

Abstract
Background
Meningiomas are the most commonly diagnosed primary intracranial neoplasms. Despite significant advances in modern therapies, the management of malignant meningioma and skull base meningioma remains a challenge. Thus, the development of new treatment modalities is urgently needed for these difficult-to-treat meningiomas. The goal of this study was to investigate the potential of build-in short interfering RNA-based Wilms' tumor protein (WT1)–targeted adoptive immunotherapy in a reproducible mouse model of malignant skull base meningioma that we recently established.
Methods
We compared WT1 mRNA expression in human meningioma tissues and gliomas by quantitative real-time reverse-transcription polymerase chain reaction. Human malignant meningioma cells (IOMM-Lee cells) were labeled with green fluorescent protein (GFP) and implanted at the skull base of immunodeficient mice by using the postglenoid foramen injection (PGFi) technique. The animals were sacrificed at specific time points for analysis of tumor formation. Two groups of animals received adoptive immunotherapy with control peripheral blood mononuclear cells (PBMCs) or WT1-targeted PBMCs.
Results
High levels of WT1 mRNA expression were observed in many meningioma tissues and all meningioma cell lines. IOMM-Lee-GFP cells were successfully implanted using the PGFi technique, and malignant skull base meningiomas were induced in all mice. The systemically delivered WT1-targeted PBMCs infiltrated skull base meningiomas and significantly delayed tumor growth and increased survival time.
Conclusions
We have established a reproducible mouse model of malignant skull base meningioma. WT1-targeted adoptive immunotherapy appears to be a promising approach for the treatment of difficult-to-treat meningiomas.
Keywords: adoptive immunotherapy, cranial nerve, skull base meningioma, Wilms' tumor 1
Recent advances in T cell immunology and gene transfer have enabled adoptive tumor immunotherapy using genetically engineered T cells in clinical medicine.1 Among a number of tumor-associated antigens, Wilms' tumor gene product 1 (WT1) is one of the most promising and universal target antigens for tumor immunotherapy. WT1-peptide vaccines have been the most widely evaluated, because they are easy to produce and are well tolerated in clinical trials with minimal toxicity.2,3 However, whether vaccine therapies induce sufficient amounts of effector cells to kill solid tumors in vivo is an issue that remains to be addressed. In contrast, the adoptive transfer of ex vivo–expanded effector cells could be more advantageous than vaccination, given the greater control of tumor-specific effector cell numbers. Thus, adoptive T cell immunotherapy using WT1-specific T cell receptor (TCR) gene transfer is an alternative direct approach. To increase the effectiveness of TCR gene therapy, we have recently developed a novel vector system that can selectively express target antigen–specific TCR, in which expression of endogenous TCR is suppressed by built-in short-interfering RNAs (siRNAs), named as siTCR vector.4 By using this siTCR vector, we previously generated WT1-specific/HLA-A*2402–restricted T cells with enhanced antitumor cytotoxicity.4,5
Meningiomas are the most commonly diagnosed of all primary intracranial neoplasms, constituting ∼30% of all primary tumors (Central Brain Tumor Registry of the United States, 2010). Approximately 75% of meningiomas are benign (World Health Organization [WHO] grade I), 20%–35% are atypical (WHO grade II), and 1%–3% are anaplastic/malignant (WHO grade III).6,7 Despite significant advances in modern therapies, surgical resection remains the treatment of choice for many patients with meningiomas.8–10 However, some histologically benign meningiomas often recur and become difficult to treat.11 Moreover, grade II and III meningiomas have high recurrence rates after surgical or radiosurgical management.12–14 In addition to the intrinsic biology, tumor location is also an important determinant of patient outcome. Skull base is a common site of origin for meningiomas. Complete resection of skull base meningioma is often not possible without a high risk of morbidity and mortality, given its surgical inaccessibility and proximity to vital brain structures, such as the cranial nerves. Cranial nerves are delicate nerves that arise directly from the brain, and meningiomas have a tendency to involve and infiltrate cranial nerves.15 The management of malignant meningioma and skull base meningioma remains a challenge, and development of new treatment modalities is urgently needed for these difficult-to-treat meningiomas.
In this study, we examined the expression of WT1 antigen in meningioma tissues and found a high level of WT1 mRNA expression in a majority of the tissues, compared with malignant gliomas. The evidence prompted us to develop adoptive transfer of WT1-specific TCR gene-engineered T cells targeting meningioma cells. In vitro studies revealed that TCR-transduced peripheral blood mononuclear cells (PBMCs) were able to secrete interferon-γ (IFN-γ) and lyse meningioma cells in an HLA-A*2402–restricted manner. To evaluate the efficacy of adoptive transfer of TCR-transduced PBMCs in meningioma in vivo, we developed a clinically relevant skull base model of malignant meningioma encasing the trigeminal nerve using the postglenoid foramen injection (PGFi) technique. To the best of our knowledge, this is the first report to describe the efficacy of adoptive immunotherapy by using genetically modified WT1-specific PBMCs in a meningioma model.
Materials and Methods
PBMCs
Whole blood samples were obtained from healthy donors who gave their informed consent. Whole blood was then diluted with the equal volume of phosphate-buffered saline (PBS) and FICOLL and centrifuged at 1600 rpm for 30 min. The buffy coat with PBMCs was carefully aspired. PBMCs were cultured in GT-T503 (Takara Bio, Shiga, Japan) supplemented with 1% autologous plasma, 0.2% human serum albumin, 2.5 mg/mL fungizone (Bristol-Myers Squibb, Tokyo, Japan), and 600 IU/mL interleukin-2 (IL-2). PBMCs obtained from the same donor and same blood sample were used to generate gene-modified PBMCs (GMCs) and non–gene-modified PBMCs (NGMCs).
Construction of Retroviral Vector and Retroviral Transduction
TCR genes were cloned from the HLA-A*2402–restricted WT1235–243–specific CD8+ CTL clone TAK-1.16–18 Partial codon optimization was performed by replacing the Cα and Cβ regions with codon-optimized TCR Cα and Cβ regions, respectively.4 Partially codon-optimized TCR-α and TCR-β genes were integrated into a novel vector encoding small-hairpin RNAs that complementarily bind to the constant regions of endogenous TCR-α and TCR-β genes (WT1-siTCR vector).4
PBMCs were stimulated with 30 ng/mL OKT-3 (Janssen Pharmaceutical, Beerse, Belgium) and 600 IU/mL IL-2 and transduced using the RetroNectin-Bound Virus Infection Method, in which retroviral solutions were preloaded onto plates coated with RetroNectin (Takara Bio), centrifuged at 2000 × g for 2 h, and rinsed with PBS. The procedure was repeated twice on days 4 and 5 after the initiation of PBMC culture. PBMCs were applied onto the preloaded plate.4 The transduced PBMCs were cultured for a total of 10 days. Control PBMCs (NGMCs) and TCR-transduced PBMCs (GMCs) were stored frozen in liquid nitrogen, thawed, and cultured in GT-T503 supplemented with 1% autologous plasma, 0.2% human serum albumin, 2.5 mg/mL fungizone, and 600 IU/mL IL-2 for 2 days to use in all the experiments below.
Cell Lines
The human meningioma cell lines IOMM-Lee (HLA-A*2402/0301),19 HKBMM (HLA-A*2402/1101),20 and KT21-MG1 (HLA-A*0207/1101)21 were used. IOMM-Lee was kindly provided by Dr. Anita Lai (University of California at San Francisco, CA), and HKBMM and KT21-MG1 were from Dr. Shinichi Miyatake (Osaka Medical University, Osaka, Japan). The T2A24 cell line was derived from the T2 cell line, which is deficient in TAP transporter proteins, after transfection with the HLA-A*2402 complementary DNA (cDNA). The human embryonic kidney cell line GP2-293 was obtained from the American Type Tissue Culture Collection (ATCC; MD). All cell lines were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and penicillin/streptomycin. Cell lines were grown at 37°C in a humidified atmosphere of 5% carbon dioxide. HLA-A genotyping was performed using polymerase chain reaction (PCR) sequence-based typing (SRL, Tokyo, Japan).
Sample Collection and RNA Extraction
Tumor specimens for molecular genetic analysis were obtained from 29 patients with meningioma and 25 patients with high-grade glioma who underwent surgical procedures at Nagoya University Hospital or affiliated hospitals. The molecular genetic analysis performed in this study was approved by the institutional ethics committee of Nagoya University, and all patients who registered for this study provided written informed consent. All tumors were histologically verified according to the WHO 2007 guidelines; 23 patients had grade-I meningioma, 5 had grade-II meningioma, 1 had grade-III meningioma, 6 had grade-III glioma, and 19 had grade-IV glioma. RNA purification was performed using the standard TRIzol (Invitrogen, Carlsbad, CA) method.
Quantitative Analysis of WT1 mRNA Expression
Total RNA was extracted from 54 tumors, 3 cell lines, and normal whole brain (Human Total RNA Master Panel II; Takara Bio, Otsu, Japan), and first-strand cDNA was synthesized using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany). The cDNA product was used in reverse-transcription (RT) PCR for the quantitation of WT1 and GAPDH mRNA levels. The primers and Taqman probes for the assay were purchased from Roche Diagnostics (Indianapolis, IN). The sequences of the primers and probe used to detect WT1 mRNA were as follows: WT1 forward primer (5′-GATAACCACACAACGCCCATC-3′), WT1 reverse primer (5′-CACACGTCGCACATCCTGAAT-3′), and WT1 probe (5′-FAM-ACACCGTGCGTGTGTATTCTGTATTGG-TAMRA-3′). The sequences of the primers used to detect GAPDH mRNA were as follows: GAPDH forward primer (5′-AGCCACATCGCTCAGACAC-3′) and GAPDH reverse primer (5′-GCCCAATACGACCAAATCC-3′). The GAPDH probe was from the Roche Human Probe Library (no. 60). RT-PCR assay was performed using the LightCycler 480 Probes Master and LightCycler 480 instrument II (Roche Diagnostics). WT1 expression was normalized to that of GAPDH in each sample.
Measurement of Proviral Copy Number in Retrovirus-Transduced PBMCs
Genomic DNA was purified from transduced PBMCs, and the mean proviral copy number per cell was quantified using the Cycleave PCR core kit (Takara Bio) and Proviral Copy Number Detection Primer Set (Takara Bio).
Flow Cytometry
PE-conjugated anti-human CD4 monoclonal antibody (mAb; eBioscience, San Diego, CA), FITC-conjugated anti-human CD8 mAb (BD Biosciences, San Diego, CA), and PE-conjugated WT1235–243/HLA-A*2402 tetramer (provided by Dr. Kuzushima, Aichi Cancer Center Research Institute) were used. Stained cells were analyzed using a FACScanto II flow cytometer (BD Biosciences).
For intracellular IFN-γ staining, PBMCs (1.0 × 106 cells) were cultured with IOMM-Lee or KT21-MG1 meningioma cells (1.0 × 106 cells) for 1 h. BD GolgiStop (0.7 μg/mL; BD Biosciences) was added, and cells were cultured for an additional 8 h. Then, the PBMCs were incubated with Fc blocker (eBioscience, San Diego, CA) and stained with FITC-conjugated anti-human CD8 mAb. After this, the PBMCs were incubated with BD Cytofix/Cytoperm solution (BD Biosciences) at 4°C for 20 min and then washed with BD Perm/Wash solution (BD Biosciences). The PBMCs were then incubated with APC-conjugated anti-human IFN-γ mAb (BD Biosciences), followed by flow cytometry.
Calcein-AM Cytotoxicity Assay
The ability of the transduced PBMCs to lyse target cells was measured using a calcein-AM (Dojindo, Kumamoto, Japan) release assay, as described previously.22 In brief, 5 × 103 calcein-AM–labeled target cells and various numbers of effector cells in 200 μL of RPMI 1640 medium containing 10% fetal bovine serum were seeded into 96-well round-bottom plates. The target cells were incubated with or without 10 nM WT1 peptide for 2 h before the addition of effector cells. After incubation with the effector cells for 4 h, 100 μL of supernatant was collected from each well. The percentage of specific lysis was calculated according to the formula [(experimental release – spontaneous release)/(maximum release – spontaneous release)] × 100.
Generation of Green Fluorescent Protein–Expressing IOMM-Lee Cells
A retrovirus expressing green fluorescent protein (GFP) was constructed using the Retro-X Universal Packaging System (Clontech, CA). GP2-293 cells were transfected with pRetroQ-AcGFP-C1 along with a pVSVG plasmid (Clontech). After 48 h, cell-free viral supernatants were obtained and stored at −80°C. IOMM-Lee cells were transduced with the retroviral vectors encoding GFP with use of the RetroNectin-bound Virus Infection Method, in which retroviral solutions were preloaded onto RetroNectin-coated plates, centrifuged at 2000 × g for 2 h, and rinsed with PBS. IOMM-Lee cells were then applied onto the preloaded plate.
Skull Base Meningioma Xenograft
NOD/Shi-SCID, IL-2Rγcnull (NOG) mice were created at the Central Institute of Experimental Animals (Kawasaki, Japan) by backcrossing γcnull mice with NOD/Shi-SCID mice, as reported previously.23 Eight-week-old mice were given intracranial injections containing 3 μL of 5.0 × 104 freshly dissociated GFP-expressing IOMM-Lee cells with use of the PGFi technique.24 In brief, the mice were anesthetized with an intraperitoneal injection of 45 mg/kg sodium pentobarbital (Dainippon Sumitomo Pharma, Osaka, Japan). A 26-gauge needle tip was positioned on the right PGF (the rostral area of the opening of the external acoustic meatus). The implantation site, the lateral part of the foramen ovale, was accessed via the following injection track: horizontal angle, 60°; sagittal angle, −45°; and insertion depth, 3 mm (Supplementary Material, Fig. S1A and B). The cells were injected over 5 s. The needle was slowly withdrawn over several seconds. Minimal finger pressure was applied for 30 s after needle withdrawal to stop the bleeding at the puncture site. After the injections, the mice were given free access to water and were examined twice per day. Corneal sensitivity was also recorded using a cotton filament, and the blinking of the right eye was compared with that of the control (left) eye.
In Vivo Anti-Meningioma Effects of WT1-siTCR Gene-Transduced CTLs
In our preliminary experiments, the median survival of untreated animals was consistently 12.5 days (data not shown). Twenty-four mice bearing established tumors were randomly assigned to 2 different experimental groups. Five days after tumor inoculation, human PBMCs (5.0 × 107 cells) were injected into the tail vein. On the twelfth day after tumor inoculation, 6 mice per group were sacrificed to evaluate tumor size and CD8+ T cell infiltration. According to statistical considerations based on our preliminary experiments, the remaining mice were monitored for signs of keratopathy and survival for up to 28 days after inoculation,
Tissue Processing and Immunohistochemistry
Mouse heads were fixed in 10% neutral buffered formalin (Wako Pure Chemical Industries, Osaka, Japan) for 48 h. GFP fluorescence in tumor cells was analyzed using a fluorescence imaging system (IVIS spectrum; Caliper Life Sciences, Alameda, CA) after the removal of the skull. Two-dimensional tumor size was calculated from the fluorescent area by using the Living Image software (Caliper Life Sciences), because the established tumors grew in a flattened pattern, similar to meningioma en plaque in humans. For histopathologic examination, formalin-fixed mouse heads were decalcified in Decalcifying Solution B (Wako Pure Chemical Industries) for 96 h and embedded in paraffin. Serial 5-μm sections were cut and processed for hematoxylin and eosin (H&E) staining, Luxol fast blue (LFB) staining, or immunohistochemistry. The sections were deparaffinized with xylene and rehydrated with ethanol. LFB staining was performed according to the method of Werner et al.25 In brief, the sections were placed in 0.1% LFB solution at 60°C for 16 h. After several washes, sections were differentiated in 0.05% lithium carbonate solution, followed by 70% ethanol. The slides were then incubated in 0.1% Cresyl echt violet solution to counterstain nuclei. Immunohistochemistry for human CD8+ T cells was performed using anti-human CD8 antibody (MBL, Nagoya, Japan). In brief, the sections were rinsed in PBS and incubated with the antibody freshly diluted at 1:100 in PBS. The Vector M.O.M. Immunodetection Kit (Vector Laboratories, Burlingame, CA) was used to perform the secondary antibody incubations. The staining was visualized with diaminobenzidine, and sections were counterstained with hematoxylin. We counted the number of both normal tissue-infiltrating (oral mucosa and submucosal soft tissues) and tumor-infiltrating CD8+ T cells in a microscopic grid 0.5 × 0.5 mm in size (0.25 mm2) at a magnification of 200×. The area with the most abundant distribution of CD8+ T cells was selected in each mouse.
Statistical Analysis
Comparisons between groups were done using paired t test or Welch's t test or Mann–Whitney exact test, where appropriate. Differences were considered to be statistically significant at P < .05. The outliers were defined as data points that were >3 times the interquartile ranges below the first quartile or above the third quartile. The Kaplan-Meier method and log-rank test were used to determine whether there was a significant difference in clinical events between the groups.
Results
WT1 Expression in Meningioma Patient Samples and Cell Lines
WT1 mRNA levels in samples from patients with meningioma and human meningioma cell lines were determined using quantitative RT-PCR and calculated relative to the WT1 expression level in the normal brain. As shown in Table 1, WT1 mRNA was expressed at high levels in samples from patients with meningioma. Of interest, a correlation was found between the WT1 expression levels and the MIB-1 labeling index (P = .0018, Fig. 1A), but there was no significant correlation with tumor location, tumor grading, and performance status (modified Rankin scale). We also examined WT1 expression in 25 high-grade glioma samples. The mean expression level of WT1 mRNA in meningioma samples was significantly higher than that in high-grade glioma samples (26.25 [18.27] vs. 5.45 [12.52]; P = .000014). The genotype, WT1 expression levels, and intracranial tumorigenicity of NOG mice implanted with the 3 meningioma cell lines are presented in Table 2. In all the 3 meningioma cell lines, the WT1 mRNA levels were >8-fold higher than that of the normal brain.
Table 1.
Data on patients with meningiomas and tumor characteristics
| Age (years)/Sex | Location of Lesion | Pathological Diagnosis (subtype) | WHO Grade | MIB-1 Index (%) | Relative Quantity of WT1 mRNA (NB = 1) | Extent of Resection (Simpson grade) | Follow-up Period (month) | Recurrence/Regrowth | mRS | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 71M | SB | meningothelial | I | ND | 19.43 | 2 | 39 | − | 0 |
| 2 | 49F | non- SB | meningothelial | I | ND | 5.06 | 2 | 55 | − | 0 |
| 3 | 69F | non- SB | fibrous | I | ND | 27.86 | 3 | 77 | − | 0 |
| 4 | 68F | non- SB | transitional | I | ND | 22.78 | 1 | 77 | − | 0 |
| 5 | 30F | SB | meningothelial | I | ND | 25.63 | 2 | 23 | − | 1 |
| 6 | 68M | SB | meningothelial | I | ND | 12.64 | 4 | 15 | − | 5 |
| 7 | 73F | SB | meningothelial | I | ND | 20.68 | 2 | 32 | − | 1 |
| 8 | 46F | SB | meningothelial | I | <1 | 7.78 | 2 | 76 | − | 0 |
| 9 | 36F | SB | meningothelial | I | ND | 22.63 | 2 | 65 | − | 1 |
| 10 | 64M | SB | meningothelial | I | ND | 12.55 | 2 | 7 | − | 2 |
| 11 | 56M | non- SB | fibrous | I | ND | 48.17 | 1 | 57 | − | 0 |
| 12 | 46F | non- SB | meningothelial | I | ND | 56.89 | 1 | 69 | − | 0 |
| 13 | 42F | non-s SB | meningothelial | I | ND | 4.50 | 1 | 89 | − | 2 |
| 14 | 58M | SB | meningothelial | I | 5 | 38.59 | 2 | 65 | + | 1 |
| 15 | 39F | non- SB | transitional | I | 3 | 39.67 | 1 | 2 | − | 3 |
| 16 | 72F | non- SB | fibrous | II | 3 | 33.13 | 2 | 72 | + | 1 |
| 17 | 63M | SB | atypical | II | 4 | 60.97 | 4 | 63 | − | 1 |
| 18 | 77M | SB | meningothelial | I | 1–2 | 4.03 | 4 | 70 | − | 0 |
| 19 | 54M | SB | meningothelial | I | 1–3 | 7.57 | 4 | 53 | − | 2 |
| 20 | 33F | non- SB | meningothelial | II | 5–6 | 47.50 | 4 | 16 | − | 2 |
| 21 | 48F | SB | meningothelial | I | ND | 28.25 | 2 | 57 | − | 1 |
| 22 | 62M | SB | meningothelial | I | ND | 47.84 | 4 | 2 | − | 1 |
| 23 | 66F | SB | meningothelial | II | ND | 58.49 | 1 | 48 | − | 2 |
| 24 | 70F | SB | atypical | II | 20–30 | 41.64 | 2 | 80 | + | 1 |
| 25 | 52M | SB | atypical | II | 10–15 | 14.22 | 3 | 37 | + | 5 |
| 26 | 55M | SB | atypical | II | 2 | 4.63 | 2 | 73 | − | 1 |
| 27 | 63M | non- SB | atypical | II | 4 | 2.79 | 2 | 83 | − | 0 |
| 28 | 45M | non- SB | clear cell | II | 1–2 | 9.06 | 1 | 92 | − | 0 |
| 29 | 68F | non- SB | anaplastic | III | 3–10 | 36.25 | 3 | 64 | + | 2 |
Abbreviations: mRS, modified Rankin Scale; NB, normal brain; ND, not done; SB, skull base.
Fig. 1.

Correlation between Wilms' tumor protein (WT1) expression and MIB-1 index (A) in vitro effector activity of Wilms' tumor protein (WT1)–targeted peripheral blood mononuclear cells (PBMCs). Gene-modified PBMCs (GMCs) were produced by transducing PBMCs with the WT1-siTCR vector. Non–gene-modified PBMCs (NGMCs) were used as a negative control. (B) CD4+ and CD8+ cells constitute 14.6% and 78.4% of NGMCs and 19.7% and 72.8% of GMCs, respectively. The proportions of WT1-tetramer–positive cells in NGMCs and GMCs are shown in the lower column. (C) Intracellular interferon-γ (IFN-γ) production by NGMCs and GMCs against human meningioma cell lines. NGMCs and GMCs were cocultured with WT1-positive and HLA-A*2402–negative KT21-MG1 cells or WT1-positive and HLA-A*2402–positive IOMM-Lee cells. The PBMCs were analyzed for intracellular IFN-γ production. (D and E) WT1-specific and HLA-A*2402–restricted cytotoxicity of GMCs. Cytotoxic activities of NGMCs and GMCs against WT1-peptide–loaded and nonloaded T2A24 cells (D) and KT21-MG1 and IOMM-Lee cells (E) were examined using the calcein-AM assay at various effector/target (E/T) ratios.
Table 2.
Characteristics of human meningioma cell lines
| Cell Line | Relative Quantity of WT1 mRNA (NB = 1) | HLA-A Genotyping | Intracranial Tumorigenicity in Immunocompromised Mice |
|---|---|---|---|
| HKBMM | 8.51 | 2402/1101 | − |
| IOMM-Lee | 8.82 | 2402/0301 | + |
| KT21-MG1 | 18.77 | 0207/1101 | − |
Abbreviation: NB, normal brain.
Cell Surface Expression of CD4, CD8, and WT1-Specific TCR in NGMCs and GMCs
PBMCs were transduced with WT1-siTCR at relatively low copy numbers to reduce the risk of insertional mutagenesis. In the GMCs used in this study, the proviral copy number was 2.24 copies per cell (data not shown). CD4+ and CD8+ cells constituted 14.6% and 78.4% of NGMCs and 19.7% and 72.8% of GMCs, respectively (Fig. 1B). About 20% of the GMCs were positive for HLA-A*2402/WT1 tetramer staining (Fig. 1B). Moreover, HLA-A*2402/WT1-tetramer positivity in CD3+CD8+ cells and CD3+CD4+ cells was 36.2% and 36.3%, respectively, suggesting that both fractions were similarly transduced (Supplementary Material, Fig. S3A). These NGMCs and GMCs were used as effector cells in subsequent assays.
Intracellular IFN-γ Production by GMCs Against Human Meningioma Cell Lines
To confirm that the freezing and thawing procedures had not affected the antigen specificity and HLA restriction of GMCs, we first investigated their intracellular IFN-γ production in response to WT1-peptide–loaded and nonloaded T2A24 cells. As demonstrated previously, GMCs exhibited specific reactivity to WT1-peptide–pulsed T2A24 cells (data not shown).4,5 We also investigated the intracellular IFN-γ production in NGMCs and GMCs against WT1-positive meningioma cell lines, KT21-MG1 (HLA-A*2402 negative) and IOMM-Lee (HLA-A*2402 positive). As shown in Fig. 1C, GMCs exhibited specific reactivity to IOMM-Lee cells. These data confirm that GMCs can recognize WT1-positive meningioma cells in an HLA-A*2402–restricted manner.
Cytotoxicity of GMCs Against Human Meningioma Cell Lines
To determine whether GMCs were able to lyse target cells, effector cells were mixed with calcein-AM–labeled target cells. As shown in Fig. 1D, GMCs lysed T2A24 cells that had been loaded with the WT1-peptide, but were not cytotoxic against nonloaded cells. The cytotoxicities of GMCs against human meningioma cell lines are shown in Fig. 1E. GMCs exhibited significant lytic activity against IOMM-Lee cells but not KT21-MG1 cells. These results strongly suggest that WT1-specific effector cells can lyse meningioma cells via recognition of their WT1-derived peptide in the context of HLA-A*2402.
Establishment of a Mouse Model of Skull Base Meningioma
Recently, we developed PGFi, a simple method that enables percutaneous injection of cells into the mouse brain. Because the PGFi technique provides access to the skull base area with minimal brain damage from needle penetration, we applied this technique to establish a mouse model of skull base meningioma. We selected the lateral part of the right foramen ovale as a tumor implantation site and used the needle trajectory shown in Supplementary Material, Fig. S1. GFP-labeled IOMM-Lee cells (IOMM-Lee-GFP) were implanted into 9 NOG mice. At 5 and 10 days after xenografting, 3 mice each were sacrificed and tumor growth was assessed. On day 14, the remaining 3 mice appeared to be sickly, and they were sacrificed for the assessment of tumor size. The overall intraoperative mortality was 0%, with a tumor induction rate of 100%. Representative macroscopic pictures, fluorescence images, and the corresponding H&E-stained sections of the IOMM-Lee-GFP skull base xenografts are shown in Fig. 2A. Tumors were induced in the right temporal fossa, enlarged rapidly and encased the ipsilateral trigeminal nerves, and extended into the contralateral skull base in the late phase. Macroscopic analysis revealed that tumors grew along the skull base and did not invade the surface of the brain (Fig. 2B). Because tumors grew in a flattened pattern, we used the 2-dimensional tumor size, which was calculated from the fluorescent area, to assess the tumor size. Fig. 2C shows the line graph representing the mean tumor size on days 5, 10, and 14 after implantation. Clinical monitoring of tumor-bearing mice revealed progressive ophthalmic signs, including decreased blink reflex and corneal aberration (Fig. 3A). These signs were consistent with a diagnosis of neurotrophic keratopathy. It is known that the trigeminal nerve provides corneal innervations, and corneal denervation abolishes the corneal blink reflex and leads to neurotrophic keratopathy. In a previous study on a mouse model of neurotrophic keratopathy, Ferrari et al damaged the mouse's trigeminal nerve at the skull base by electrolysis to induce ipsilateral neurotrophic keratopathy.26 They reported that electrode insertion did not cause keratopathy and that electrocoagulation was required to induce keratopathy. Similarly, no keratopathy was caused by needle insertion alone in our model, because the needle was inserted to the skull base just lateral to the foramen ovale (Supplementary Material, Fig. S1). We performed histopathologic analysis on the symptomatic mice. The right trigeminal nerves were encased and infiltrated by tumor cells and exhibited extensive disruption compared with the contralateral nerves (Fig. 3B). Thus, in our mouse model, keratopathy was considered to be caused by skull base meningioma, and it provided an indirect indication of trigeminal nerve damage caused by the tumor.
Fig. 2.

Representative histologic images of skull base tumor formation at days 5, 10, and 14. (A) White-light imaging, fluorescence imaging, and the corresponding H&E staining of the tumors are shown. The tumor formed from the implanted IOMM-Lee tumor cells labeled with green fluorescent protein (GFP) is seen in the right skull base. Green light emitted from the GFP was captured by a charge-coupled device camera. After fluorescence imaging, the skull base specimens were processed for H&E staining. Scale bar = 2 mm. (B) White-light imaging and fluorescence imaging of the whole brain of a tumor-bearing mouse at day 14. (C) Line graph representing the mean tumor size on days 5, 10, and 14 after tumor implantation.
Fig. 3.
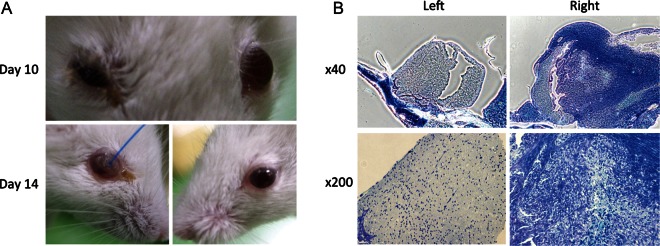
Skull base meningioma xenograft induces neurotrophic keratopathy by damaging the trigeminal nerve. (A) Representative photographs of the face of a mouse demonstrating the development of neurotrophic keratopathy in the right eye at days 10 and 14 after tumor inoculation in the right skull base. (B) Representative Luxol fast blue staining of the trigeminal nerves of a skull base meningioma-bearing mouse. The right trigeminal nerve is encased and infiltrated by tumor cells and exhibits extensive disruption, compared with the contralateral side. Original magnification: 40× (upper) and 200× (lower).
Effects of GMCs on WT1-Expressing Meningioma In Vivo
We used our newly developed skull base meningioma model to evaluate the in vivo efficacy of GMCs. Five days after intracranial injection of IOMM-Lee-GFP cells, NGMCs or GMCs were adoptively transferred via the tail vein. Although complete tumor eradication was not observed on day 12, tumor growth was significantly retarded in GMC-injected mice, compared with the control group (P = .0062; Fig. 4A–C). In both groups, we counted the number of infiltrating CD8+ T cells in the tumor and normal mesectoderm tissues, including the oral mucosa and submucosal soft tissues (Fig. 4D–F). There was no significant difference in the number of CD8+ T cells infiltrating the normal tissue in the 2 groups. In contrast, the number of CD8+ T cells infiltrating the tumor was greater in the GMC-treated group than in the NGMC-treated group (P = .0040). The number of CD4+ T cells infiltrating the normal tissue and the tumor was limited in both the NGMC- and GMC-treated groups (Supplementary Material, Fig. S3B). Moreover, the survival time was remarkably prolonged in GMC-treated mice (log rank test, P = .0055; Fig. 5A). Although there were no survivors among the NGMC-treated mice, there were 3 survivors (50%) among the GMC-treated mice by day 28. However, all 3 survivors on day 28 harbored a small size of tumor (Fig. 5B). Consistent with the tumor growth retardation, GMCs decreased the incidence and delayed the onset of neurotrophic keratopathy during the observation period (P = .014; Fig. 5C). Two GMC-treated mice (33%) survived with no symptoms of keratopathy until the experiment was terminated at day 28 after tumor inoculation. Therefore, these 2 mice were excluded from the statistical analysis of time to onset.
Fig. 4.

Inhibition of skull base meningioma growth in NOG mice after adoptive transfer of GMCs. (A and B) Fluorescence images of skull base meningiomas in mice treated with NGMCs (A) and GMCs (B). The lower graph (C) is a summary of the 2-dimensional tumor size in mice treated with NGMCs and GMCs. The black line in the box indicates the median and the open circle indicates the outlier. (D and E) Representative photomicrographs of immunohistochemical staining (human CD8+ T cells) of tumors from NGMC-treated mouse (D) and GMC-treated mouse (E). Original magnification: 200×. The lower table (F) is a summary of normal tissue- and tumor-infiltrating CD8+ T cell counts in mice treated with NGMCs and GMCs.
Fig. 5.

(A) Survival analysis of skull base meningioma-bearing mice treated with NGMCs and GMCs. The mice received an intravenous injection of NGMCs or GMCs on day 5 after tumor inoculation. There are no survivors among the NGMC-treated mice but 3 survivors among GMC-treated mice by day 28. (B) Three survivors have a small tumor. (C) Effect of adoptive transfer of GMCs on the incidence and time to onset of neurotrophic keratopathy in skull base meningioma-bearing mice. All NGMC-treated mice exhibited neurotrophic keratopathy throughout the observation period. Two GMC-treated mice survived with no symptoms of keratopathy until the experiment was terminated at day 28 after tumor inoculation. These 2 mice were excluded from the statistical analysis of time to onset.
Discussion
The principal findings of this study are (1) WT1 is highly expressed in menigiomas and (2) unmanageable skull base meningiomas are markedly treated with adoptive transfer of T cells retrovirally transduced with WT1-specific TCR gene that were also designed to prevent miscoupling with endogenous TCR.
WT1-Targeted Cell Therapy
We investigated the use of WT1 as a target for meningioma immunotherapy. To date, there have been no reports on the relationship between WT1 and meningioma. In the present study, we observed high levels of WT1 mRNA in meningioma tissues and cultured cell lines. WT1 is highly expressed in various types of tumors, and clinical trials in WT1-targeted immunotherapy have confirmed the safety and clinical efficacy of major histocompatibility complex class I–restricted WT1 epitope peptides.27,28 Of note, in a recent study, WT1 was selected from 75 defined tumor antigens as the most promising antigen.29
Immunotherapy is a conceptually attractive approach for malignant skull base meningioma, because it is highly specific and can deal with adherent and invasive tumor cells with minimal impact on normal vital brain structures. Induction of tumor-specific effector T cells is critical for eradicating bulky solid tumors, and it is the final goal of tumor immunotherapy approaches. Tumor-specific cytotoxic T cells can be genetically engineered to express altered or totally artificial TCRs, but the limited efficacy of TCR gene therapy has been reported to be associated with insufficient surface expression of the transduced TCRs.30–33 The existence of endogenous TCR is one of the major reasons for this insufficient cell surface expression, because endogenous TCRs compete with the introduced TCRs for CD3 molecules, and the endogenous TCR chains have been reported to mispair with the transduced TCR chains.33–37 To address this problem, our group has previously constructed a number of siRNAs to knock down the endogenous TCR α and β chains, and we have measured the knock-down efficiency. We used a tetramer assay to show that the vector knocking down the endogenous TCRs most efficiently achieved the highest expression of engineered WT1-TCR.4 We have also shown that the introduction of WT1-siTCR to HBZ-specific CTLs resulted in an upregulation of WT1-TCR and a downregulation of HBZ-TCR.5 Using this retroviral vector system, we transduced PBMCs with the HLA-A*2402–restricted and WT1-specific TCR. In a recent preclinical study, Ochi et al reported marked antileukemic reactivity and safety of WT-siTCR–transduced T cells.5 In the present study, we purposely used PBMCs transduced with WT1-siTCR at relatively low copy numbers with a view to clinical application, because restricting the copy number per cell is ideal for reducing the risk of insertional mutagenesis.38,39 We first demonstrated that GMCs exhibited a strong cytotoxic effect against human meningioma cells in an HLA-class І–restricted manner. Then, we investigated the in vivo efficacy of a single injection of GMCs. Although complete tumor eradication was not observed, GMCs significantly retarded tumor growth and prolonged the overall survival of treated mice. Moreover, GMC treatment significantly retarded the progression of trigeminal nerve damage caused by meningioma. Immunohistochemistry revealed robust accumulation of human CD8+ cells in meningioma lesions, which is a critical factor governing the success of tumor immunotherapy. Our results suggest that gene immunotherapy using WT1-siTCR is a promising new modality for the treatment of difficult-to-treat meningiomas. Before translating the proposed project into a clinical trial, off-target effects on normal tissues are major concerns. We have previously reported that WT1-siTCR CTLs had no cytolytic effects on CD34+ cells.5 These issues should be addressed in a phase I clinical trial.
A Novel Skull Base Meningioma Model
In addition to the intrinsic biology of meningiomas, tumor location is also an important factor in determining the outcome in patients with meningioma. Skull base is one of the most common locations for meningiomas. Resection of skull base meningiomas can lead to high rates of morbidity and mortality because of their deep locations and the possible involvement of vital brain structures, such as cranial nerves. Cranial nerves arise directly from the brain and are so delicate as to be susceptible to damage by surgical procedures or radiation. Meningiomas have a tendency to involve and infiltrate cranial nerves.15 It is very difficult to preserve the anatomical and functional integrity of the cranial nerves involved in tumors, particularly in hard lesions, such as meningiomas. If a new treatment modality for meningioma is to be of clinical value, it must be therapeutically effective against malignant meningioma and skull base meningioma involving and infiltrating cranial nerves.
To test the effectiveness of a new treatment modality in skull base meningioma, a patient-like orthotropic model of unresectable meningioma is needed. Several studies have reported xenograft tumor models of skull base meningioma, and IOMM-Lee is the most commonly used cell line. In conventional xenograft meningioma models, tumor cells are implanted using a stereotactic head frame and a bur hole drilled in the frontal bone.40–44 We implanted IOMM-Lee cells into the lateral part of the foramen ovale in NOG mice to establish meningioma involving the trigeminal nerve. Trigeminal nerve is suitable for histopathologic analysis because it is the largest cranial nerve in rodents. In addition, the integrity of the trigeminal nerve can be evaluated using the corneal reflex and neurotrophic keratopathy.26 The trigeminal nerve lies in the medial part of the temporal fossa and has 3 branches, one of which passes through the foramen ovale; the others are located on the medial side of this foramen (Supplementary Material, Fig. S1C and D). In rodents, the PGF is a natural cavity in the rostral area of the opening of the external acoustic meatus, communicating with the temporal fossa (Supplementary Material, Fig. S1C). Thus, the lateral part of the foramen ovale can be easily accessed using the PGFi technique (Supplementary Material, Fig. S1D).24 The PGFi has technical and anatomical advantages over the conventional implantation technique. The operation time for PGFi is short, requiring ∼1 min. In this study, there were no operation-related complications, and skull base meningiomas involving trigeminal nerves were established in all mice. Of intrigue, IOMM-Lee cells infiltrated trigeminal nerve fibers, mimicking the human meningioma infiltration into cranial nerves. Loss of corneal reflex and neurotrophic keratopathy reflected the trigeminal nerve injury caused by tumor infiltration in this mouse model.
In summary, we established a clinically relevant orthotropic model of unresectable meningioma involving the trigeminal nerve that is suitable for preclinical studies. We have shown that WT1 in meningioma cells is a potential target for immunotherapy. WT1-specific T cells recognized and killed meningioma cells in vitro. They retarded the growth of experimental meningioma and the accompanying progression of cranial nerve damage in vivo. Thus, adoptive transfer of WT1-redirected T cells may be an attractive therapeutic approach for difficult-to-treat meningiomas.
Supplementary Material
Funding
This work was supported by a Grant-in-Aid (B) for Scientific Research from the Japan Society for the Promotion of Science (A.N.).
Conflict of interest statement. None declared.
Supplementary Material
References
- 1.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oka Y, Tsuboi A, Oji Y, Kawase I, Sugiyama H. WT1 peptide vaccine for the treatment of cancer. Curr Opin Immunol. 2008;20(2):211–220. doi: 10.1016/j.coi.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Ohno S, Kyo S, Myojo S, et al. Wilms’ tumor 1 (WT1) peptide immunotherapy for gynecological malignancy. Anticancer Res. 2009;29(11):4779–4784. [PubMed] [Google Scholar]
- 4.Okamoto S, Mineno J, Ikeda H, et al. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res. 2009;69(23):9003–9011. doi: 10.1158/0008-5472.CAN-09-1450. [DOI] [PubMed] [Google Scholar]
- 5.Ochi T, Fujiwara H, Okamoto S, et al. Novel adoptive T-cell immunotherapy using a WT1-specific TCR vector encoding silencers for endogenous TCRs shows marked antileukemia reactivity and safety. Blood. 2011;118(6):1495–1503. doi: 10.1182/blood-2011-02-337089. [DOI] [PubMed] [Google Scholar]
- 6.Rogers L, Gilbert M, Vogelbaum MA. Intracranial meningiomas of atypical (WHO grade II) histology. J Neurooncol. 2010;99(3):393–405. doi: 10.1007/s11060-010-0343-1. [DOI] [PubMed] [Google Scholar]
- 7.Hanft S, Canoll P, Bruce JN. A review of malignant meningiomas: diagnosis, characteristics, and treatment. J Neurooncol. 2010;99(3):433–443. doi: 10.1007/s11060-010-0348-9. [DOI] [PubMed] [Google Scholar]
- 8.Black P, Kathiresan S, Chung W. Meningioma surgery in the elderly: a case-control study assessing morbidity and mortality. Acta Neurochir (Wien) 1998;140(10):1013–1016. doi: 10.1007/s007010050209. discussion 1016–1017. [DOI] [PubMed] [Google Scholar]
- 9.Black PM, Morokoff AP, Zauberman J. Surgery for extra-axial tumors of the cerebral convexity and midline. Neurosurgery. 2008;62(6 suppl 3):1115–1121. doi: 10.1227/01.neu.0000333778.66316.38. discussion 1121–1113. [DOI] [PubMed] [Google Scholar]
- 10.Sanai N, Sughrue ME, Shangari G, Chung K, Berger MS, McDermott MW. Risk profile associated with convexity meningioma resection in the modern neurosurgical era. J Neurosurg. 2010;112(5):913–919. doi: 10.3171/2009.6.JNS081490. [DOI] [PubMed] [Google Scholar]
- 11.Monleón D, Morales JM, Gonzalez-Segura A, et al. Metabolic aggressiveness in benign meningiomas with chromosomal instabilities. Cancer Res. 2010;70(21):8426–8434. doi: 10.1158/0008-5472.CAN-10-1498. [DOI] [PubMed] [Google Scholar]
- 12.Harris AE, Lee JY, Omalu B, Flickinger JC, Kondziolka D, Lunsford LD. The effect of radiosurgery during management of aggressive meningiomas. Surg Neurol. 2003;60(4):298–305. doi: 10.1016/s0090-3019(03)00320-3. discussion 305. [DOI] [PubMed] [Google Scholar]
- 13.Huffmann BC, Reinacher PC, Gilsbach JM. Gamma knife surgery for atypical meningiomas. J Neurosurg. 2005;102(suppl):283–286. doi: 10.3171/jns.2005.102.s_supplement.0283. [DOI] [PubMed] [Google Scholar]
- 14.Stafford SL, Pollock BE, Foote RL, et al. Meningioma radiosurgery: tumor control, outcomes, and complications among 190 consecutive patients. Neurosurgery. 2001;49(5):1029–1037. doi: 10.1097/00006123-200111000-00001. discussion 1037–1028. [DOI] [PubMed] [Google Scholar]
- 15.Larson JJ, van Loveren HR, Balko MG, Tew JM. Evidence of meningioma infiltration into cranial nerves: clinical implications for cavernous sinus meningiomas. J Neurosurg. 1995;83(4):596–599. doi: 10.3171/jns.1995.83.4.0596. [DOI] [PubMed] [Google Scholar]
- 16.Ohminami H, Yasukawa M, Fujita S. HLA class I-restricted lysis of leukemia cells by a CD8(+) cytotoxic T-lymphocyte clone specific for WT1 peptide. Blood. 2000;95(1):286–293. [PubMed] [Google Scholar]
- 17.Makita M, Hiraki A, Azuma T, et al. Antilung cancer effect of WT1-specific cytotoxic T lymphocytes. Clin Cancer Res. 2002;8(8):2626–2631. [PubMed] [Google Scholar]
- 18.Tsuji T, Yasukawa M, Matsuzaki J, et al. Generation of tumor-specific, HLA class I-restricted human Th1 and Tc1 cells by cell engineering with tumor peptide-specific T-cell receptor genes. Blood. 2005;106(2):470–476. doi: 10.1182/blood-2004-09-3663. [DOI] [PubMed] [Google Scholar]
- 19.Lee WH. Characterization of a newly established malignant meningioma cell line of the human brain: IOMM-Lee. Neurosurgery. 1990;27(3):389–395. discussion 396. [PubMed] [Google Scholar]
- 20.Tanaka K, Sato C, Maeda Y, et al. Establishment of a human malignant meningioma cell line with amplified c-myc oncogene. Cancer. 1989;64(11):2243–2249. doi: 10.1002/1097-0142(19891201)64:11<2243::aid-cncr2820641110>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 21.Ishiwata I, Ishiwata C, Ishiwata E, et al. In vitro culture of various typed meningiomas and characterization of a human malignant meningioma cell line (HKBMM) Hum Cell. 2004;17(4):211–217. doi: 10.1111/j.1749-0774.2004.tb00045.x. [DOI] [PubMed] [Google Scholar]
- 22.Neri S, Mariani E, Meneghetti A, Cattini L, Facchini A. Calcein-acetyoxymethyl cytotoxicity assay: standardization of a method allowing additional analyses on recovered effector cells and supernatants. Clin Diagn Lab Immunol. 2001;8(6):1131–1135. doi: 10.1128/CDLI.8.6.1131-1135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito M, Hiramatsu H, Kobayashi K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100(9):3175–3182. doi: 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- 24.Iwami K, Momota H, Natsume A, Kinjo S, Nagatani T, Wakabayashi T. A novel method of intracranial injection via the postglenoid foramen for brain tumor mouse models. J Neurosurg. 2012;116(3):630–635. doi: 10.3171/2011.10.JNS11852. [DOI] [PubMed] [Google Scholar]
- 25.Werner SR, Dotzlaf JE, Smith RC. MMP-28 as a regulator of myelination. BMC Neurosci. 2008;9:83. doi: 10.1186/1471-2202-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrari G, Chauhan SK, Ueno H, et al. A novel mouse model for neurotrophic keratopathy: trigeminal nerve stereotactic electrolysis through the brain. Invest Ophthalmol Vis Sci. 2011;52(5):2532–2539. doi: 10.1167/iovs.10-5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutchings Y, Osada T, Woo CY, Clay TM, Lyerly HK, Morse MA. Immunotherapeutic targeting of Wilms’ tumor protein. Curr Opin Mol Ther. 2007;9(1):62–69. [PubMed] [Google Scholar]
- 28.Sugiyama H. Cancer immunotherapy targeting Wilms’ tumor gene WT1 product. Expert Rev Vaccines. 2005;4(4):503–512. doi: 10.1586/14760584.4.4.503. [DOI] [PubMed] [Google Scholar]
- 29.Cheever MA, Allison JP, Ferris AS, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15(17):5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273(5271):104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 31.Debets R, Willemsen R, Bolhuis R. Adoptive transfer of T-cell immunity: gene transfer with MHC-restricted receptors. Trends Immunol. 2002;23(9):435–436. doi: 10.1016/s1471-4906(02)02290-1. author reply 436–437. [DOI] [PubMed] [Google Scholar]
- 32.Jorritsma A, Gomez-Eerland R, Dokter M, et al. Selecting highly affine and well-expressed TCRs for gene therapy of melanoma. Blood. 2007;110(10):3564–3572. doi: 10.1182/blood-2007-02-075010. [DOI] [PubMed] [Google Scholar]
- 33.Stauss HJ, Cesco-Gaspere M, Thomas S, et al. Monoclonal T-cell receptors: new reagents for cancer therapy. Mol Ther. 2007;15(10):1744–1750. doi: 10.1038/sj.mt.6300216. [DOI] [PubMed] [Google Scholar]
- 34.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66(17):8878–8886. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67(8):3898–3903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas S, Xue SA, Cesco-Gaspere M, et al. Targeting the Wilms tumor antigen 1 by TCR gene transfer: TCR variants improve tetramer binding but not the function of gene modified human T cells. J Immunol. 2007;179(9):5803–5810. doi: 10.4049/jimmunol.179.9.5803. [DOI] [PubMed] [Google Scholar]
- 37.Heemskerk MH, Hagedoorn RS, van der Hoorn MA, et al. Efficiency of T-cell receptor expression in dual-specific T cells is controlled by the intrinsic qualities of the TCR chains within the TCR-CD3 complex. Blood. 2007;109(1):235–243. doi: 10.1182/blood-2006-03-013318. [DOI] [PubMed] [Google Scholar]
- 38.Kustikova OS, Wahlers A, Kuhlcke K, et al. Dose finding with retroviral vectors: correlation of retroviral vector copy numbers in single cells with gene transfer efficiency in a cell population. Blood. 2003;102(12):3934–3937. doi: 10.1182/blood-2003-05-1424. [DOI] [PubMed] [Google Scholar]
- 39.Sadelain M. Insertional oncogenesis in gene therapy: how much of a risk? Gene Ther. 2004;11(7):569–573. doi: 10.1038/sj.gt.3302243. [DOI] [PubMed] [Google Scholar]
- 40.Ragel BT, Couldwell WT, Gillespie DL, Wendland MM, Whang K, Jensen RL. A comparison of the cell lines used in meningioma research. Surg Neurol. 2008;70(3):295–307. doi: 10.1016/j.surneu.2007.06.031. discussion 307. [DOI] [PubMed] [Google Scholar]
- 41.Ragel BT, Elam IL, Gillespie DL, et al. A novel model of intracranial meningioma in mice using luciferase-expressing meningioma cells. Laboratory investigation. J Neurosurg. 2008;108(2):304–310. doi: 10.3171/JNS/2008/108/2/0304. [DOI] [PubMed] [Google Scholar]
- 42.McCutcheon IE, Friend KE, Gerdes TM, Zhang BM, Wildrick DM, Fuller GN. Intracranial injection of human meningioma cells in athymic mice: an orthotopic model for meningioma growth. J Neurosurg. 2000;92(2):306–314. doi: 10.3171/jns.2000.92.2.0306. [DOI] [PubMed] [Google Scholar]
- 43.Baia GS, Dinca EB, Ozawa T, et al. An orthotopic skull base model of malignant meningioma. Brain Pathol. 2008;18(2):172–179. doi: 10.1111/j.1750-3639.2007.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salhia B, Rutka JT, Lingwood C, Nutikka A, Van Furth WR. The treatment of malignant meningioma with verotoxin. Neoplasia. 2002;4(4):304–311. doi: 10.1038/sj.neo.7900243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.