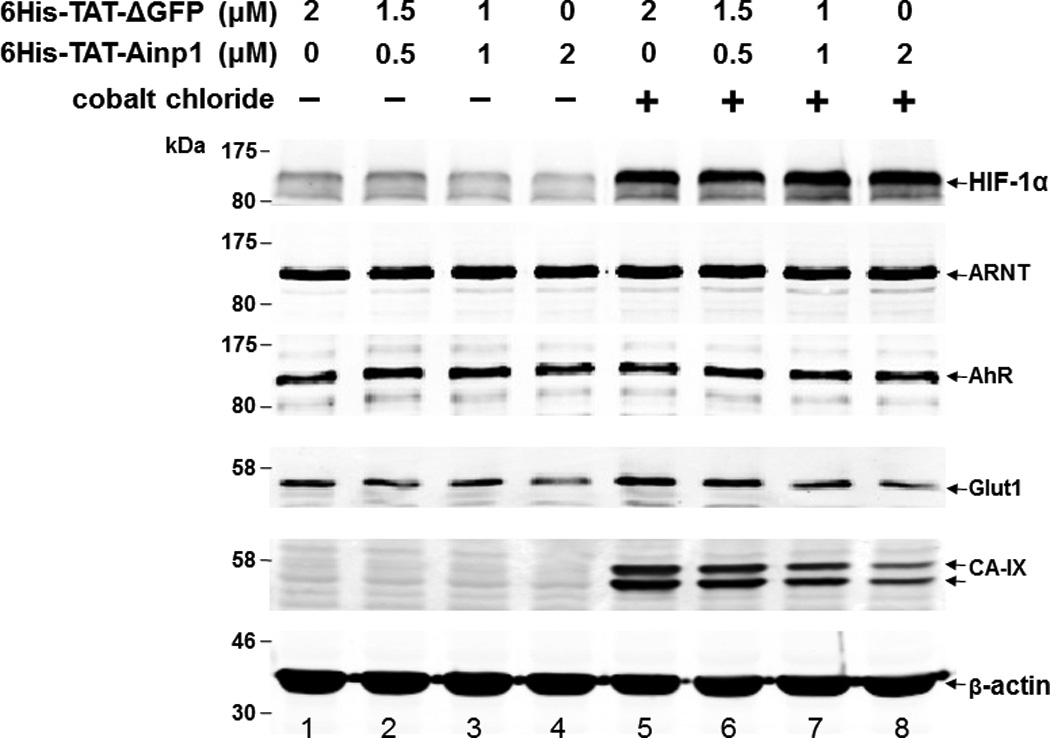
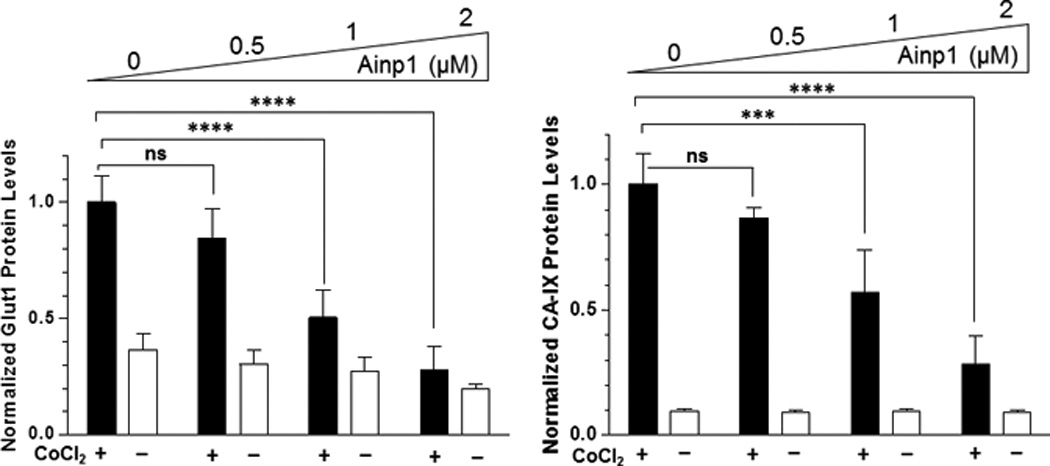
Suppression of the cobalt chloride-dependent HIF-1 target gene expression by 6His-TAT-Ainp1. The cobalt chloride (CoCl2)-activated, HRE-driven luciferase activity was measured in the HeLa (A), MCF-7 (B) and Hep3B (C) cells (± 100 µM cobalt chloride) treated with different combinations of 6His-TAT-ΔGFP and 6His-TAT-Ainp1. The condition with cobalt chloride and no Ainp1 in each case was arbitrarily set as 1. This experiment was repeated once with similar results. (D) RT-qPCR showing vegf, aldolase C and arnt message levels ± 100 µM cobalt chloride (CoCl2) in the presence or absence of PBS, 6His-TAT-ΔGFP (2 µM) or 6His-TAT-Ainp1 (2 µM). The condition with cobalt chloride and PBS in each case was arbitrarily set as 1. This experiment was repeated twice with similar results. Error bars (A–D) indicate the variations of the means (mean ± SD, n = 3). *p < 0.05; **p <0.01; ***p < 0.001; ns, not significant. (E) Western analysis of HeLa cell lysate ± 200 µM cobalt chloride treated with different combination of 6His-TAT-ΔGFP and 6His-TAT-Ainp1. Arrows indicate the bands of interest. This experiment was repeated twice with similar results. (F) Graph showing the quantified Glut1 and CA-IX protein levels ± Ainp1 (0–2 µM) as shown in lanes 5 to 8 of Fig. 5E. The protein levels were normalized by β-actin. The condition with cobalt chloride and no Ainp1 in each case was arbitrarily set as 1. Error bars indicate the variations of the means (mean ± SD, n = 3). ***p < 0.001; ****p < 0.0001; ns, not significant.
3. Results
3.1. Ainp1 interacts with the HLH domain of ARNT
We previously utilized the bacterially expressed thioredoxin fusion of ARNT CΔ418 as the bait to identify ARNT-interacting peptides by a phage display method [10] and subsequently showed that Ainp1 interacts with ARNT but not HIF-1α in vitro [11]. CΔ418 contains the N-terminal 356 amino acids of ARNT, which includes NLS, bHLH and PAS-A domains (Fig. 1A). Here we performed deletion mapping studies to determine the ARNT location where Ainp1 binds. All of the thioredoxin fusions of ARNT deletion were analyzed by western using anti-Thio mouse IgG (Fig. 1A and 1B). Initially, we examined whether Ainp1 would interact with ARNT at its NLS/bHLH (D1) or PAS-A domain (D2). We performed co-immunoprecipitation experiment using anti-Ainp1 mouse IgG to co-precipitate ARNT deletions (D1 and D2) using 6His-Ainp1 as the bait. We observed that thioredoxin fusion of D1, but not thioredoxin fusion of D2, was co-immunoprecipitated in an Ainp1-dependent manner (Fig. 1C). The antibody itself interacted minimally, if any, with the thioredoxin fusions or the thioredoxin control, validating that Ainp1 specifically interacted with the N-terminal 160 amino acids of ARNT. Based on the D1 structure, three additional ARNT deletions were used to fine map the ARNT region where Ainp1 binds – D1A, D1B, and D1C. D1A contains the NLS region (aa 1–75); D1B contains the bHLH domain (aa 70–140); D1C contains the connecting sequencing between HLH and PAS-A (aa 130–160) (Fig. 1A). We observed that among the three deletions, only D1B was co-immunoprecipitated in an Ainp1-dependent manner (Fig. 1D). Neither anti-Ainp1 mouse IgG nor mouse IgG directly interacted with D1A, D1B or D1C. Based on the D1B structure, two smaller ARNT deletions corresponding to the basic (aa 75–87) and HLH (aa 88–128) regions were generated (Fig. 1A). Results from the co-immunoprecipitation experiment revealed that only thioredoxin fusion of HLH, but not thioredoxin fusion of basic, was co-immunoprecipitated in an Ainp1-dependent manner (Fig. 1E). Collectively, we concluded that Ainp1 binds to the ARNT HLH domain.
Fig. 1.
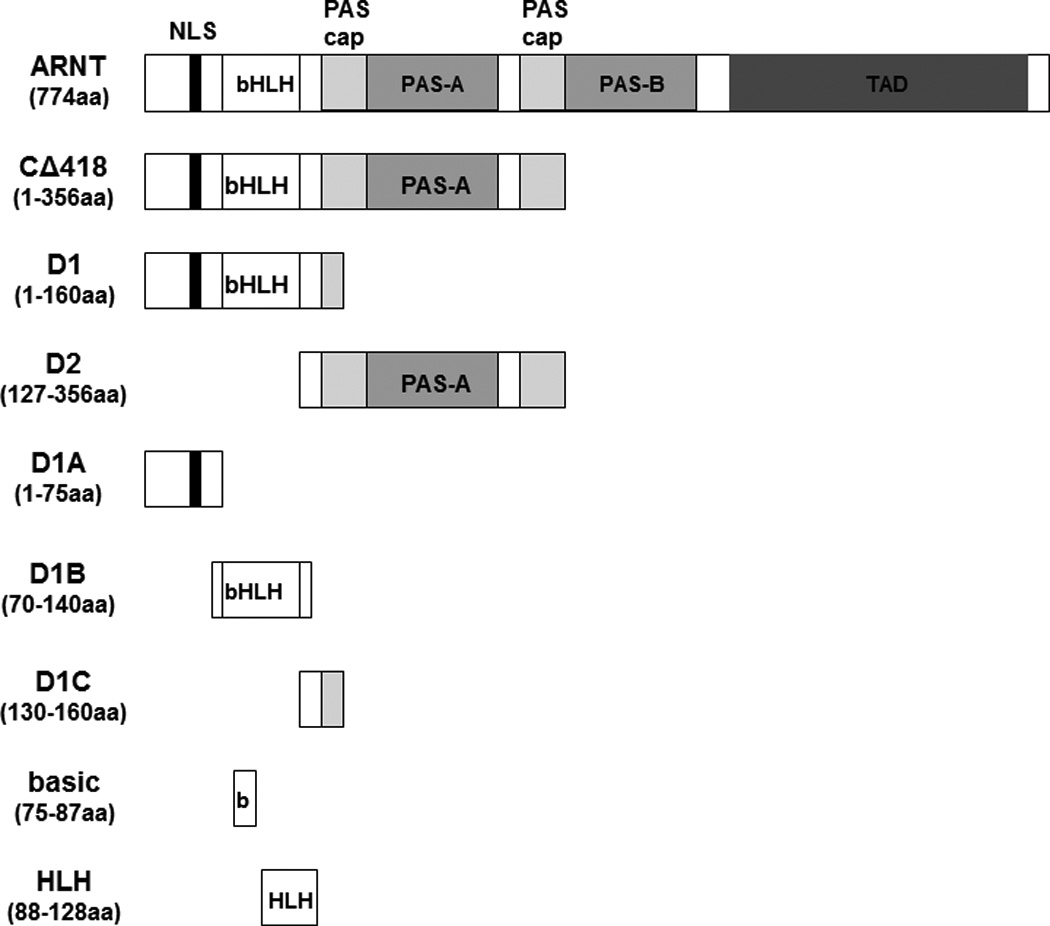
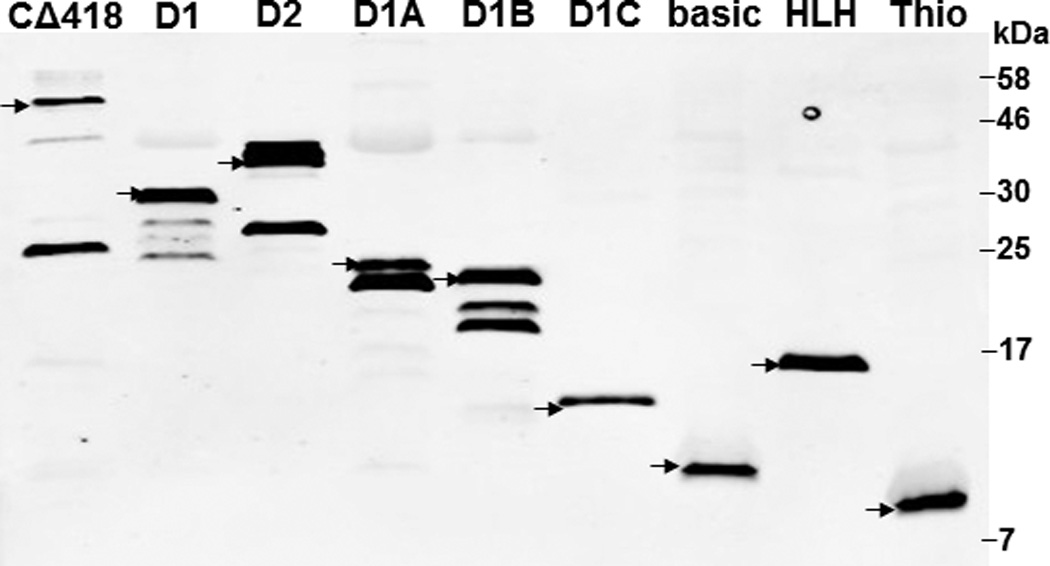
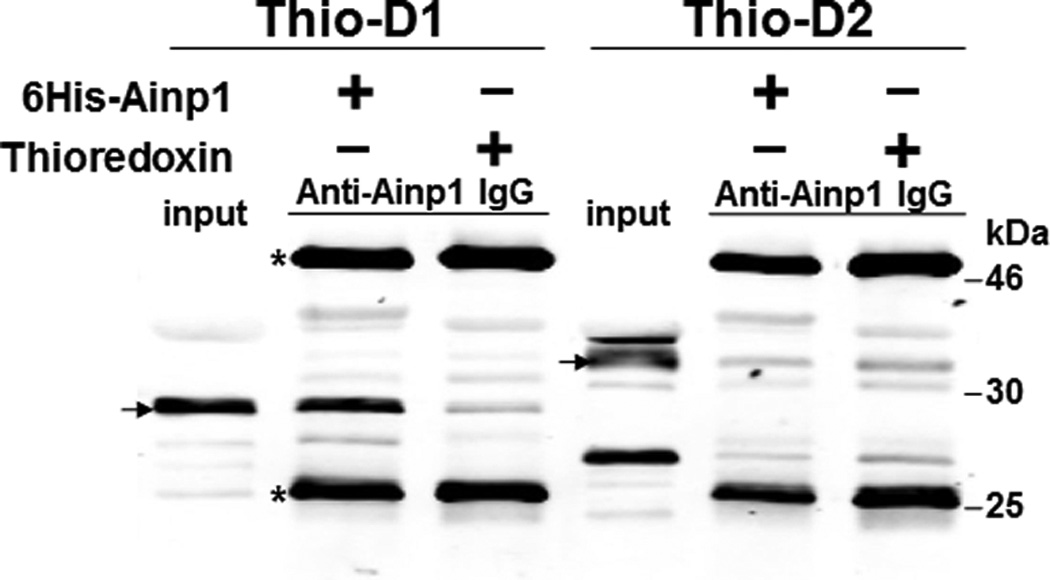
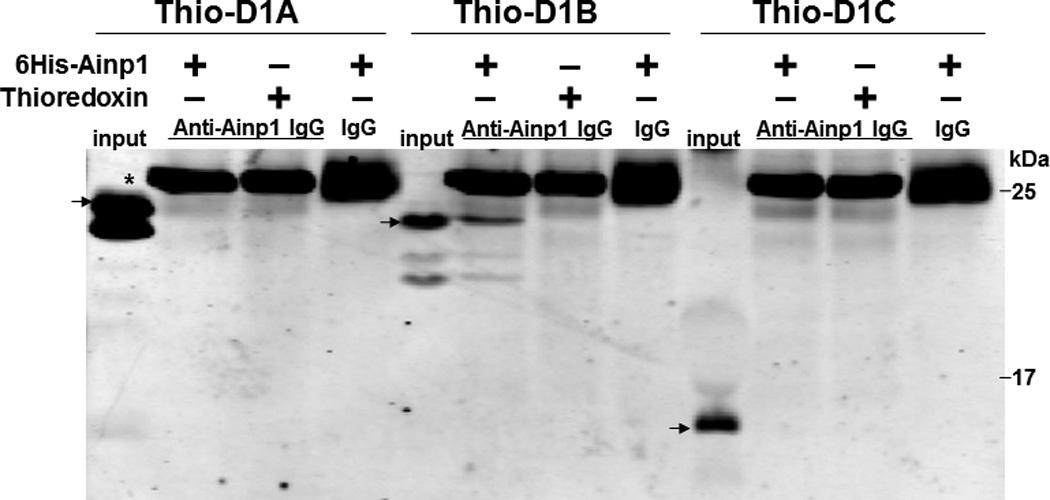
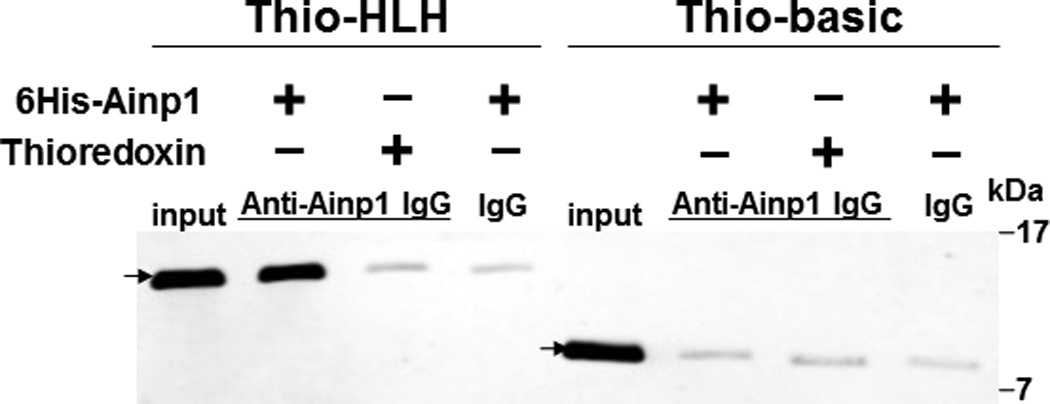
Deletion studies showing that Ainp1 interacted with the HLH domain of ARNT. (A) Human ARNT deletion constructs used for this study. (B) Western using anti-Thio mouse IgG to show expression of all thioredoxin (Thio) fusions of ARNT deletions. Arrows indicate the bands of interest. Anti-Ainp1 mouse IgG was used to co-precipitate bacterially expressed Thio-ARNT D1/D2 (C) or Thio-ARNT D1A/B/C (D) or Thio-ARNT basic/HLH (E) using 6His-Ainp1 as bait. Anti-Thio mouse IgG was used to detect Thio-ARNT deletions. Thioredoxin and mouse IgG were used as negative controls. Arrows indicate the bands of interest. Asterisks (*) in C and D indicate the heavy and light chains of mouse IgG of the IP antibodies (anti-Ainp1 mouse IgG) detected by the anti-mouse secondary antibodies. Amount of protein used: +, 40 µg of protein used. These experiments (C–E) were repeated twice with similar results.
3.2. Refolded TAT fusion of Ainp1 interacts with the HLH domain of ARNT and colocalizes with ARNT in the nucleus
Since Ainp1 is a 59-amino-acid peptide, we generated a GFP deletion (ΔGFP) containing N-terminal 58 amino acids of GFP as the control peptide of similar size. Both of these TAT fusions were purified by 8 M urea and refolded using limited dialysis. We examined whether the refolded TAT fusion of Ainp1 is functional by performing co-immunoprecipitation experiments using 6His-TAT-ΔGFP and 6His-Ainp1 as controls. We observed that these proteins were quite pure in the Coomassie staining gel and western analysis showed that anti-6His mouse IgG could be used to detect these proteins in co-immunoprecipitation experiment (Fig. 2A). Results from the co-immunoprecipitation experiment using HeLa lysate showed that ARNT interacted similarly with native 6His-Ainp1 and refolded 6His-TAT-Ainp1, but not with 6His-TAT-ΔGFP (Fig. 2B). Therefore, the TAT sequence did not seem to alter the Ainp1∷ARNT interaction, and the limited dialysis was able to refold the 6His-TAT-Ainp1 that was in the inclusion bodies. Next, we examined whether the TAT moiety would alter the specific binding of Ainp1 at the HLH region of ARNT. We observed that thioredoxin fusion of ARNT HLH was similarly co-immunoprecipitated in the presence of either 6His-Ainp1 or 6His-TAT-Ainp1, but not 6His-TAT-ΔGFP, using anti-Ainp1 mouse IgG (Fig. 2C). In addition, both 6His-Ainp1 and 6His-TAT-Ainp1, but not TAT-ΔGFP, were co-immunoprecipitated in the presence of thioredoxin fusion of ARNT HLH using anti-Thio mouse IgG (Fig. 2D). These results supported that 6His-TAT-Ainp1 and 6His-Ainp1 bind to the HLH domain of ARNT in a similar fashion. Knowing that ARNT is exclusively localized in the nucleus in HeLa cells [14, 15], we examined whether 6His-TAT-Ainp1 would reside with ARNT in the nucleus. Results from our immunofluorescence staining experiment showed that 4 h after peptide transduction, 6His-TAT-Ainp1 was primarily localized to the cell nucleus, while 6His TAT-ΔGFP was equally distributed in both cytoplasm and nucleus (Fig. 2E), revealing that 6His-TAT-Ainp1 interacts with ARNT in the nucleus of HeLa cells. Similar nuclear localization of 6His-TAT-Ainp1 was also observed in Hep3B cells (Fig. 2F).
Fig. 2.
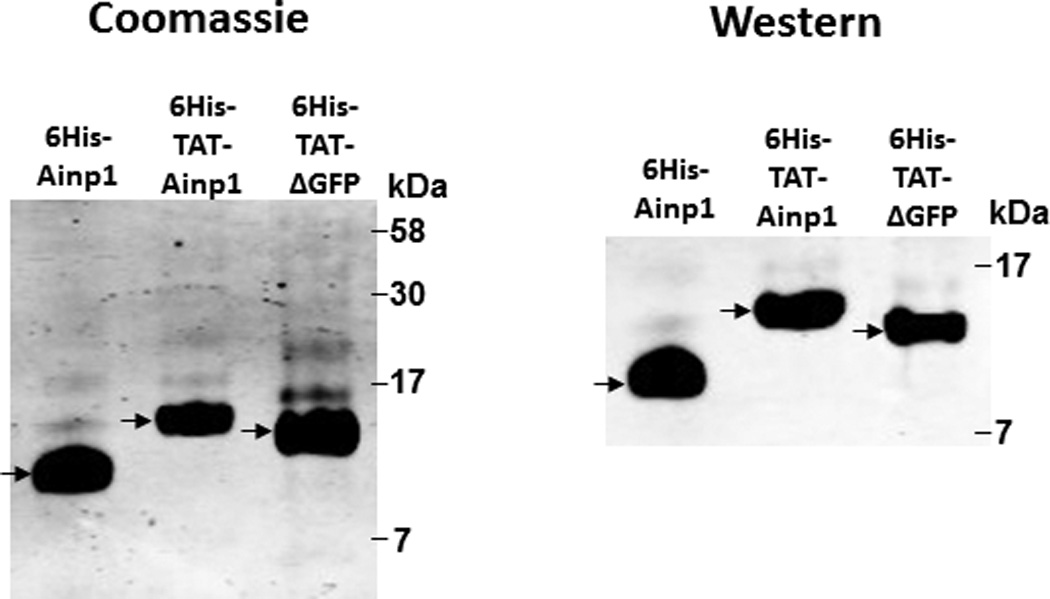
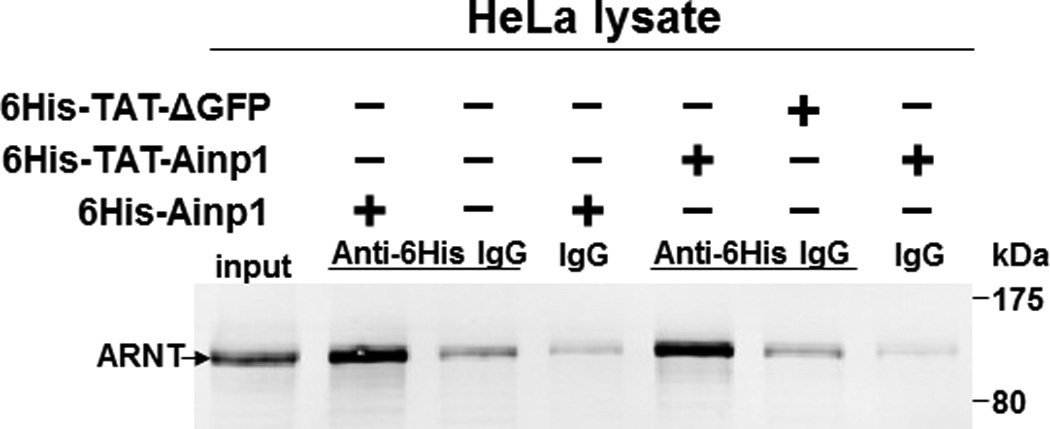
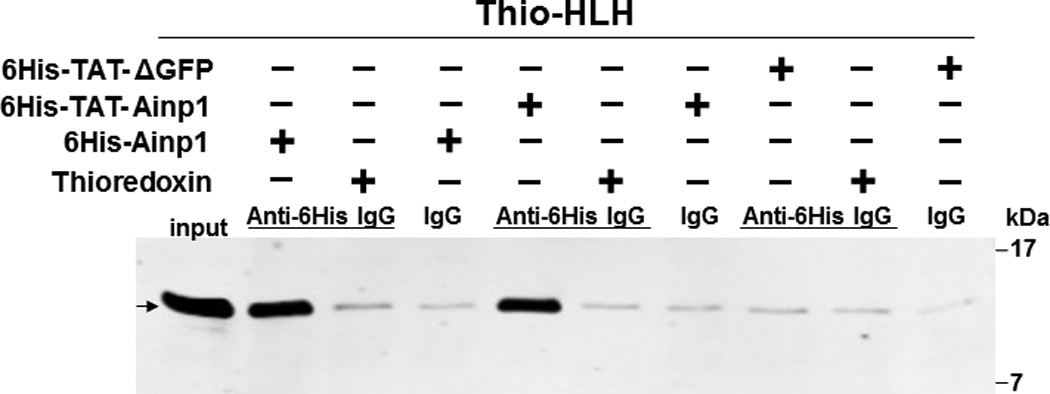
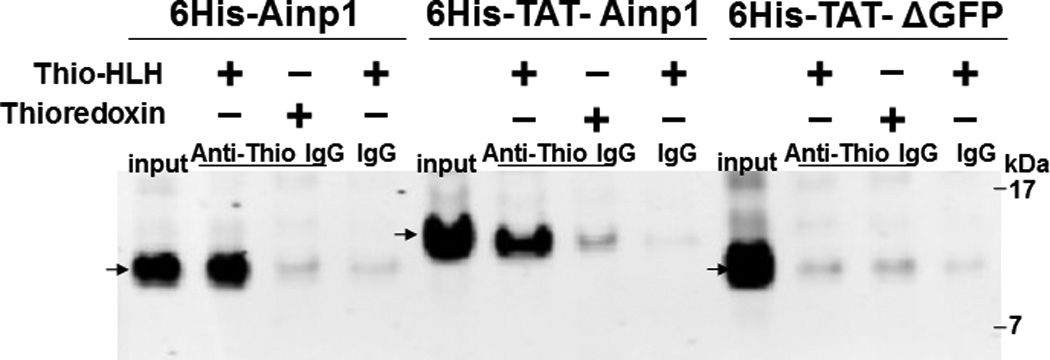
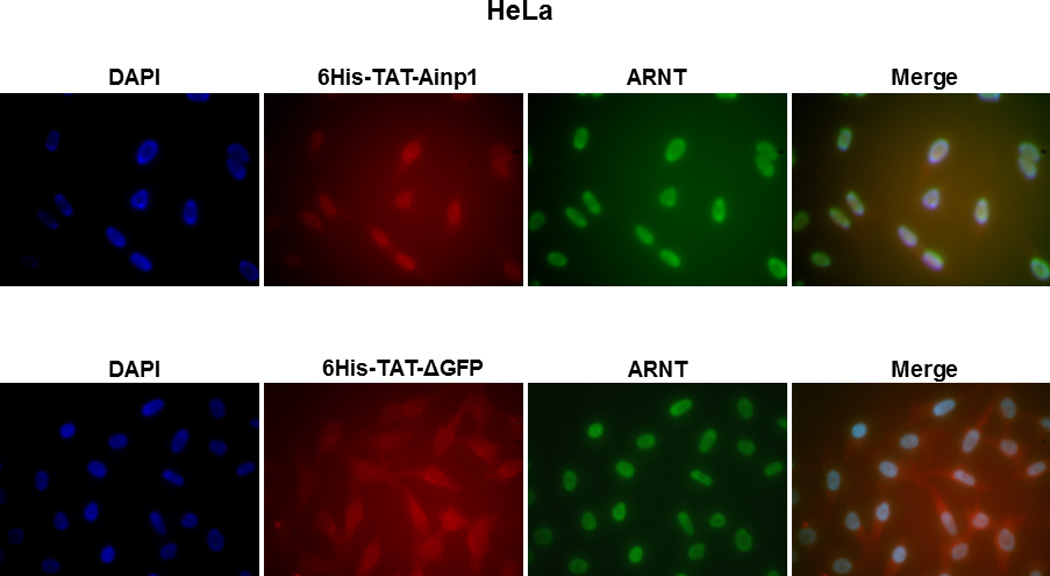
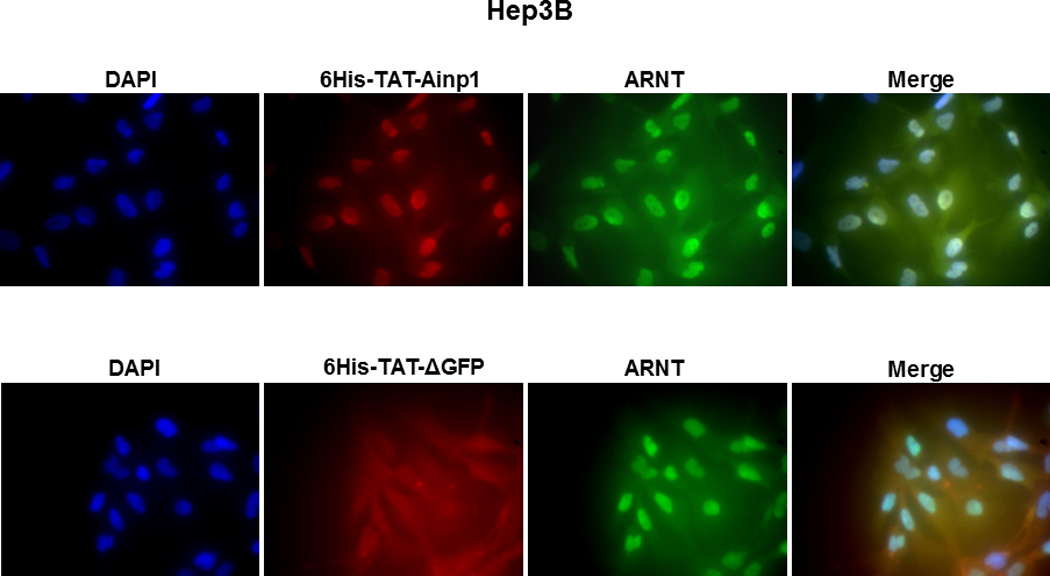
Co-immunoprecipitation studies showing that refolded 6His-TAT-Ainp1 interacted with the HLH domain of ARNT and colocalized with ARNT in the HeLa cell nucleus. Coomassie staining (A, left panel) and western analysis (A, right panel) of soluble 6His-Ainp1 and refolded 6His-TAT-Ainp1 and 6His-TAT-ΔGFP. Each sample was 10 µg for Coomassie and 2 µg for western. 16% tricine gel was used for analysis [39]. Arrows indicate the band of interest. Anti-6His mouse IgG was used to co-immunoprecipitate ARNT from HeLa cell lysate (B) or thioredoxin fusion of ARNT HLH (Thio-HLH) (C). Soluble 6His-Ainp1 (positive control) and refolded 6His-TAT-Ainp1 were used as baits; refolded 6His-TAT-ΔGFP, thioredoxin and mouse IgG as negative controls. (D) Anti-Thio mouse IgG was used to co-immunoprecipitate soluble 6HisAinp1, refolded 6His-TAT-Ainp1 and refolded 6His-TAT-ΔGFP. Thioredoxin fusion of ARNT HLH (Thio-HLH) was used as bait: thioredoxin and mouse IgG were negative controls. Amount of protein used: +, 40 µg of protein. These experiments (B–D) were repeated twice with similar results. Immunofluorescence microscopy of HeLa (E) and Hep3B (F) cells stained for 6His-TAT-Ainp1 (red), 6His-TAT-ΔGFP (red) and ARNT (green). Before the immunofluorescence staining, the cells were treated with either 6His-TAT-Ainp1 (2 µM) or 6His-TAT-ΔGFP (2 µM) for 4 h. DAPI (blue) staining was used to visualize DNA stained nuclei. Merge, image merging analysis. This experiment was repeated once with similar results.
3.3. TAT fusion of Ainp1 reaches the maximum levels in 2 h after transduction and remains detectable up to 96 h in HeLa cells
Since TAT fusions must internalize into mammalian cells by endocytosis and then undergo lysosomal degradation, we examined whether we could quantify intracellular 6His-TAT-Ainp1 protein levels by western analysis and subsequently determined its disposition in HeLa cells. Realizing that a significant amount of TAT fusion would adhere to the cell surface, which might cause overestimation of its transduction efficiency, we followed the recommended practice of removing the adherent protein by trypsinization, followed by thorough washing before analysis [16]. This trypsinization step caused partial cell detachment as observed under light microscope, and subsequent PBS washes (three times) appeared to get rid of nonspecific binding of 6His-TAT fusions on the cell surface. Results from our western data showed that 6His-TAT-Ainp1 could be detected as early as 30 min after transduction, and reached the maximum intracellular levels after about 2 h (Fig. 3A). Longer transduction time up to 4 h did not show higher intracellular 6His-TAT-Ainp1 content in a statistically significant manner. After reaching the maximum levels in 2 h, 6His-TAT-Ainp1 was degraded to around 50% after 24 h and maintained detectable up to 96 h (Fig. 3B, left panel). The same experiment was repeated with the 6His-TAT-ΔGFP control peptide: we observed that 6His-TAT-ΔGFP was degraded to 50% between 12 h and 24 h, and maintained detectable up to 96 h (Fig. 3B, right panel), which was slightly, but not significantly, different from what we observed for 6His-TAT-Ainp1.
3.4. TAT fusion of Ainp1 does not have significant cytotoxicity in HeLa and MCF-7 cells, but causes cell death in Hep3B cells
We examined whether 6His-TAT-Ainp1 is toxic to human cells at the concentration used to suppress the HIF-1 function. The cell viability was determined by the absorbance at 560 nm using the Promega CellTiter kit. HeLa cells grew to 270% after 72 h while only to 160% in the presence of cobalt chloride (Fig. 4A). This observed slower growth rate was consistent with the cell cycle arrest during acute hypoxia exposure [17]. In the presence or absence of cobalt chloride, neither 6His-TAT-ΔGFP nor 6His-TAT-Ainp1 had any effect on HeLa cell viability at 2 µM concentration (Fig. 4A). Our previous data showed that significant Hep3B cell death occurred after transfection of the plasmid carrying the Ainp1 cDNA, and cell death was partially rescued by exogenous ARNT [11]. We examined whether direct introduction of the Ainp1 peptide would cause the same consequence in Hep3B cells. Realizing that 6His-TAT-Ainp1 is not toxic to HeLa cells, we examined three human cell lines – HeLa, MCF-7, and Hep3B – under a light microscope to address whether cell death could be cell line specific. It appeared that the timecourse of 6His-TAT-Ainp1 transduction was similar among HeLa, Hep3B, and MCF-7 cells (Fig. 3A and 4B). Forty-eight h after 6His-TAT-Ainp1 transduction, HeLa and MCF-7 cells were indistinguishable from the controls (PBS or 6His-TAT-ΔGFP treatment) (Fig. 4C). However Hep3B cells showed significant morphological changes such as rounding and shrinkage 48 h after treatment with the same concentration (2 µM) of 6His-TAT-Ainp1. These results suggested that although 6His-TAT-Ainp1 is not toxic to HeLa and MCF-7 cells, Hep3B cells have less tolerance to the 6His-TAT-Ainp1 peptide.
3.5. TAT fusion of Ainp1 suppresses the cobalt chloride-driven HIF-1 downstream gene expression
We previously reported that transfection of the Ainp1-expressing plasmid interferes with the ARNT∷HIF-1α heterodimerization and suppresses the HIF-1 function in Hep3B cells [11]. In an effort to address whether the Ainp1 peptide is responsible for the suppression, we examined whether transduction of the 6His-TAT-Ainp1 peptide would suppress the HIF-1 downstream gene expression. Results from our luciferase reporter assay showed that, upon cobalt chloride treatment, 6His-TAT-Ainp1 suppressed the hypoxia response element (HRE)-driven luciferase expression by up to 70% in a dose-dependent manner in HeLa cells (Fig. 5A). Similar dose-dependent suppression was also observed in both MCF-7 and Hep3B cells (Fig. 5B and C), suggesting that TAT fusion of Ainp1 is capable of suppressing the HIF-1 signaling function regardless of cell types. Next, we examined whether transduction of the 6His-TAT-Ainp1 peptide would suppress the transcription of two endogenous HIF-1 target gene – vegf and aldolase c [18]. We observed that transcription of the vegf and aldolase c genes was effectively upregulated by 5- and 10-fold, respectively, after cobalt chloride treatment in HeLa cells (Fig. 5D). Transduction of 2 µM 6His-TAT-Ainp1 suppressed the cobalt chloride-induced vegf and aldolase c message levels by 50 and 30%, respectively, without affecting the transcription of the arnt gene, showing that 6His-TAT-Ainp1 specifically suppressed the cobalt chloride-induced HIF-1 target gene transcription (Fig. 5D). In addition, we observed that 2 µM 6His-TAT-Ainp1 specifically suppressed the cobalt chloride-dependent expression of two HIF-1 target proteins – CA-IX [19] and Glut1 [18] – in a dose-dependent manner (Fig. 5E). This suppression was not a general effect on proteins since 6His-TAT-Ainp1 did not alter the HIF-1α, ARNT, and AhR protein levels. Taken together, we concluded that the Ainp1 peptide suppresses the HIF-1α signaling not by lowering the HIF-1α and ARNT protein levels, but by inhibiting the formation of the HIF-1 complex.
4. Discussion
Tumor hypoxia is one of the central issues in tumor physiology. It is associated with malignant tumor progression and therapeutic resistance in both radio- and chemotherapy [20]. The hypoxia-induced pathophysiological changes are primarily mediated through HIF-1; thus, many potential anticancer agents have been developed to target HIF-1. These agents suppress the HIF-1 function by various mechanisms: decreasing the HIF-1α mRNA levels, decreasing the HIF-1α protein levels, suppressing the binding of HIF-1 to DNA, and down-regulating the HIF-1-mediated transactivation [21]. In addition, small molecules have been discovered to interfere with the heterodimerization of HIF-1α and ARNT specifically at the PAS-A [22] and PAS-B [23] regions. We are interested in potential peptide candidates which may suppress the HIF-1 function. In our case, Ainp1 uniquely suppresses the HIF-1 function by targeting the HLH domain of ARNT, which appears to be very effective in suppressing the formation of the HIF-1 complex without affecting the PAS domain. This Ainp1 phage display peptide may serve as a prototype of a new line of anticancer agents since many phage display-derived peptides have been extensively developed as alternatives to antibodies for therapeutic purpose [24]. In particular, a few peptides which interact with either HIF-1 or HIF-1 downstream targets have been developed using the phage display method for diagnostic or therapeutic purpose [14, 25].
We previously reported that Ainp1 suppresses the HIF-1 function in Hep3B cells in transient transfection experiments [11]. Although Ainp1 appeared to suppress the interaction between HIF-1α and ARNT, the precise mechanism causing this suppression was unclear. In addition, we suspect that this Ainp1 nucleotide sequence obtained from the human liver cDNA library may be non-coding (nc) RNA, since a large portion of the mammalian transcriptome does not code for proteins and the Ainp1 cDNA does not match any of the characterized genes and proteins [26–28]. Realizing that ncRNAs are involved in many endogenous processes, namely translational repression, germline transposon silencing, epigenetic regulation, and alternative splicing [29], transient transfection of an Ainp1 expressing plasmid might conceivably cause RNA-mediated effects leading to suppression of the HIF-1 function. In order to clarify the cellular function of the Ainp1 peptide, we utilized the peptide delivery approach to address whether the Ainp1 peptide itself suppresses the HIF-1 function. We proved that TAT fusion of Ainp1 can effectively penetrate into cells and achieve a concentration that suppresses the HIF-1 signaling without causing cell death or changes in cell morphology.
Both ARNT and HIF-1α translocate into the cell nucleus to form the HIF-1α heterodimer [30, 31]. We previously showed that Ainp1 interferes with the formation of the HIF-1α ∷ARNT complex in vitro by interacting with ARNT but not with HIF-1α [13]. In addition, we observed that the HIF-1α and ARNT protein levels are essentially unaffected in the presence of Ainp1 in HeLa cells, suggesting that Ainp1 does not affect HIF-1α directly but formation of the HIF-1 complex is competitively inhibited by Ainp1 in HeLa cells. If the Ainp1 peptide competes with HIF-1α for ARNT binding within a cell, one would expect that Ainp1 should be present in the cell nucleus. We previously observed that after transfecting the plasmid carrying the GFP fusion of the Ainp1 cDNA into Hep3B cells, both fluorescence imaging and subcellular fractionation data showed that GFP-Ainp1 was equally localized to both cytoplasm and nucleus. On the contrary, the immunofluorescence results from this study clearly revealed that 6His-TAT-Ainp1 was primarily co-localized with ARNT in the cell nucleus in both HeLa and Hep3B cells. It is known that the arbitrary cutoff limit for passive diffusion across the cell nuclear pore is 40 kDa [32], and large protein tags such as GFP have been reported to sterically interfere with protein trafficking and function [33, 34]. We believe that the lack of colocalization of GFP-Ainp1 and ARNT in Hep3B cells could be caused by difficult diffusion of GFP-Ainp1 (37 kDa) into the nucleus whereas the smaller 6His-TAT-Ainp1 peptide (12 kDa) without the GFP moiety diffuses readily into the nucleus. Nevertheless, we observed that the Ainp1 peptide is capable of reaching the nucleus and inhibiting the interaction between HIF-1α and ARNT. Previously, we showed that Ainp1 suppresses the cobalt chloride-dependent recruitment of ARNT to the vegf promoter in Hep3B cells [11]. However, we were still able to detect some ARNT at the vegf promoter in the presence of Ainp1. It is known that the PAS-B domain of ARNT recruits a number of coactivators – namely TRIP230 [35], CoCoA [35], and TACC3 [36] – which involve in transcriptional regulation. We cannot rule out the possibility that binding of Ainp1 to the HLH domain of ARNT would suppress the coactivator recruitment by ARNT which in turn would contribute to inhibition of the HIF-1 function.
Although protein transduction efficacy varies extensively depending on conditions and cell types, TAT-mediated protein delivery offers many distinct advantages: (1) transduction of TAT fusion is a simple process; (2) transduction of TAT fusion is not sensitive to the size of the cargo and (3) transfection with the use of protein transduction reagents, which often cause unwanted cellular effects, is not needed. Functional 6His-Ainp1 can be readily purified under native condition. However, TAT fusion of 6His-Ainp1 was primarily in the inclusion bodies, possibly caused by the highly positive-charged TAT moiety. Methods of purifying recombinant protein using urea or guanidine hydrochloride followed by protein refolding have been used to generate functional recombinant proteins [26, 37]. Our affinity purification coupled with limited dialysis is capable of generating functional 6His-TAT-Ainp1 from the inclusion bodies to close to 100% activity when compared with the soluble 6His-Ainp1. This refolded 6His-TAT-Ainp1 peptide binds to the HLH domain of ARNT in the nucleus and suppresses the HIF-1-dependent gene expression, proving that 6His-TAT-Ainp1 can be a model peptide for developing therapeutic peptides targeting HIF-1 in tumors.
Since the late 20th century, numerous diagnostic and therapeutic proteins and peptides have been developed and emerging in the market, but their safety and efficacy are limited by the short half-lives, instability, and immunogenicity [38]. In our case, the timecourse studies showed that 6His-TAT-Ainp1 reaches the maximum intracellular levels within 2 h, and has a half-life of about 24 h. This relatively short half-life of 6His-TAT-Ainp1 might compromise its efficiency on the sustained suppression of the HIF-1 function with a single dose treatment. In order to achieve faster onset, longer half-life and reduced immunogenicity, further modification of 6His-TAT-Ainp1 is desirable and necessary. Nevertheless, our data revealed that the HLH domain of ARNT is a potential target for suppression of the HIF-1 function, and the 6His-TAT-Ainp1 peptide binds to the endogenous ARNT and suppresses the HIF-1 function in multiple human cell lines.
Highlights.
-
-
Ainp1 is an ARNT-interacting peptide
-
-
functional TAT fusion of Ainp1 can be obtained from inclusion bodies by limited dialysis
-
-
TAT fusion of Ainp1 penetrates into HeLa, MCF-7, and Hep3B cells
-
-
TAT fusion of Ainp1 suppresses the HRE-driven luciferase gene expression
-
-
TAT fusion of Ainp1 suppresses the cobalt-chloride-dependent HIF-1 target gene activation
Acknowledgement
This work is supported by the National Institutes of Health (R01 ES014050).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jiang B, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 1996;271:17771–17778. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- 2.Massari ME, Murre C. Helix-loop-helix protein: regulators of transcription in eucaryotic organisms. Mol. Cell Biol. 2000;20:429–440. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ivan M, Kondo L, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 4.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim Av, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 5.Semenza GL. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 6.Hoffmann A, Mori R, Vallbohmer D, Brabender J, Klein E, Drebber U, Baldus SE, Cooc J, Azuma M, Metzger R, Hoelscher AH, Danenberg KD, Prenzel KL, Danenberg PV. High expression of HIF1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA, VEGF, bFGF. Neoplasia. 2008;10:674–679. doi: 10.1593/neo.08292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.dos Santos M, da Cunha Mercante AM, Louro ID, Goncalves AJ, de Carvalho MB, da Silva EHT, da Silva AMA. HIF1-alpha expression predicts survival of patients with squamous cell carcinoma of the oral cavity. PLoS One. 2012;7:e45228. doi: 10.1371/journal.pone.0045228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindsay MA. Peptide-mediated cell delivery: application in protein target validation. Curr. Opin. Pharmacol. 2002;2:587–594. doi: 10.1016/s1471-4892(02)00199-6. [DOI] [PubMed] [Google Scholar]
- 9.Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Luu TC, Chan WK. A novel Arnt-interacting protein Ainp2 enhances the aryl hydrocarbon receptor signalling. Arch. Biochem. Biophys. 2005;441:84–95. doi: 10.1016/j.abb.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Li Y, Wang D, Li Y, Chang A, Chan WK. Suppression of the hypoxia inducible factor-1 function by redistributing the aryl hydrocarbon receptor nuclear translocator from nucleus to cytoplasm. Cancer Lett. 2012;320:111–121. doi: 10.1016/j.canlet.2012.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jensen KA, Luu TC, Chan WK. A truncated Ah receptor blocks the hypoxia and estrogen receptor signaling pathways: a viable approach for breast cancer treatment. Mol. Pharm. 2006;3:695–703. doi: 10.1021/mp0600438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 14.Askoxylakis V, Garcia-Boy R, Rana S, Kramer S, Hebling U, Mier W, Altmann A, Markert A, Debus J, Haberkorn U. A new peptide ligand for targeting human carbonic anhydrase IX, identified through the phage display technology. PLoS One. 2010;5:e15962. doi: 10.1371/journal.pone.0015962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elbi C, Misteli T, Hager GL. Recruitment of dioxin receptor to active transcription sites. Mol. Biol. Cell. 2002;13:2001–2015. doi: 10.1091/mboc.13.6.mk0602002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Becker-Hapak M, Dowdy SF. Protein transduction: generation of full-length transducible proteins using the TAT system. Curr. Protoc. Cell Biol. 2003;Chapter 20(Unit 20.2) doi: 10.1002/0471143030.cb2002s18. [DOI] [PubMed] [Google Scholar]
- 17.Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-inducible factor 1 is essential for cell cycle arrest during hypoxia. Mol. Cell. Biol. 2003;23:359–369. doi: 10.1128/MCB.23.1.359-369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit. Rev. Biochem. Mol. Biol. 2000;35:71–103. doi: 10.1080/10409230091169186. [DOI] [PubMed] [Google Scholar]
- 19.Pantuck AJ, Zeng G, Belldegrun AS. Pathobiology, prognosis, and targeted therapy for renal cell carcinoma: exploiting the hypoxia-induced pathway. Clin. Cancer Res. 2003;9:4641–4652. [PubMed] [Google Scholar]
- 20.Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical biologic, and molecular aspects. J. Natl. Cancer Inst. 2001;93:266–276. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- 21.Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov. Today. 2007;12:853–859. doi: 10.1016/j.drudis.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 22.Park EJ, Kong D, Fisher R, Cardellina J, Shoemaker RH, Melillo G. Targeting the PAS-A domain of HIF-1α for development of small molecule inhibitors of HIF-1. Cell Cycle. 2006;5:1847–1853. doi: 10.4161/cc.5.16.3019. [DOI] [PubMed] [Google Scholar]
- 23.Cardoso R, Love R, Nilsson CL, Bergqvist S, Nowlin D, Yan J, Liu KK, Zhu J, Chen P, Deng YL, Dyson HJ, Greig MJ, Brooun A. Identification of cys255 in HIF-1α as a novel site for development of covalent inhibitors of HIF-1α/ARNT PAS-B domain protein-protein interaction. Protein Sci. 2012;21:1885–1896. doi: 10.1002/pro.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ladner RC, Sato AK, Gorzelany J, de Souza M. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discov. Today. 2004;9:525–529. doi: 10.1016/S1359-6446(04)03104-6. [DOI] [PubMed] [Google Scholar]
- 25.Groot AJ, Verheesen P, Westerlaken EJ, Gort EH, van der Groep P, Bovenschen N, van der Wall E, van Diest PJ, Shvarts A. Identification by phage display of single-domain antibody fragments specific for the ODD domain in hypoxia-inducible factor 1alpha. Lab Invest. 2006;86:345–356. doi: 10.1038/labinvest.3700395. [DOI] [PubMed] [Google Scholar]
- 26.Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, Yamanaka I, Kiyosawa H, Yagi K, Tomaru Y, Hasegawa Y, Nogami A, Schonbach C, Gojobori T, Baldarelli R, Hill DP, Bult C, Hume DA, Quackenbush J, Schriml M, Kanapin A, Matsuda H, Batalov S, Beisel KW, Balke JA, Bradt D, Brusic V, Chothia C, Corbani LE, Cousins S, Dalla E, Dragani TA, Fletcher CF, Forrest A, Frazer KS, Gaasterland T, Gariboldi M, Gissi C, Godzik A, Gough J, Grimmond S, Gustincich S, Hirokawa N, Jackson IJ, Jarvis ED, Kanai A, Kawaji H, Kawasawa Y, Kedzierski RM, King BL, Konagaya A, Kurochkin IV, Lee Y, Lenhard B, Lyons PA, Maglott DR, Maltais L, Marchionni L, McKenzie L, Miki H, Nagashima T, Numata K, Okido T, Pavan WJ, Pertea G, Pesole G, Petrovsky N, Pillai R, Pontius JU, Qi D, Ramachandran S, Ravasi T, Reed JC, Reed DJ, Reid J, Ring BZ, Ringwald M, Sandelin A, Schneider C, Semple CAM, Setou M, Shimada K, Sultana R, Takenaka Y, Taylor MS, Teasdale RD, Tomita M, Verardo R, Wagner L, Wahlestedt C, Wang Y, Watanabe Y, Wells C, Wilming LG, Wynshaw-Boris A, Yanagisawa M, Yang I, Yang L, Yuan Z, Zavolan M, Zhu Y, Zimmer A. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- 27.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amaral PP, Clark MB, Gascoigne DK, Dinger ME, Mattick JS. lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res. 2011;39:D146–D151. doi: 10.1093/nar/gkq1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Esteller M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 30.Eguchi H, Ikuta T, Tachibana T, Yoneda Y, Kawajiri K. A nuclear localization signal of human aryl hydrocarbon receptor nuclear translocator/hypoxia-inducible factor 1β is a novel bipartite type recognized by the two components of nuclear pore-targetting complex. J. Biol. Chem. 1997;272:17640–17647. doi: 10.1074/jbc.272.28.17640. [DOI] [PubMed] [Google Scholar]
- 31.Chilov D, Camenisch G, Kvietikova I, Ziegler U, Gassmann M, Wenger RH. Induction and nuclear translocation of hypoxia-inducible factor-1 (HIF-1): heterodimerization with ARNT is not necessary for nuclear accumulation of HIF-1alpha. J. Cell Sci. 1999;112:1203–1212. doi: 10.1242/jcs.112.8.1203. [DOI] [PubMed] [Google Scholar]
- 32.Naim B, Brumfeld V, Kapon R, Kiss V, Nevo R, Reich Z. Passive and facilitated transport in nuclear pore complexes is largely uncoupled. J. Biol. Chem. 2007;282:3881–3888. doi: 10.1074/jbc.M608329200. [DOI] [PubMed] [Google Scholar]
- 33.Marsh DR, Holmes KD, Dekaban GA, Weaver LC. Distribution of an NMDA receptor: GFP fusion protein in sensory neurons is altered by a C-terminal construct. J. Neurochem. 2001;77:23–33. doi: 10.1046/j.1471-4159.2001.t01-1-00182.x. [DOI] [PubMed] [Google Scholar]
- 34.Moritz OL, Tam BM, Papermaster DS, Nakayama T. A functional rhodopsin-green fluorescent protein fusion protein localizes correctly in transgenic Xenopus laevis retinal rods and is expressed in a time-dependent pattern. J. Biol. Chem. 2001;276:28242–28251. doi: 10.1074/jbc.M101476200. [DOI] [PubMed] [Google Scholar]
- 35.Partch CL, Card PB, Amezcua CA, Gardner KH. Molecular basis of coiled coil coactivator recruitment by ARNT. J. Biol. Chem. 2009;284:15184–15192. doi: 10.1074/jbc.M808479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Partch CL, Gardner KH. Coactivators necessary for transcriptional output of the hypoxia inducible factor, HIF, are directly recruited by ARNT PAS-B. Proc. Natl. Acad. Sci. USA. 2011;108:7739–7744. doi: 10.1073/pnas.1101357108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Z, Zhang L, Zhang Y, Zhang T, Feng Y, Lu X, Lan W, Wang J, Wu H, Cao C, Wang X. Highly efficient production of soluble proteins from insoluble inclusion bodies by a two-step-denaturing and refolding method. PLoS One. 2011;6:e22981. doi: 10.1371/journal.pone.0022981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pisal DS, Kosloski MP, Balu-Iyer SV. Delivery of therapeutic proteins. J. Pharm. Sci. 2010;99:2557–2575. doi: 10.1002/jps.22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schagger H. Tricine-SDS-PAGE. Nat. Protoc. 2007;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]