

Abstract
Motivation: Text-mining mutation information from the literature becomes a critical part of the bioinformatics approach for the analysis and interpretation of sequence variations in complex diseases in the post-genomic era. It has also been used for assisting the creation of disease-related mutation databases. Most of existing approaches are rule-based and focus on limited types of sequence variations, such as protein point mutations. Thus, extending their extraction scope requires significant manual efforts in examining new instances and developing corresponding rules. As such, new automatic approaches are greatly needed for extracting different kinds of mutations with high accuracy.
Results: Here, we report tmVar, a text-mining approach based on conditional random field (CRF) for extracting a wide range of sequence variants described at protein, DNA and RNA levels according to a standard nomenclature developed by the Human Genome Variation Society. By doing so, we cover several important types of mutations that were not considered in past studies. Using a novel CRF label model and feature set, our method achieves higher performance than a state-of-the-art method on both our corpus (91.4 versus 78.1% in F-measure) and their own gold standard (93.9 versus 89.4% in F-measure). These results suggest that tmVar is a high-performance method for mutation extraction from biomedical literature.
Availability: tmVar software and its corpus of 500 manually curated abstracts are available for download at http://www.ncbi.nlm.nih.gov/CBBresearch/Lu/pub/tmVar.
Contact: zhiyong.lu@nih.gov
1 INTRODUCTION
In the past 10 years, the focus of biological research has shifted from individual genes and proteins toward the study of entire biological systems. One of the most important research issues is gene/protein and disease relationship analysis. Sequence variation plays the key role between gene and disease. Therefore, identifying sequence variation is one of the major approaches for characterizing gene–disease relationships (Erdogmus and Sezerman, 2007; Schenck et al., 2012), with many study results subsequently reported in scientific publications. As such, text-mining mutation-related information from the literature has become an increasingly important task in many downstream bioinformatics applications, such as the curation of mutation-related biological databases (Gyimesi et al., 2012; Kuipers et al., 2012), the systematic study of biological effects of protein mutations (Izarzugaza et al., 2012; Winnenburg et al., 2009) and the interpretation of individual genomes toward personalized medicine in pharmacogenomics research (Capriotti et al., 2012).
Despite some reported success in identifying specific mutation types or identifiers, such as dbSNP RS numbers (Yu et al., 2009), mutation identification from free text in general remains a challenge because most mutations are not described in accordance with standard nomenclature (<25% in our corpus) and only few are mentioned with standard database identifiers, such as dbSNP RS numbers (<10% in our corpus). To the opposite, it is common to see the same mutation described in many different non-standard ways in the literature, which makes it similar to the named entity recognition task in biomedicine (Lu et al., 2011; Morgan et al., 2008).
In response, recently a number of automatic systems have been developed for extracting mutation mentions from the biomedical literature (Caporaso et al., 2007; McDonald et al., 2004) and some investigated further with respect to mutations’ associations with genes (Horn et al., 2004; Rebholz-Schuhmann et al., 2004), diseases (Doughty et al., 2011; Furlong et al., 2008; Yeniterzi and Sezerman, 2009) and other related information (Kanagasabai et al., 2007; Naderi and Witte, 2012). Despite different scopes, with regard to methods for mutation detection, most systems rely on manually derived regular expressions. For instance, for detecting protein point mutations (e.g. A42G) from text, Caporaso et al. (2007) developed MutationFinder, which contains >700 regular expression patterns and achieves state-of-the-art performance of 90% in F-measure. Compared with the overwhelmingly used rule-based systems, VTag (McDonald et al., 2004) stands out with a machine-learning approach and reports an F-measure of 0.82 on their evaluation data. We refer readers to Izarzugaza et al. (2012) for a summary of previous implementations for mutation extraction. Following VTag and other previous studies on biomedical named entities (Doğan and Lu, 2012b; Hsu et al., 2008; Leaman and Gonzalez, 2008), we formulated the problem of named entity recognition as a sequence-labeling problem. Therefore, a conditional random field (CRF) model (Lafferty et al., 2001) was naturally chosen as our learning algorithm. However, as detailed in our method description, we developed a new CRF model with 11 labels (as opposed to the traditional BIO model), as well as a set of novel features in this work. As shown in Section 4, both designs helped improve extraction performance and made our method compare favorably with the state of the art.
In addition, our work is also unique in extracting mutations of many types that are not considered by previous methods. Existing methods, such as MutationFinder either exclusively aim for extracting point mutations in proteins or are limited to a few mutation types, such as substitution and deletion in both proteins and genes. To our best knowledge, this work is the first attempt to identify various mutation types according to a standard nomenclature endorsed by the Human Genomic Variation Society (HGVS) for the description of sequence variants (mutations).
Finally, similar to MutationFinder, along with a public tool for mutation extraction, we also contribute to the text-mining community a large corpus (500 PubMed abstracts) of manually annotated raw and normalized mutation mentions. A raw mutation extraction is normalized when individual mutation components are identified and standardized when applicable. For instance, ‘Arg987Ter’ (PMID: 22188495) is normalized as ‘p|R|987|X’ to denote the replacement of an arginine residue at position 573 by termination codon, where a single letter ‘p’ is added to indicate the mutation type, and the standard one-letter codes are used (with their respective positions in the normalized notation) to represent the wild-type and mutant residue. As noted earlier in the text, our corpus covers many kinds of mutations not previously considered, such as ‘p.Pro246HisfsX13’ (PMID: 21738389) and ‘IVS3+1G/A’ (PMID: 15111599).
2 METHODS
As shown in Figure 1, our method first performs tokenization on the input text as pre-processing. Next, our method extracts mutation mentions from text using a CRF-based approach, followed by some post-processing steps. As illustrated in the figure, instead of extracting a mutation mention such as c.2708_2711delTTAG as a whole, our CRF module identifies each mutation component (e.g. ‘del’ as the mutation type) individually. Finally, we have implemented a post-processing module to handle some rare mutation formulas and nature language mentions that are not curated in our own corpus. We describe details of each step later in the text.
Fig. 1.

The system overview that includes three major components: pre-processing (tokenization), mutation identification (CRF) and post-processing (regular expression patterns)
2.1 Pre-processing: tokenization
A tokenizer divides text input into a sequence of tokens, which generally correspond to ‘words’. However, to capture individual components within a mutation mention, we performed tokenization on a finer level than traditional methods (Webster and Kit, 1992) that separate input text by space or punctuation. Specifically, special characters (e.g. ‘-’, ‘*’, ‘+’), numbers, lowercase letters and uppercase letters are divided as separate tokens. For instance, instead of regarding the mention ‘c.2708_2711delTTAG’ in Figure 1 as one token, we split it into seven pieces as shown in the top row of Table 1.
Table 1.
An example of mutation component labels in an excerpt ‘… (c.2708_2711delTTAG, p.V903GfsX905) …’ in PMID: 22042570
Each cell in the top row represents a token in our processing.
2.2 Mutation identification: CRF module
As aforementioned, we regarded the mutation identification problem as a sequence-labeling task. In particular, each mutation component was considered as an individual label (Table 1) such that every mutation mention becomes a sequence of labels. Accordingly, we adapted a probability-based sequence detection CRF model (Lafferty et al., 2001), which defines the conditional probability distribution 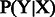 of label sequence Y given observation sequence X.
of label sequence Y given observation sequence X.
| (1) |
where  is a label sequence from Y and
is a label sequence from Y and 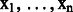 is a token sequence from X.
is a token sequence from X. 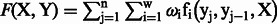 is a global feature vector for label sequence Y and observation sequence X and
is a global feature vector for label sequence Y and observation sequence X and  is a feature weight vector.
is a feature weight vector.
CRF is a log-linear model based on a set of the feature functions 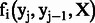 . This function determines the pair of state and observation tokens to a binary value and associates with the weight
. This function determines the pair of state and observation tokens to a binary value and associates with the weight 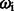 . The weight presents the importance of the tag and can be obtained from the training data by a limited-memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) (Liu and Nocedal, 1989) method. This model can combine the effects of many meaningful features. We then followed Wallach (2004) to design the observation function
. The weight presents the importance of the tag and can be obtained from the training data by a limited-memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) (Liu and Nocedal, 1989) method. This model can combine the effects of many meaningful features. We then followed Wallach (2004) to design the observation function 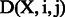 and feature function:
and feature function:
| (2) |
The observation function returns true, if the token in j position matches the criteria of feature i and vice versa. For example, if the token at xj is ‘glycine’, then the observation function for the Protein symbols feature would return true. Consider the following feature function:
| (3) |
The 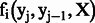 would return true if the two labels of previous
would return true if the two labels of previous 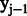 and current
and current 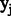 positions belong to one of our designed labels. The CRF model is determined by the features
positions belong to one of our designed labels. The CRF model is determined by the features 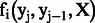 and their corresponding weight
and their corresponding weight 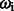 .
.
In this work, the  indicate the label for the corresponding tokens. Unlike the traditional BIO labeling models, which label each token as being the beginning of (B), the inside of (I) or entirely outside (O) of a span of interest, we designed 10 different labels (Table 2) for describing mutation elements (i.e. tokens within the mutation mentions) based on the HGVS nomenclature, and one additional label ‘O’ for all tokens outside a mention (see Table 1 for an example). In this work, we used the CRF++ (http://crfpp.googlecode.com/svn/trun-k/doc/index.html#download) for the actual implementation.
indicate the label for the corresponding tokens. Unlike the traditional BIO labeling models, which label each token as being the beginning of (B), the inside of (I) or entirely outside (O) of a span of interest, we designed 10 different labels (Table 2) for describing mutation elements (i.e. tokens within the mutation mentions) based on the HGVS nomenclature, and one additional label ‘O’ for all tokens outside a mention (see Table 1 for an example). In this work, we used the CRF++ (http://crfpp.googlecode.com/svn/trun-k/doc/index.html#download) for the actual implementation.
Table 2.
We defined 10 different labels for tokens within mutation mentions: reference sequence (A); mutation position (P); mutation type (T); wild-type (W); mutant (M); frame shift (F); frame shift position (S); duplication time (D); SNP (R); other inside mutation tokens (I)
| Mutation types | A | P | T | W | M | F | S | D | R | I |
|---|---|---|---|---|---|---|---|---|---|---|
| Substitution | • | • | • | • | • | • | ||||
| Deletion | • | • | • | • | • | |||||
| Insertion | • | • | • | • | • | |||||
| Insertion/deletion | • | • | • | • | • | |||||
| Duplication | • | • | • | • | • | • | ||||
| Frame shift | • | • | • | • | • | • | • | • | ||
| RS number | • |
Each mutation type has its own set of labels (e.g. substitution corresponds to six labels).
2.3 Features for CRF
We engineered six different types of features for this problem:
Dictionary features. We followed the HGVS mutation nomenclature and developed 11 (7 for genomic and 4 for protein mutations) regular expressions patterns as shown in Table 3. When there is a match, each token in the corresponding matched text will be assigned to one of the three values (‘B/I/E’) for that feature (B for the beginning token; E for the last token and I for any other tokens in between B and E). Any token that is not matched against these patterns will have the value of ‘O’ for this feature.
General linguistic features. Sometimes, the mutation mentions may include brief nature language, such as ‘G–>A at nucleotide position 2141’. To capture such mutations, we included the original tokens (e.g. nucleotide), as well as stemmed tokens (e.g. nucleotide), as features using the Porter’s stemmer.
Character features. We noticed that many mutation mentions contain numbers and special characters (e.g. the greater sign ‘>’ is often used to represent amino acid substitution). Therefore, for each token, we calculated several statistics as its features, including its number of digitals, number of uppercase and lower letters, number of all characters and presence or absence of mutation-specific characters (;,. - > + − _ /?).
- Semantic features. We created several semantic classes for describing mutation-specific characteristics. All the following features are binary: 1 when a corresponding word (e.g. del) is present; 0 otherwise.
- Reference sequence type: c (for coding DNA sequence), g (for genomic sequence), r (for RNA sequence), m (for mitochondrial sequence), p (for protein sequence)
- Exon/intron: IVS, Intron, Ex, Exon
- Mutation type: del, ins, dup, tri, delins, indel
- Frame shift mutation: fs, fsX, fsx
- DNA/RNA nucleotide: A, T, C, G, a, c, g, u
- Protein amino acid: e.g. glutamine, glu, E
- Mutation-type indicating word: deletion(s), insertion(s), repeat(s)
- Mutation unit: amino acid, acid(s), codon, position(s), bp, nucleotide(s), residue(s)
- Word preceding mutation mention: intron, exon, promoter, 5′-UTR, 3′-UTR
Case pattern features. A pattern is constructed to represent case shifting in the token, and this pattern is included as an additional feature. As such, each character in the token is represented in a simplified form. Any upper case alphabetic character is replaced by ‘A’ and any lower case one is replaced by ‘a’. Likewise any number (0–9) is replaced by ‘0’. Thus, the token ‘TTAG’ generates the case pattern feature ‘AAAA’, and the token ‘2711’ generates the pattern feature ‘0000’. Moreover, we also merged consecutive letters and numbers and generated additional single letter ‘a’ and number ‘0’ features.
Contextual features. We observed that the tokens in the mutation mention are highly correlated with each other. Take the letter ‘G’ for example; only six possible suffixes (‘lycine’, ‘lutamic’, ‘lutamine’, ‘ln’, ‘ly’ and ‘lu’) can be associated with it to be an amino acid. To take advantage of contextual information, for a given token, we included the dictionary and linguistic features of three neighboring tokens from each side.
Table 3.
Regular expression patterns of genomic and protein mutations based on examination of HGVS mutation nomenclature
| Type | Regular expression patterns | Example |
|---|---|---|
| Genomic | ([cgrm]\.[ATCGatcgu \/\>\<\?\(\)\[\]\;\:\*\_\-\+0-9]+(inv|del|ins|dup|tri|qua|con|delins|indel)[ATCGatcgu0-9\_\.\:]*) | c.2708_2711delTTAG |
| Genomic | (IVS[ATCGatcgu \/\>\<\?\(\)\[\]\;\:\*\_\-\+0-9]+(del|ins|dup|tri|qua|con|delins|indel)[ATCGatcgu0-9\_\.\:]*) | IVS2-58_55insT |
| Genomic | ([cgrm]\.[ATCGatcgu \/\>\?\(\)\[\]\;\:\*\_\-\+0-9]+) | c.467C>A |
| Genomic | (IVS[ATCGatcgu \/\>\?\(\)\[\]\;\:\*\_\-\+0-9]+) | IVS3+18C>T |
| Genomic | ([cgrm]\.[ATCGatcgu][0-9]+[ATCGatcgu]) | c.A436C |
| Genomic | ([ATCGatcgu][0-9]+[ATCGatcgu]) | A436C |
| Genomic | ([0-9]+(del|ins|dup|tri|qua|con|delins|indel)[ATCGatcgu]*) | 912delTA |
| Protein | ([p]\.[CISQMNPKDTFAGHLRWVEYX \/\>\<\?\(\)\[\]\;\:\*\_\-\+0-9]+(inv|del|ins|dup|tri|qua|con|delins|indel|fsX|fsx|fs x|fs)[CISQMNPKDTFAGHLRWVEYX \/\>\<\?\(\)\[\]\;\:\*\_\-\+0-9]*) | p.G204VfsX28 |
| Protein | ([p]\.[CISQMNPKDTFAGHLRWVEYX \/\>\?\(\)\[\]\;\:\*\_\-\+0-9]+) | p.G204V |
| Protein | ([p]\.[A-Z][a-z]{0,2}[\W\-]{0,1}[0-9]+[\W\-]{0,1}[A-Z][a-z]{0,2}) | p.Ser157Ser |
| Protein | ([p]\.[A-Z][a-z]{0,2}[\W\-]{0,1}[0-9]+[\W\-]{0,1}(fs|fsx|fsX)) | p.Ser119fsX |
2.4 Post-processing: regular expression rules
Despite our best efforts, the CRF model may still miss a few mentions. To minimize the number of false negatives within an article, we took the mentions extracted by the CRF module and translated them into regular expression patterns for finding additional mentions of similar kind in the same article. Two rules were applied to make the translated patterns more generalizable: (i) all numerical digitals become ‘[0-9]+’; (ii) all lowercase and uppercase letters become ‘[a-z]’ and ‘[A-Z]’, respectively, except three special tokens IVS, EX and RS. As a result, ‘c.IVS64+5C>G’ is translated to ‘[a-z]\.IVS[0-9]+\+[0-9]+[A-Z]\>[A-Z]’. In addition, we also added several regular expression patterns based on those of MutationFinder. But instead of directly adding their 759 specific patterns such as ‘[Wild type]-to-[Mutant] substitution at position [Position]’, we built more robust and general patterns. As a result, only a few regular expression patterns (<10) were needed. The inclusion of such patterns also complements our CRF-based approach in extracting those long natural-language mutation mentions in the article.
Based on examination of our method development data, we also developed several additional rules for matching irregular and rare mention formats such as ‘glycine-594-valine’ and ‘dup33bp’. In addition, our post-processing step also helps adjust text spans of mutation mentions, such as adding a missing closing parenthesis p.(Asp569Valfs*93 → p.(Asp569Valfs*93) or separating two consecutive mutations by semicolon rs1573496;C>G’ → ‘rs1573496’ and ‘C>G’.
2.5 Corpus construction
As a result of past research on this topic(Bonis et al., 2006; Caporaso et al., 2007; Furlong et al., 2008; Naderi and Witte, 2012), several mutation corpora are publicly available, but they are either limited in size and/or scope (protein point mutation) or lack mention-level annotations. Hence, we decided to develop our own corpus in this work.
We used PubMed to obtain a corpus of MEDLINE abstracts that contained a large number of mutation mentions. To construct a corpus containing numerous mentions of the types of mutations we were interested in, we included many facets in our query, which is composed of both MeSH (Medical Subject Heading) terms and other search field terms. To ensure we returned abstracts containing mentions of clinically relevant mutations, the MeSH ‘Diseases Category’ was an essential component. We chose the MeSH terms ‘Mutation’ and ‘Polymorphism, Genetic’ to obtain abstracts that had been indexed as pertaining to genomic variation.
The ‘Title/Abstract’ field was searched for terms identified to retrieve information pertaining to sequence type, mutation type and mutation location (see query later in the text). This strategy was chosen after reviewing example abstracts containing desired mutation mentions and attempting to retrieve abstracts with similar characteristics. Finally, we included non-genetic facets that accounted for the other preferences we had for our potential corpus. These aspects included needing abstracts in English and specifically about humans. We also wanted to ensure that only results containing abstracts were retrieved, and that we did not retrieve review articles because we preferred articles about novel mutations, as mutations are likely to be mentioned in formulaic fashion when they are first described. The PubMed query is shown later in the text:
‘Diseases Category’[Mesh] AND (mutation[MeSH Terms] OR polymorphism, genetic[MeSH Terms]) AND (DNA[Title/Abstract] OR nucleotide[Title/Abstract]) AND (deletion[Title/Abstract] OR substitution[Title/Abstract] OR insertion[Title/Abstract] OR duplication[Title/Abstract] OR indel[Title/Abstract] OR delin[Title/Abstract] OR conversion[Title/Abstract] OR translocation[Title/Abstract] OR inversion[Title/Abstract]) AND (codon[Title/Abstract] OR exon[Title/Abstract] OR intron[Title/Abstract] OR allele[Title/Abstract] OR gene[Title/Abstract] OR sequence[Title/Abstract]) AND (genotyp*[Title/Abstract] OR homozyg*[Title/Abstract] OR heterozyg*[Title/Abstract]) AND hasabstract[text] AND ‘humans’[MeSH Terms] AND English[lang] NOT Review[ptyp]’
We submitted this query and obtained 5116 abstracts from PubMed in June 2012. We randomly selected 500 abstracts among those published after 2001 for manual annotation using PubTator (Wei et al., 2012a, 2012b), a Web-based annotation tool (see a tool screenshot in Fig. 2). Our corpus was developed in stages by human annotators who have domain expertise as well as experience in NLP corpus development. In the first phase, three human annotators annotated 50 abstracts individually. They then compared their results and only obtained inter-annotator agreement of 46%. Most of the annotation discrepancies were found to be because of boundary issues (text spans of mutation mentions are overlapping but not identical) and varying conceptions of how to annotate nature language mentions. After discussion, a set of annotation guidelines was drafted (available with tmVar software and corpus). In the second phase, two of the annotators finished another set of 50 abstracts, reaching 88% agreement this time. In the final round, one annotator continued and finished the remaining 400 abstracts.
Fig. 2.

A screenshot of our applied curation system
2.6 Baseline approach and additional gold standard for evaluation
As shown in the Section 3 later in the text, we compared our method with MutationFinder. As pointed out by Izarzugaza et al. (2012), MutationFinder is ‘very competitive for recall and precision when compared to other strategies’, and over the years it has been widely adopted by many others for extracting protein point mutation (Gyimesi et al., 2012; Schenck et al., 2012; Witte and Baker, 2007). In addition, along with its public software, MutationFinder has a large corpus where both raw mentions and normalized annotations are available, which allowed us to perform cross-comparisons of both methods on two different gold-standard datasets.
3 RESULTS
3.1 Gold-standard evaluation data
As mentioned, our mutation corpus contains 500 articles. As shown in Figure 3, one mutation type (substitution) alone accounts for ∼70% of total annotations. Hence, we further divided the annotations in this group into two subgroups (i.e. amino acid substitution versus nucleic acid substitution). Despite the fact that substitution plays a dominant role, there are a few other types of sequence variations seen in our corpus, including deletion, insertion and others.
Fig. 3.

The percentage of different mutation types in our corpus (500 abstracts)
To use it as the gold standard for the method development and evaluation purposes, we randomly divided the whole set into two subsets. Detailed statistics about our corpus are shown in Table 4. Additionally, we also used the MutationFinder corpus in benchmarking. Those statistics are also shown in the table later in the text.
Table 4.
Statistics of benchmarking datasets
| Dataset | Abstracts | All mutations | Normalized mutations |
|---|---|---|---|
| Our training set | 334 | 967 | 604 |
| Our test set | 166 | 464 | 311 |
| MutationFinder corpus | 507 | 907 | 480 |
3.2 System performance
Following Caporaso et al. (2007), we computed precision, recall and F-measure on all mentions (including duplicates), as well as on normalized mentions, which emphasizes an evaluating system’s ability of extracting different mutations. In all cases, our method was compared with MutationFinder. Tables 5 and 6 show results on both our and MutationFinder corpus, respectively. Because MutationFinder was designed exclusively for detecting protein point mutation, we report its performance on all mutations, as well as just protein point mutations, when using our test corpus. As such, there are two rows of results in Table 5 for MutationFinder.
Table 5.
Results on the test set of our corpus in terms of precision (P), recall (R) and F-measure (F)
| Methods | P (%) | R (%) | F (%) | |
|---|---|---|---|---|
| All mutations | MutationFinder | 91.66 | 33.21 | 48.76 |
| MutationFindera | 89.66 | 69.15 | 78.08 | |
| tmVar | 91.38 | 91.40 | 91.39 | |
| Normalized mutations | MutationFinder | 84.21 | 25.29 | 38.90 |
| MutationFindera | 84.09 | 63.25 | 72.20 | |
| tmVar | 87.74 | 87.46 | 87.60 |
aProtein point mutations only.
Table 6.
Results on the MutationFinder corpus in terms of precision (P), recall (R) and F-measure (F)
| Methods | P (%) | R (%) | F (%) | |
|---|---|---|---|---|
| All mutations | MutationFinder | 98.41 | 81.92 | 89.41 |
| tmVar | 98.80 | 89.62 | 93.98 | |
| Normalized mutations | MutationFinder | 98.47 | 80.63 | 88.66 |
| tmVar | 97.58 | 83.96 | 90.26 |
As can been seen in Tables 5 and 6, our method tmVar achieved consistently higher F-measures than MutationFinder (P < 0.05; two-sided t-test) on two independent datasets. On the other hand, when benchmarked on our corpus, MutationFinder’s results (Table 5) dropped significantly from the performance on its own corpus (Table 6), especially in recall, even though we limited our evaluation to its extraction scope (protein point mutation). Our analysis shows that slight drop in precision was mainly because its patterns incorrectly identified DNA substitutions that are protein substitution-like (e.g. C35322T in PMID: 21054465) in our corpus. In terms of recall, most missed mentions are due to the lack of its patterns to recognize nonsense point mutations (e.g. V561X in PMID: 15749661) and point mutations preceded with a sequence type (e.g. p.A150P in 15880727).
Finally, we find our method tmVar is as fast as MutationFinder in extracting mutation mentions from text. When tested on a typical modern desktop computer with Core 2 Duo 3.16 GHz CPU and 4 GB random access memory, the required time for processing 5000 PubMed abstracts is comparable: 21.75 (tmVar) versus 24.17 min (MutationFinder).
4 DISCUSSION
4.1 Evaluation of post-processing step
As stated earlier, for optimal performance, the results of CRF were further supplemented by a set of manually derived rules for handling issues, such as mention boundaries, rare form mentions and so forth. As shown in table later in the text, when the post-processing module was removed altogether, modest drop in overall performance was found because of the loss in recall.
As can be seen in Table 7, experimental results using all mentions versus normalized mentions yields similar findings. Hence, in the following analysis, only results using all mentions are reported.
Table 7.
Evaluation of post-processing steps using our corpus in terms of precision (P), recall (R) and F-measure (F)
| Methods | P (%) | R (%) | F (%) | |
|---|---|---|---|---|
| All mutations | tmVar | 91.38 | 91.40 | 91.39 |
| Removing post-processing | 92.01 | 83.72 | 87.67 | |
| Normalized mutations | tmVar | 87.74 | 87.46 | 87.60 |
| Removing post-processing | 88.40 | 82.17 | 85.17 |
4.2 Performance comparison with different CRF labeling models
As aforementioned, unlike previous rule-based approaches (Caporaso et al., 2007; Doughty et al., 2011; Furlong et al., 2008), we developed a CRF-based method for extracting different elements of a mutation. Furthermore, different from the common labeling models, such as B(beginning), I(inside), O(outside) or B(beginning), I(inside), E(end), O(outside) (Settles, 2004), for named entity recognition, our approach used a finer-grained design including 11 different labels. As shown in Table 8, such a design not only allowed us to recognize individual mutation components, but also led to higher accuracy in extracted results. Note that results in Table 8 do not involve the use of post-processing patterns (i.e. we show results directly derived from the CRF module).
Table 8.
Performance comparison between different CRF labeling models using all mutations in our test set in terms of precision (P), recall (R) and F-measure (F)
| CRF labeling model | P (%) | R (%) | F (%) |
|---|---|---|---|
| Three labels (B, I, O) | 85.81 | 80.82 | 83.24 |
| Four labels (B, I, E, O) | 86.18 | 81.59 | 83.82 |
| Eleven labels (A, T, P, W, M, F, S, D, R, I, O) | 92.01 | 83.72 | 87.67 |
4.3 Evaluation of different features
To examine the contribution of individual feature types, we performed a feature ablation study where different types of features were removed from the entire set of features one at a time. As shown in Table 9, the largest drop in performance was due to the removal of general linguistic features, followed by character and semantic features. On the other hand, the removal of case pattern or contextual features had little effect on final performance. Same as results in Table 8, no post-processing patterns were used in these experiments.
Table 9.
Performance decrease when removing features in terms of precision (P), recall (R) and F-measure (F)
| Features | P (%) | R (%) | F (%) |
|---|---|---|---|
| All features | 92.01 | 83.72 | 87.67 |
| General linguistic features | 86.62 | 58.02 | 69.49 |
| Character features | 87.78 | 80.44 | 83.95 |
| Semantic features | 87.12 | 80.66 | 83.77 |
| Dictionary features | 88.64 | 83.65 | 86.07 |
| Contextual features | 89.81 | 83.62 | 86.60 |
| Case pattern features | 91.02 | 82.97 | 86.81 |
All mentions in our test set were used in this study.
4.4 Error analysis
Despite our best efforts, there are still errors in our mutation extraction results. We examined all the extraction errors from the test set and classified them into several major categories. As shown in Figure 4, majority of our errors were due to boundary issues (shown in red), as we require exact string offset match in our evaluation. Further analysis shows that in many cases, our method only extracts partial mutation information, such as ‘A > G’ in ‘A > G polymorphism at position −670’. This kind of errors affect both precision and recall. The second largest error category (shown in green) affects recall only, as our method simply missed true positive mutations like ‘p.S522fs 525stop’. Our method was also confused by some genotype descriptions (shown in blue) that have similar appearance to mutations by incorrectly predicting some genotypes (e.g. IVS9 + 459 GA + GG in PMID: 19880293) as mutations (false positive errors). Meanwhile, our specific genotype-filtering rule removes all mentions with identical wide type and mutant information during the post-processing, resulting in the loss of silent mutations (false negative errors). These three classes of errors accounted for >80% of the errors made by our method. Other smaller error types (shown in gray) include identifying mentions that look like mutations (e.g. ‘Gly-X-Y’) or the ones that are not included in the gold standard (e.g. we excluded natural language mutations).
Fig. 4.

Mutation extraction error types. False positive (FP) and negative (FN) errors are shown, respectively
5 CONCLUSION
In summary, we introduced a CRF-based machine-learning method for mutation extraction from text with high performance. Our method complements and extends existing methods in extracting a wide range of different types of sequence variants in scientific publications. There are several future directions for extending this work. First, tmVar is shown to be accurate on PubMed abstracts in this study. Although tmVar is also applicable to full text, future study is warranted to determine its actual performance on different text genres. Second, despite the fact this work in itself can be used and integrated by other research, just like any other standalone mutation extraction tools (e.g. MutationFinder), it is useful to extract and associate contextual information (e.g. gene/protein sequence) to the extracted mutations. Third, given our high extraction performance, we are interested in exploring direct applications of this work in the framework of computer-assisted biocuration (Lu and Hirschman, 2012; Névéol et al., 2011, 2012; Wei et al., 2012a). Indeed, we have started discussing potential opportunities of contributing our computer-generated results to dbSNP (L.Phan, personal communication). Finally, together with our ability to recognize proteins and diseases (Doğan and Lu, 2012a; 2012b; Lu et al., 2011; Wei et al., 2012c), we would like to investigate automatic methods for characterizing de facto relationships between sequence variations in complex diseases buried in the biomedical literature.
ACKNOWLEDGEMENT
The authors would like to thank Robert Leaman for his helpful discussion on the manuscript.
Funding: NIH Intramural Research Program, National Library of Medicine; NLM Associate Fellowship Program sponsored by the National Library of Medicine and administered by the Oak Ridge Institute for Science and Education (to B.H. in part).
Conflict of Interest: none declared.
REFERENCES
- Bonis J, et al. OSIRIS: a tool for retrieving literature about sequence variants. Bioinformatics. 2006;22:2567–2569. doi: 10.1093/bioinformatics/btl421. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, et al. MutationFinder: a high-performance system for extracting point mutation mentions from text. Bioinformatics. 2007;23:1862–1865. doi: 10.1093/bioinformatics/btm235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E, et al. Bioinformatics for personal genome interpretation. Brief. Bioinform. 2012;13:495–512. doi: 10.1093/bib/bbr070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doğan RI, Lu Z. Proceedings of the ACL Workshop on Biomedical Natural Language Processing. Stroudsburg, PA, USA: Association for Computational Linguistics; 2012a. An improved corpus of disease mentions in PubMed citations; pp. 91–99. [Google Scholar]
- Doğan RI, Lu Z. Proceddings of the Association for the Advancement of Artificial Intelligence. 2012b. An inference method for disease name normalization; pp. 8–13. [Google Scholar]
- Doughty E, et al. Toward an automatic method for extracting cancer - and other disease-related point mutations from the biomedical literature. Bioinformatics. 2011;27:408–415. doi: 10.1093/bioinformatics/btq667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdogmus M, Sezerman OU. Application of automatic mutation-gene pair extraction to diseases. J. Bioinform. Comput. Biol. 2007;5:1261–1275. doi: 10.1142/s021972000700317x. [DOI] [PubMed] [Google Scholar]
- Furlong LI, et al. OSIRISv1.2: a named entity recognition system for sequence variants of genes in biomedical literature. BMC Bioinformatics. 2008;2008:84. doi: 10.1186/1471-2105-9-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyimesi G, et al. ABCMdb: a database for the comparative analysis of protein mutations in ABC transporters, and a potential framework for a general application. Hum. Mutat. 2012;33:1547–1556. doi: 10.1002/humu.22138. [DOI] [PubMed] [Google Scholar]
- Horn F, et al. Automated extraction of mutation data from the literature: application of MuteXt to G protein-coupled receptors and nuclear hormone receptors. Bioinformatics. 2004;20:557–568. doi: 10.1093/bioinformatics/btg449. [DOI] [PubMed] [Google Scholar]
- Hsu CN, et al. Integrating high dimensional bi-directional parsing models for gene mention tagging. Bioinformatics. 2008;24:i286–i294. doi: 10.1093/bioinformatics/btn183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izarzugaza JMG, et al. Interpretation of the consequences of mutations in protein kinases: combined use of bioinformatics and text mining. Front. Physiol. 2012;3:323. doi: 10.3389/fphys.2012.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanagasabai R, et al. A workflow for mutation extraction and structure annotation. J. Bioinform. Comput. Biol. 2007;5:1319–1337. doi: 10.1142/s0219720007003119. [DOI] [PubMed] [Google Scholar]
- Kuipers R, et al. Novel tools for extraction and validation of disease-related mutations applied to Fabry disease. Hum. Mutat. 2012;31:1026–1032. doi: 10.1002/humu.21317. [DOI] [PubMed] [Google Scholar]
- Lafferty J, et al. Proceedings of the International Conference on Machine Learning (ICML 01) San Francisco, CA, USA: Morgan Kaufmann Publishers Inc.; 2001. Conditional random fields: probabilistic models for segmenting and labeling sequence data; pp. 282–289. [Google Scholar]
- Leaman R, Gonzalez G. Proceedsing of the Pacific Symposium on Biocomputing. Singapore: World Scientific Publishing Co. Pte. Ltd.; 2008. BANNER: an executable survey of advances in biomedical named entity recognition; pp. 652–663. [PubMed] [Google Scholar]
- Liu DC, Nocedal J. On the limited memory BFGS method for large scale optimization. Math. Prog. B. 1989;45:503–528. [Google Scholar]
- Lu Z, et al. The gene normalization task in BioCreative III. BMC Bioinformatics. 2011;12:S9. doi: 10.1186/1471-2105-12-S8-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Hirschman L. Biocuration workflows and text mining: overview of the BioCreative 2012 Workshop Track II. Database. 2012;2012 doi: 10.1093/database/bas043. article ID bas043; doi:10.1093/database/bas043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald RT, et al. An entity tagger for recognizing acquired genomic variations in cancer literature. Bioinformatics. 2004;20:3249–3251. doi: 10.1093/bioinformatics/bth350. [DOI] [PubMed] [Google Scholar]
- Morgan AA, et al. Overview of BioCreative II gene normalization. Genome Biol. 2008;9:S3. doi: 10.1186/gb-2008-9-s2-s3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naderi N, Witte R. Automated extraction and semantic analysis of mutation impacts from the biomedical literature. BMC Genomics. 2012;13(Suppl. 4):S10. doi: 10.1186/1471-2164-13-S4-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Névéol A, et al. Semi-automatic semantic annotation of PubMed queries: a study on quality, efficiency, satisfaction. J. Biomed. Inform. 2011;44:310–318. doi: 10.1016/j.jbi.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Névéol A, et al. Improving links between literature and biological data with text mining: a case study with GEO, PDB and MEDLINE. Database. 2012;2012 doi: 10.1093/database/bas026. article ID bas026; doi:10.1093/database/bas026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebholz-Schuhmann D, et al. Automatic extraction of mutations from Medline and cross-validation with OMIM. Nucleic Acids Res. 2004;32:135–142. doi: 10.1093/nar/gkh162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck M, et al. Extraction of genetic mutations associated with cancer from public literature. J. Health Med. Informat. 2012:S2. doi:10.4172/2157-7420.S2-002. [Google Scholar]
- Settles B. Proceedings of the International Joint Workshop on Natural Language Processing in Biomedicine and its Applications. Stroudsburg, PA, USA: Association for Computational Linguistics; 2004. Biomedical named entity recognition using conditional random fields and rich feature sets; pp. 104–107. [Google Scholar]
- Wallach HM. Technical Report MS-CIS-04-21. Philadelphia, PA, USA: Department of Computer and Information Science, University of Pennsylvania; 2004. Conditional random fields: an introduction. [Google Scholar]
- Webster JJ, Kit C. Proceedings of the 14th conference on computational linguistics. 1992. Tokenization as the initial phase in NLP; pp. 1106–1110. [Google Scholar]
- Wei CH, et al. Accelerating literature curation with text-mining tools: a case study of using PubTator to curate genes in PubMed abstracts. Database. 2012a;2012 doi: 10.1093/database/bas041. article ID bas041; doi:10.1093/database/bas041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CH, et al. Proceedings of the International BioCreative 2012 workshop. Washington DC, USA: 2012b. PubTator: a PubMed-like interactive curation system for document triage and literature curation; pp. 145–150. [Google Scholar]
- Wei CH, et al. SR4GN: a species recognition software tool for gene normalization. PLoS One. 2012c;7:e38460. doi: 10.1371/journal.pone.0038460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnenburg R, et al. Improved mutation tagging with gene identifiers applied to membrane protein stability prediction. BMC Bioinformatics. 2009;10:S3. doi: 10.1186/1471-2105-10-S8-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte R, Baker CJO. Towards a systematic evaluation of protein mutation extraction systems. J. Bioinform. Comput. Biol. 2007;5:1339–1359. doi: 10.1142/s0219720007003193. [DOI] [PubMed] [Google Scholar]
- Yeniterzi S, Sezerman U. EnzyMiner: automatic identification of protein level mutations and their impact on target enzymes from PubMed abstracts. BMC Bioinformatics. 2009;10:S2. doi: 10.1186/1471-2105-10-S8-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, et al. The need for genetic variant naming standards in published abstracts of human genetic association studies. BMC Res. Notes. 2009;2:56. doi: 10.1186/1756-0500-2-56. [DOI] [PMC free article] [PubMed] [Google Scholar]